

Abstract
Background
Fungal pathogens of plants produce diverse repertoires of secondary metabolites, which have functions ranging from iron acquisition, defense against immune perturbation, to toxic assaults on the host. The wheat pathogen Zymoseptoria tritici causes Septoria tritici blotch, a foliar disease which is a significant threat to global food security. Currently, there is limited knowledge of the secondary metabolite arsenal produced by Z. tritici, which significantly restricts mechanistic understanding of infection. In this study, we analyzed the genome of Z. tritici isolate IP0323 to identify putative secondary metabolite biosynthetic gene clusters, and used comparative genomics to predict their encoded products.
Results
We identified 32 putative secondary metabolite clusters. These were physically enriched at subtelomeric regions, which may facilitate diversification of cognate products by rapid gene rearrangement or mutations. Comparative genomics revealed a four gene cluster with significant similarity to the ferrichrome-A biosynthetic locus of the maize pathogen Ustilago maydis, suggesting this siderophore is deployed by Z. tritici to acquire iron. The Z. tritici genome also contains several isoprenoid biosynthetic gene clusters, including one with high similarity to a carotenoid/opsin producing locus in several fungi. Furthermore, we identify putative phytotoxin biosynthetic clusters, suggesting Z. tritici can produce an epipolythiodioxopiperazine, and a polyketide and non-ribosomal peptide with predicted structural similarities to fumonisin and the Alternaria alternata AM-toxin, respectively. Interrogation of an existing transcriptional dataset suggests stage specific deployment of numerous predicted loci during infection, indicating an important role of these secondary metabolites in Z. tritici disease.
Conclusions
We were able to assign putative biosynthetic products to numerous clusters based on conservation amongst other fungi. However, analysis of the majority of secondary metabolite loci did not enable prediction of a cluster product, and consequently the capacity of these loci to play as yet undetermined roles in disease or other stages of the Z. tritici lifecycle is significant. These data will drive future experimentation for determining the role of these clusters and cognate secondary metabolite products in Z. tritici virulence, and may lead to discovery of novel bioactive molecules.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3969-y) contains supplementary material, which is available to authorized users.
Keywords: Zymoseptoria tritici, Mycosphaerella graminicola, Secondary metabolism, Gene cluster, Toxin, Siderophore
Background
The fungal pathogen Zymoseptoria tritici (previously Mycosphaerella graminicola) causes Septoria tritci blotch, an important foliar disease of wheat. Average yield losses attributed to this disease range from 5 to 10% annually, which can rise to 50% in some conditions [1]. Approximately 70% of European fungicides are deployed to control Z. tritici [2], which is likely to drive emergence of drug resistance in fungal pathogens of humans [3].
The infectious propagules for Z. tritici disease are multicellular, haline pycnidiospores (asexual) or ascospores (sexual), which are dispersed via rain splash or wind. After germination on the leaf surface, polarized hyphae enter the mesophyll through stomatal openings within 12–24 h [4], followed by a 9–16 day asymptomatic phase with very limited fungal growth. Subsequently, there is rapid onset of host cell death, a dramatic increase in fungal biomass, and finally production of spore-bearing reproductive structures (pycnidia) in necrotic plant tissue [5–7]. It was assumed that during the initial asymptomatic phase, a biotrophic interaction occurs, where the fungus acquires nutrients from the host tissue, and consequently, Z. tritici has been considered a hemibiotroph (i.e. that the lifecycle consists of both biotrophic and necrotrophic phases). Recent transcriptional profiling [6] has challenged the notion of fungal nutrient acquisition during the asymptomatic phase, suggesting nutrient acquisition predominantly occurs from dead host tissue, and consequently Z. tritici may more accurately be classified as a latent necrotroph [7, 8].
With regards to the molecular basis of Z. tritici disease, recent work has strongly suggested Z. tritici utilizes effector proteins to orchestrate infection. Fungal effectors are small molecular weight, cysteine rich, secreted proteins that manipulate or subvert host immunity. Sequencing of the Z. tritici genome revealed hundreds of genes predicted to encode fungal effectors [9], and transcriptional profiling has revealed phase-specific deployment of numerous putative effectors throughout infection, notably during the switch from asymptomatic to necrotrophic stages [6]. Functional characterization has revealed that secreted proteins protect Z. tritici pathogen associated molecular patterns from host recognition [10]. Additionally, the small secreted protein encoded by the AvrStb6 gene is recognized by wheat cultivars carrying the Stb6 resistance gene [7]. Such gene-for-gene interactions are a product of an evolutionary arms race between pathogen and host, resulting in either effector mediated disease susceptibility or effector triggered immunity. Taken together, these data indicate that small secreted effectors are likely a critical component of the Z. tritici virulence arsenal, and much work has been invested in functional analysis of these genes and their encoded products [7, 10–12].
In addition to secreted effectors, plant infecting fungi also utilize a diverse range of secondary metabolites during disease and other lifecycle stages. However, Z. tritici secondary metabolites remain largely unexplored, even though they may play crucial roles in the molecular basis of infection. Pathogens from the Dothideomycetes class are known to produce numerous secondary metabolite phytotoxins. For example, the HC-toxin produced by the maize pathogen Cochliobolus carbonum is a non-ribosomal peptide that functions as a histone deacetylase inhibitor, which enables pathogen modification of host chromatin structure and gene expression, and ultimately causes host cell death [13]. The AM-toxin produced by Alternaria alternata apple pathotype targets plasma membranes and chloroplast function, and is necessary for disease in susceptible cultivars [14]. In addition to toxins, secondary metabolites can play diverse roles during disease [15]. This includes structural functions, for example melanins in condial cell walls, as well as iron acquisition by siderophores, or modulation of host responses by production of plant hormones.
Most fungal secondary metabolites are produced by biosynthetic gene clusters, consisting of key enzymes, such as polyketide synthases (PKSs) or non-ribosomal peptide synthetases (NRPSs), and contiguous genes encoding tailoring enzymes and transporters [16]. Following a drastic increase in the number of available fungal genome sequences and improved bioinformatics tools, it is now possible to postulate the biosynthetic product of some clusters in silico. Previously, these in silico approaches have often relied solely on homology between key enzymes. For example, a Z. tritici gene predicted to encode a PKS (Mycgr3g96592) was hypothesized to catalyze the first step in the biosynthesis of the toxin dothistromin [17]. However, this gene has recently been demonstrated to be involved in melanin biosynthesis using quantitative trait locus mapping [18], highlighting the limitations of approaches based exclusively on key genes. A new approach for in silico predictions of secondary metabolite products is based on MultiGene Basic Local Alignment Search Tools (BLASTS), which identify significant conservation of gene cluster loci across the fungal kingdom based on conservation of gene architecture for both key biosynthetic and tailoring genes [19]. Where significant conservation is identified between a predicted cluster in a genome of interest, and a second locus for which the secondary metabolite has been experimentally defined in another species, it is possible to postulate a comparable biosynthetic product [20].
In this study we conducted in silico analysis of Z. tritici secondary metabolite loci. We firstly used existing bioinformatics pipelines to predict secondary metabolite loci in the sequenced isolate IP0323 [9], identifying 32 putative clusters. These loci are enriched at chromosome subtelomeres, and often contain genes encoding putative metabolite efflux proteins, strongly suggesting a diverse range of secreted metabolites. Subsequently, we used MultiGeneBLASTs to predict biosynthetic products of various clusters, and postulate roles of these metabolites during the Z. tritici lifecycle based on existing experimentation in other pathogenic fungi. Finally, we analyzed existing transcriptomic datasets for Z. tritici to study expression profiles of the predicted gene clusters during infection.
Methods
The Z. tritici IP0323 genome [9] was retrieved from Genbank (accession number GCA_000219625.1). Secondary metabolite clusters were predicted using AntiSMASH [21] and SMURF [22] based domain searchers. 34 clusters predicted by identification of genes encoding putative biosynthetic enzymes (e.g. polyketide synthases, nonribosomal peptide synthases, or geranylgeranyl diphosphate synthases) and associated genes were identified, which were refined to a total of 32 following manual interrogation of gene content.
A MultiGeneBLAST algorithm [19] was used to detect homologous clusters across all Genbank genomes [23]. MultiGeneBLAST architecture searches were carried out as described previously [20], with the percent identity threshold set to 25%, the synteny weight set to 0 and the maximum intergenic distance set to 110% of the span of the corresponding Z. tritici cluster with a minimum bound of 25 kb. Other parameters were set to default. The input for each search was a multiFASTA file of the amino acid sequences of proteins predicted to be encoded by the genes in the region of the Z. tritici predicted cluster, erring on the side of AntiSMASH over-inclusion to detect cluster boundaries. Amino acid sequences were received from Ensembl database [24]. Both input and output files for MultiGeneBLAST are given in Additional file 1.
Subtelomeric regions were defined within 300 kb of the chromosome end, an approach used in other analyses of filamentous ascomycetes [25]. Any predicted clusters with one or more genes residing at these loci were considered subtelomeric (Additional file 2).
For assessment of gene cluster co-expression, we mined an existing RNA seq dataset [6] which previously profiled Z. tritici gene expression from two in vitro conditions (growth on potato dextrose or Czapek- Dox broth) and during key stages of disease (1, 4, 9, 14, 21 post infection). Only genes with detectable transcripts at all time points were considered. Average fragments per kilobase for each gene per million fragments (FPKM) at each timepoint ([6], Additional file 3) were analyzed using the FunGeneClusteRs programme using default parameters [26]. Only clusters with genes encoding 3 or more co-expressed transcripts were considered co-regulated. This analysis identified a total of 397 genes residing in 99 contiguous clusters, which were then mapped to the predicted secondary metabolite loci (Additional file 3).
Predicted paralogues for putative secondary metabolite genes were retrieved from the Ensemble database, which were limited to same-species paralogies [27]. Only paralogues that also resided in secondary metabolite loci were further analyzed.
Results and discussion
The Z. tritici genome contains 32 putative secondary metabolite clusters that are enriched at subtelomeric loci
Analysis of the Z. tritici genome identified numerous putative secondary metabolite clusters containing a predicted 682 genes (Table 1 and Additional file 4). When comparing both AntiSMASH and SMURF genome analysis pipelines (Table 1), AntiSMASH resulted in a greater number of clusters (33 vs 19), containing a greater number of total genes (669 vs 143) respectively, which is consistent with other studies [28]. SMURF identified only a single cluster that was not predicted by AntiSMASH (cluster 13, Table 1). While it is likely that these approaches overestimate the number of genes which are resident in secondary metabolite biosynthetic clusters [20], we did not manually curate cluster boundaries for two reasons. Firstly, we reasoned that for subsequent MultiGeneBLAST analysis, large cluster boundaries would maximize the chance of identifying homologous clusters from other species with a defined biosynthetic product. Secondly, fungal genomes are known to contain secondary metabolite super clusters composed of >50 genes [29].
Table 1.
Predicted secondary metabolite loci in Z. tritici
| Cluster number | Secondary metabolite class | Predicted key biosynthetic gene(s) | Predicted transporter | Resident transcription factor | Subtelomeric | Evidence of transcriptional co-expression? | AntiSMASH | SMURF | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Cluster boundary | No. of genes | Cluster boundary | No. of genes | |||||||
| 1 | NRPS-Like | Mycgr3G107072 | YES | NO | NO | NO | 1:2,172,553:2,212,738 | 12 | 1:2,189,682:2,216,244 | 8 |
| 2 | Transporter | Na | YES | NO | NO | NO | 1:2,306,586:2,310,597 | 2 | Na | Na |
| 3 | Terpene | Mycgr3G34236 | NO | NO | NO | YES | 1:2,366,583:2,438,578 | 28 | Na | Na |
| 4 | Terpene | Mycgr3G33174 | YES | NO | NO | NO | 1:3,518,416:3,535,837 | 9 | Na | Na |
| 5 | PKS | Mycgr3G83965 | NO | NO | NO | NO | 1:3,821,874:3,860,702 | 14 | Na | Na |
| 6 | NRPS | Mycgr3G16590 | YES | YES | NO | NO | 1:5,128,643:5,192,491 | 23 | Na | Na |
| 7 | PKS | Mycgr3G67477 | YES | YES | NO | YES | 1:5,513,274:5,566,181 | 17 | 1:5,524,934:5,578,932 | 20 |
| 8 | PKS | Mycgr3G9788 | YES | YES | YES | YES | 2:31,258:149,728 | 37 | 2:123,001:157,676 | 11 |
| 9 | NRPS | Mycgr3G19958 | NO | NO | YES | NO | 2:222,377:310,014 | 19 | 2:260,654:284,183 | 5 |
| 10 | NRPS | Mycgr3G36951 | YES | NO | NO | NO | 2:420,337:471,819 | 14 | 2:433,742:453,243 | 2 |
| 11 | NRPS | Mycgr3G90558 | YES | NO | NO | NO | 2:1,008,626:1,058,615 | 13 | 2:1,026,842:1,052,480 | 6 |
| 12 | Terpene | Mycgr3G99148 | NO | YES | NO | YES | 2:1,934,751:2,046,004 | 31 | Na | Na |
| 13 | PKS-Like | Mycgr3G39149 | YES | NO | YES | YES | Na | Na | 3:3,181,477:3,213,160 | 13 |
| 14 | NRPS | Mycgr3G39931, Mycgr3G40534 | YES | NO | YES | NO | 4:12,877:43,949 | 7 | Na | Na |
| 15 | PKS | Mycgr3G100089 | YES | YES | YES | YES | 5:85,463:149,465 | 22 | 5:94,206:129,292 | 12 |
| 16 | NRPS-Like | Mycgr3G93235, Mycgr3G100227 | NO | YES | NO | YES | 5:1,291,545:1,380,182 | 31 | 5:1,309,417:1,324,422 | 4 |
| 17 | PKS | Mycgr3G72709 | YES | YES | YES | YES | 6:72,339:170,251 | 36 | 6:106,974:121,853 | 4 |
| 18 | NRPS | Mycgr3G72768 | YES | NO | YES | YES | 6:283,399:352,537 | 24 | 6:310,922:346,260 | 11 |
| 19 | NRPS | Mycgr3G44313 | NO | NO | NO | YES | 6:1,808,394:1,845,239 | 16 | 6:1,821,703:1,841,259 | 9 |
| 20 | NRPS | Mycgr3G109989 | YES | YES | NO | NO | 6:2,243,873:2,278,779 | 9 | Na | Na |
| 21 | PKS | Mycgr3G45348 | YES | NO | YES | NO | 7:2,566,479:2,608,068 | 12 | 7:2,577,096:2,606,813 | 8 |
| 22 | NRPS | Mycgr3G110642 | YES | NO | YES | NO | 8:2,346,378:2,374,211 | 8 | 8:2,352,913:2,362,665 | 2 |
| 23 | NRPS-Like | Mycgr3G75370 | YES | YES | NO | YES | 9:483,924:714,190 | 65 | Na | Na |
| 24 | PKS | Mycgr3G47832 | YES | YES | NO | NO | 9:1,474,679:1,516,611 | 12 | 9:1,494,054:1,506,383 | 3 |
| 25 | Terpene | Mycgr3G76129 | YES | YES | NO | NO | 10:341,276:419,193 | 29 | Na | Na |
| 26 | Hybrid PKS- NRPS | Mycgr3G62978 | YES | NO | NO | YES | 10:425,702:485,734 | 20 | 10:439,622:460,865 | 6 |
| 27 | PKS | Mycgr3G101493 | YES | NO | YES | YES | 10:1,274,562:1,480,511 | 54 | 10:1,436,111:1,437,155 | 7 |
| 28 | NRPS-Like | Mycgr3G49555 | NO | NO | NO | YES | 11:393,046:429,601 | 12 | 11:409,058:423,584 | 5 |
| 29 | PKS | Mycgr3G96592 | NO | YES | NO | NO | 11:566,694:611,143 | 14 | Na | Na |
| 30 | NRPS-Like | Mycgr3G50095 | YES | YES | NO | NO | 11:823,532:917,882 | 30 | 11:849,800:868,675 | 7 |
| 31 | NRPS | Mycgr3G96900 | YES | NO | YES | YES | 12:157,368:195,467 | 15 | Na | Na |
| 32 | NRPS-Like | Mycgr3G77312 | NO | NO | NO | NO | 12:434,096:471,899 | 9 | Na | Na |
| 33 | PKS | Mycgr3G51018 | NO | NO | NO | YES | 13:656,107:712,077 | 17 | Na | Na |
| 34 | Terpene | Mycgr3G101921, Mycgr3G12838 | YES | NO | YES | NO | 13:1,116,277:1,139,970 | 8 | Na | Na |
AntiSMASH and SMURF analysis pipelines were used to predict secondary metabolite clusters in Z. tritici isolate IP0323, identifying a total of 34 putative loci. Ensembl gene identifiers for predicted key biosynthetic genes identified by these analyses are given. Clusters were numbered in ascending numerical order based on their respective location in the Z. tritici genome. In order to identify genes encoding ABC or MFS transporters at each locus, genes were interrogated based on GO-terms GO:0055052 (ATP-binding cassette transporter complex) and GO:0055085 (transmembrane transporter). Transcription factors were identified by screening genes for GO-terms GO:0003677 (DNA binding) and GO:0003700 (transcription factor activity). Subtelomeric clusters were defined as any loci with predicted genes residing within 300 kb of the chromosome end. Interrogation of each individual cluster suggests the key gene of cluster 13 encodes a ketoacyl synthase domain-containing protein, and consequently is likely involved in fatty acid synthesis. Cluster 2 consists of two transport proteins, and lacks a gene encoding a putative key biosynthetic enzyme
Genes predicted to encode key biosynthetic enzymes resident in each cluster include 10 NRPSs, 6 NRPS-like enzymes, 10 PKSs and 1 hybrid PKS-NRPS (Table 1). Further interrogation of individual clusters suggested that the PKS of cluster 13 is actually a ketoacyl synthase domain-containing protein, and consequently is probably involved in fatty acid synthesis rather that production of a secondary metabolite. Our analysis also identified an additional 2 gene locus containing genes encoding ferric reductase like transmembrane transporters (cluster 2), which lacked any key biosynthetic genes. Consequently, clusters 2 and 13 are unlikely to be involved in secondary metabolite biosynthesis, giving a total of 32 predicted secondary metabolite biosynthetic clusters in the Z. tritici IP0323 genome. We did not identify any dimethylallyl tryptophan synthases (DMATs) which suggests Z. tritici does not produce any DMAT derived alkaloids (e.g. ergot alkaloid [30]). However, we identified 5 clusters with putative roles in isoprenoid biosynthesis based on the presence of genes predicted to encode geranylgeranyl diphosphate synthases and other key enzymes (Table 1).
The majority of the 32 putative clusters (n = 22) contain genes encoding a predicted ATP-binding cassette (ABC) transporter and/or major facilitator superfamily (MFS) transporter (Table 1 and Additional file 5). These are the main classes of transporters responsible for secondary metabolite efflux from fungal cells [31], and are often contiguously clustered with genes necessary for product biosynthesis [32, 33]. Our data therefore suggests that the products of these 22 gene clusters are extracellular, and consequently may biosynthesize molecules which mediate host-pathogen interactions during infection.
We identified 13 clusters that contain a predicted transcription factor (Table 1 and Additional file 6). Regulation of secondary metabolite gene expression in filamentous fungi is an multifaceted integrated system composed of epigenetic regulators, such as the velvet complex, which function at the level of chromatin remodeling, global transcription factors, including StuA and PacC, which link gene expression to development or environmental changes, and cluster-specific transcription factors which are physically located in respective clusters and control transcription of contiguous biosynthetic genes [34]. The velvet complex has been demonstrated to regulate secondary metabolism in Z. tritici [35], and our analyses has identified several genes that are likely important components for comprehensive understanding of cluster regulation in this pathogen.
With regards to physical distribution on the Z. tritici chromosomes (Fig. 1), all 32 clusters are located on core chromosomes [9], with 11 located at subtelomeric loci, representing 34% of the predicted clusters, and containing 36% of the putative secondary metabolite genes (Table 1). Given that the total number genes residing at subtelomeric loci in Z. tritici is 2501, or 22.8% of the genome (Additional file 2), our analysis suggests that subtelomeres and telomere proximal regions are enriched with secondary metabolite gene clusters, an observation consistent with the genomes of other ascomycetes [25, 36]. Subtelomeres of filamentous fungi are rich in repeat regions and transposable elements, and consequently undergo frequent chromosomal rearrangements. Additionally, repeat regions can lead to DNA polymerase ‘slippage’, resulting in elevated mutations in gene coding sequences when compared to telomere distal regions [37]. This had led to the duplication, diversification and differential gene loss (DDL) hypothesis, which suggests that subtelomeres are important for rapid evolution, gene expansion, and niche adaptation [38]. DDL events at subtelomeric secondary metabolite loci might result in novel biosynthetic products that could conceivably enhance Z. tritici virulence or expand pathogen host range. We therefore interrogated gene duplication amongst putative Z. tritici secondary metabolite loci in order to test if these events are more frequent at subtelomeres. We identified 72 genes residing in predicted biosynthetic gene clusters that had at least one or more paralogues at other secondary metabolite loci (Fig. 1 and Additional file 7). In support of the DDL hypothesis, 44% of these genes (n = 32) resided in subtelomeric loci, which represents enrichment of gene duplication relative to the total amount of secondary metabolite genes that are found at these loci (i.e. 36%). In several instances, local gene duplication events were found within specific subtelomeric clusters (Additional file 8). Other studies have also supported DDL in Z. tritici. For example, gene diversification is evidenced by the discovery of the avirulence gene AvrStb6, which resides in the subtelomere of chromosome 5, and encodes a secreted effector with numerous single nucleotide polymorphisms among tested isolates [7]. Additionally, PKS genes from clusters 7 and 8 (Table 1) are absent in certain field isolates, indicating gene loss also occurs at telomere proximal and subtelomeric loci [39]. Consequently, Z. tritici secondary metabolite clusters identified in this study are likely undergoing DDL processes, which may be more frequent at subtelomeric loci. Ultimately, this may result in modification or loss of cognate secondary metabolite products, which could result in enhanced virulence or expand pathogen cultivar or host range.
Fig. 1.

Physical distribution of predicted secondary metabolite clusters on the Z. tritici genome. Putative loci on the 13 Z. tritici core chromosomes are shown, and no clusters were predicted on the 8 dispensable chromosomes. Manual interrogation of clusters 2 and 13 demonstrated they are unlikely to biosynthesize a secondary metabolite (see main text), resulting in a total of 32 putative secondary metabolite clusters. Line boxes indicate boundaries of putative secondary metabolite cluster as predicted by AntiSMASH. Numbers in parentheses are the cluster number (Table 1). Shaded regions are loci that demonstrate evidence of gene co-regulation in transcriptional profiling. Red lines below secondary metabolite loci indicate regions containing genes that have one or more paralogues that also reside within a predicted biosynthetic gene cluster
In order to postulate putative cluster products from the identified loci we conducted MultiGeneBLAST analyses of all clusters across the genomes present in the Genbank archive. This identified several clusters with high homology to loci in other fungi for which the biosynthetic product has been experimentally determined, allowing us to predict several metabolites that are produced by Z. tritici.
Putative ferrichrome A biosynthetic gene cluster
We identified a putative cluster (number 14, Table 1, Fig. 2a) which had significant similarity to the ferrichrome A biosynthetic locus (a total of 4 conserved genes) found in the basidiomycete pathogen of maize, Ustilago maydis [40]. Many fungi utilize small molecular weight, high-affinity iron-chelating NRPs termed siderophores for both internal iron storage and uptake from the external environment [41]. The most common fungal siderophore types are of the hydroxamate class, and include ferrichromes, coprogens or fusarinines.
Fig. 2.
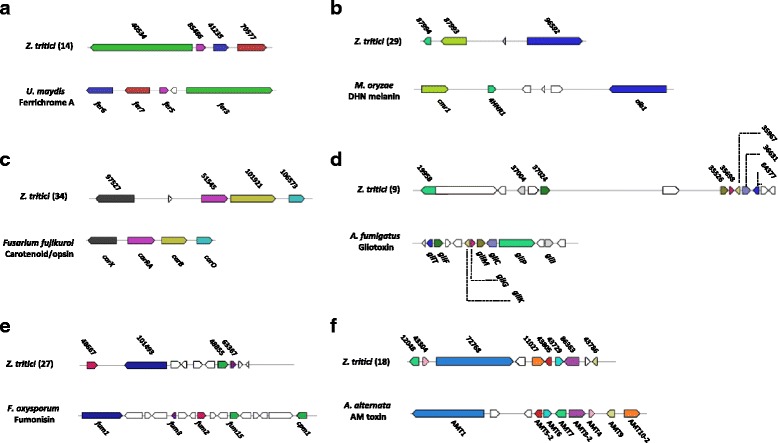
Schematic representation of conserved gene architecture between predicted Z. tritici loci and clusters from other fungi (a-f). Predicted gene boundaries are depicted by arrows and assigned Ensemble identifiers (Z. tritici) or gene names (other species). Identical colors between top and bottom loci are indicative of significant similarity at the level of encoded amino acid sequence (see Table 2). Non-colored arrows are predicted genes that lack an orthologue at the other respective locus. Numbers in parentheses are Z. tritici cluster number (see Table 1)
In U. maydis, ferrichrome A production firstly requires synthesis of the precursor hydroxymethylglutaryl-CoA (HMG-CoA), by the HMG-CoA synthase Hcs1 [40]. Z. tritici also contains an orthologue of this gene (Mycgr3G54740, Table 2), and as in U. maydis, this gene is not part of the contiguous cluster. Comparable architecture between the U. maydis ferrichrome A biosynthetic gene cluster [40] and cluster 14 identified in this study included genes encoding the NRPS Fer3 and acylase Fer5, both of which are essential for production of this siderophore (Table 1 and Fig. 2a). Additionally, we found genes predicted to encode a multidrug resistance protein (fer6) and a siderophore transporter (fer7) which currently have uncharacterized functions but are co-regulated during iron perturbation in U. maydis [40, 42]. Interestingly, the enoyl-CoA reductase encoding gene fer4, which is essential for biosynthesis of this metabolite in U. maydis, is not present in the putative Z. tritici cluster, although there are orthologues of this gene at other genomic loci (e.g. Mycgr3G76805, Table 2), an observation that may reflect DDL events due to this clusters subtelomeric locus. These data suggest that despite this deviation in cluster architecture between these species, the Z. tritici genome still contains the necessary gene repertoire for biosynthesis of a ferrichrome A-like siderophore.
Table 2.
BLAST analyses of predicted Z. tritici secondary metabolite loci across the Genbank sequence archive
| Putative cluster in Z. tritici | Hit cluster with experimentally defined biosynthetic product | BLAST Results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Putative product (cluster number) | Ensembl Gene ID | GenBank ID | Annotation in Z. tritici | Species Name | Ensembl Gene ID | GenBank ID | Annotation | % similarity | Sequence coverage | E value |
| Siderophore (14) | Mycgr3G40534 | EGP88586 | Non-ribosomal peptide synthetase | Ustilago maydis | UMAG_01433 | 23562457 | Ferrichrome siderophore peptide synthetase fer3 | 48% | 82% | 8,00E-61 |
| Mycgr3G85486 | EGP87766 | putative siderophore biosynthesis protein | UMAG_01432 | 23562456 | Putative lysine N-acyltransferase fer5 | 43% | 89% | 8,00E-103 | ||
| Mycgr3G41235 | EGP87768 | putative ABC transporter | UMAG_01431 | 23562455 | Multidrug resistance-associated protein 1 fer6 | 42% | 100% | 0,00E + 00 | ||
| Mycgr3G70577 | EGP87767 | putative siderophore-dependent iron transporter | UMAG_01439 | 23562463 | Siderophore iron transporter 3 fer7 | 39% | 93% | 3,00E-114 | ||
| (Mycgr3G76805) | Hypothetical protein | UMAG_01433 | putative enoyl-CoA hydratase/isomerase fer4 | 39% | 83% | 2e-46 | ||||
| (Mycgr3G5470) | HMG-CoA synthase | (UMAG_05362) | HMG-CoA synthase | 54% | 98% | 3,00E-180 | ||||
| DHN melanin (29) | Mycgr3G87993 | EGP83310 | Hypothetical protein | Magnaporthe oryzae | MGG_07215 | EHA55622 | Transcription factor cmr1 | 40% | 100% | 0,00E + 00 |
| Mycgr3G87994 | EGP83311 | Hypothetical protein | MGG_07216 | EHA55623 | Versicolorin reductase 4HNR | 48% | 97% | 2,00E-73 | ||
| Mycgr3G96592 | EGP83620 | Polyketide synthase | MGG_07219 | EHA55627 | Conidial pigment polyketide synthase alb1 | 45% | 100% | 0,00E + 00 | ||
| Carotenoid (34) | Mycgr3G97527 | EGP82655 | Hypothetical protein | Fusarium fujikuroi IMI 58289 | FFUJ_11801 | CCT75764 | related to lignostilbene alphabeta-dioxygenase I | 57% | 90% | 0,00E + 00 |
| Mycgr3G51545 | EGP82649 | Hypothetical protein | FFUJ_11802 | CCT76070 | probable geranylgeranyl-diphosphate geranylgeranyltransferase | 53% | 99% | 0,00E + 00 | ||
| Mycgr3G101921 | EGP82650 | Hypothetical protein | FFUJ_11803 | CCT75765 | probable phytoene dehydrogenase AL-1 | 58% | 96% | 0,00E + 00 | ||
| Mycgr3G106573 | EGP82651 | Hypothetical protein | FFUJ_11804 | CCT75766 | related to HSP30 heat shock protein Yro1p | 68% | 97% | 0,00E + 00 | ||
| (Mycgr3G72713) | Hypothetical protein | (FFUJ_07962) | AM418467.1 | Torulene oxygenase | 41% | 95% | 2,00E-130 | |||
| ETP (9) | Mycgr3G19958 | EGP89696 | Hypothetical protein | Aspergillus fumigatus | AFUA_6G09660 | EAL88817 | NRPS GliP | 28% | 100% | 1,00E-146 |
| Mycgr3G37004 | EGP89694 | aminocyclopropane-1-carboxylate synthase-like protein | AFUA_6G09640 | EAL88815 | Aminotransferase gliI | 34% | 96% | 4,00E-61 | ||
| Mycgr3G37024 | EGP90779 | putative P450 monooxygenase | AFUA_6G09730 | EAL88824 | cytochrome P450 oxidoreductase GliF | 51% | 97% | 3,00E-168 | ||
| Mycgr3G35526 | EGP90781 | putative O-methyltransferase | AFUA_6G09680 | EAL88819 | O-methyltransferase GliM | 42% | 90% | 2,00E-102 | ||
| Mycgr3G35698 | EGP90782 | putative glutathione S-transferase | AFUA_6G09690 | EAL88820 | glutathione S-transferase GliG | 41% | 87% | 7,00E-55 | ||
| Mycgr3G35967 | EGP89693 | gliotoxin biosynthesis protein | AFUA_6G09700 | EAL88821 | gliotoxin biosynthesis protein GliK | 35% | 96% | 1,00E-41 | ||
| Mycgr3G36631 | EGP90783 | putative P450 monooxygenase | AFUA_6G09670 | EAL88818 | Cytochrome P450 oxidoreductase gliC | 34% | 96% | 7,00E-94 | ||
| Mycgr3G84377 | EGP89692 | putative pyridine nucleotide-disulfide oxidoreductase | AFUA_6G09740 | EAL88825 | Thioredoxin reductase gliT | 44% | 99% | 3,00E-82 | ||
| Fumonisin (27) | Mycgr3G101493 | EGP83950 | Polyketide synthase | Fusarium oxysporum FRC O-1890 | ACB12550 | Fum1 polyketide synthase | 36% | 87% | 0,00E + 00 | |
| Mycgr3G63387 | EGP84006 | Hypothetical protein | ACB12552 | Fum3 cytochrome p450 | 52% | 90% | 8,00E-110 | |||
| Mycgr3G48687 | EGP84002 | putative P450 monooxygenase | ACB12551 | Fum2 cytochrome p450 | 36% | 97% | 2,00E-79 | |||
| Mycgr3G48855 | EGP84005 | putative P450 monooxygenase | ACB12560 | Fum15 cytochrome p450 | 28% | 100% | 4,00E-59 | |||
| Mycgr3G48855 | EGP84005 | putative P450 monooxygenase | ACB12565 | Cpm1 cytochrome p450 | 38% | 98% | 3,00E-118 | |||
| AM-toxin (18) | Mycgr3G72768 | EGP86944 | Hypothetical protein | Alternatia alternata | BAI44759 | nonribosomal peptide synthetase AMT1 | 40% | 84% | 0,00E + 00 | |
| Mycgr3G43805 | EGP86331 | Hypothetical protein | BAI44762 | similar to branched-chain-amino-acid AMT5-2aminotransferase | 55% | 98% | 5,00E-31 | |||
| Mycgr3G43729 | EGP86946 | Hypothetical protein | BAI44741 | similar to 3-isopropylmalate dehydrogenase AMT6 | 60% | 95% | 3,00E-150 | |||
| Mycgr3G12048 | EGP86333 | Hypothetical protein | BAI44764 | similar to 2-isopropylmalate synthase AMT7 | 58% | 95% | 0,00E + 00 | |||
| Mycgr3G86363 | EGP86330 | Hypothetical protein | BAI44765 | aconitase family protein | 58% | 100% | 0,00E + 00 | |||
| Mycgr3G43304 | EGP86943 | Hypothetical protein | BAI44766 | thioesterase domain containing protein AMT4 | 38% | 89% | 2,00E-43 | |||
| Mycgr3G43786 | EGP86329 | Hypothetical protein | BAI44767 | similar to methyltransferase AMT9 | 28% | 100% | 2,00E-13 | |||
| Mycgr3G11027 | EGP86945 | Hypothetical protein | BAI44768 | nonribosomal peptide synthetase AMT10–2 | 44% | 100% | 0,00E + 00 | |||
MultiGeneBLAST analyses of predicted cluster loci (Table 1) were conducted across all available sequences in the Genbank archive (Clark et al. 2016). Loci with statistically significant similarity to Z. tritici query sequences were manually interrogated to identify biosynthetic gene clusters for which the secondary metabolite product has been experimentally confirmed. Results for statistically significant BLAST alignment between predicted amino acid sequences encoded by genes at each locus are reported. Gene identifiers in parentheses are not contiguously located at each respective cluster, but are necessary for biosynthesis of the experimentally confirmed product (see main text for details). These genes were identified by individual protein BLAST searches between respective genomes using the NCBI BLAST tool. Both Genbank and Ensembl gene identifiers are given where available. Gene annotations were retrieved from Ensembl, or where unavailable from Genbank
Residence of several transporters within Z. tritici cluster 14 (Mycgr3G99766, Mycgr3G41235) suggests that the putative siderophore product is also likely to be extracellular. In U. maydis, localization studies using fluorescently labeled siderophore analogs suggests ferrichrome A may function as both an extracellular and intracellular siderophore for iron scavenging and storage respectively [43]. Given this dual role in U. maydis, the putative ferrichrome A-like NRP produced by this locus in Z. tritici may also function as an extra and intracellular siderophore.
Our data suggests that Z. tritici has an unusual iron scavenging and/or storage strategy when compared to many pathogenic Dothideomycetes, which use the intracellular siderophore ferricrocin for iron storage and the extracellular siderophore triacetyl fusarine C (TAFC) for iron acquisition from the external environment [44, 45]. Indeed, previous comparative genomic analyses of 18 Dothideomycetes fungi revealed that Z. tritici is unique in lacking orthologues of the NRPS encoding genes required for ferricrocin and TAFC biosynthesis [46]. Our analysis supports these data, as we did not identify putative biosynthetic clusters for siderophores ferricrocin or TAFC. With regards to postulating a biological explanation for the lack of common Dothideomycetes siderophores in Z. tritici, it has recently been suggested that these structurally conserved, extracellular molecules may function as pathogen associated molecular patterns (PAMPs) which are recognized by host pattern recognition receptors (PRRs), resulting in subsequent activation of immune responses. Indeed, bacterial ferritin, and more recently fungal coprogen, have been demonstrated to activate host immunity in Arabidopsis thaliana and maize, respectively [47, 48]. Consequently, the absence of a TAFC biosynthetic gene cluster in Z. tritici may confer a selective advantage due to avoidance of host immune activation.
Putative dihydroxynaphthalene melanin biosynthetic gene cluster
Our analyses identified a 14 gene cluster (cluster number 29, Table 1) containing a 3 gene sub-cluster which had significant similarity to putative or experimentally confirmed dihydroxynaphthalene (DHN) melanin biosynthetic gene clusters in numerous plant pathogens, including Cochliobolus heterostrophus, Bipolaris maydis, Leptosphaeria maculans, and Magnaporthe oryzae (Fig. 2b). This is the best studied secondary metabolite locus in Z. tritici, and our data is consistent with recent work by Lendenmann and colleagues, who used quantitative trait locus mapping to suggest that several genes required for DHN melanin biosynthesis reside at this locus [18]. Cluster architecture analysis identified a PKS encoding gene (Mycgr3G96592) with significant similarity to the M. oryzae alb1 (Table 2, a total of 3 conserved genes). In the DHN melanin polyketide biosynthetic pathway this PKS synthesizes 1,3,6,8 tetrahydroxynaphthalene (1,3,6,8-THN) from acetyl-CoA and malonyl-CoA precursors [49]. Additionally, an orthologue for the M. oryzae tetrahydroxynaphthalene melanin reductase (4HNR, Table 2) required for reduction of 1,3,6,8-THN to form scytalone [50] is also present in this cluster (Fig. 2b and Table 2). Finally, the cluster also contains a gene encoding an orthologue of the transcription factor cmr1 (Table 2, Fig. 2b), which regulates melanin production in several pathogenic fungi [51]. Interestingly, ∆cmr1 strains in M. oryzae and Colletotrichum lagenarium were able to produce melanin in appressoria but not vegetative hyphae [51], and consequently we speculate this transcription factor may also regulate development and melanization in Z. tritici. Our analysis did not identify clusters responsible for the biosynthesis of other melanins in fungi, such as pyomelanin [52], suggesting that DHN-melanin might be the only melanin biosynthesized by this pathogen. This is consistent by work by Beltrán-García and colleagues, who demonstrated DHN-melanin was the only melanin in mycelium and culture filtrate of the closely related pathogen of banana Mycosphaerella fijiensis [53].
With regards to postulating a function of Z. tritici DHN melanin, in other fungi this molecule provides protection from various environmental stresses, such as antifungal agents, UV light, heavy metals, extreme temperatures and reactive oxygen species [54–56]. DHN melanin also plays diverse roles in fungal virulence. In M. oryzae, it is required for the high osmotic pressure in appressoria and consequently host penetration [57]. In the fungal pathogen of humans, Aspergillus fumigatus, DHN melanin inhibits acidification of phagolysosomes [58], and may shield pathogen associated molecular patterns from host pattern recognition receptors [59]. In addition to structural or defensive roles during fungal disease, recent work in M. fijiensis demonstrated that DHN-melanin generates highly reactive oxygen species that may facilitate host cell death [53]. We therefore predict that this cluster has important implications for Z. tritici infection.
Putative carotenoid and opsin biosynthetic gene cluster
Our analysis also identified a putative carotenoid biosynthetic gene cluster in Z. tritici (cluster 34, Table 1), which is highly conserved in a variety of plant pathogens, including Botryotinia fuckeliana, L. maculans, C. heterostrophus, and Fusarium fujikuroi, amongst others. Carotenoid pigments may protect fungi from UV stress, and are also important intermediates for the biosynthesis of physiologically active apocarotenoids, such as retinal [60]. This cluster has been well characterized in F. fujikuroi (Fig. 2c, a total of 4 conserved genes), and contains genes encoding a bifunctional phytoene synthase/carotene cyclase (carRA) and a phytoene dehydrogenase (carB), which are required for biosynthesis of the red carotenoid torulene [61, 62]. Both these genes are conserved in cluster 34 (Fig. 2c), suggesting that this pigment may be part of the secondary metabolite content of Z. tritici, which may account for the light red/pink color of Z. tritici conidia when grown on rich agar. In subsequent secondary metabolic steps in both F. graminearum and F. fujikuroi, a carotenoid oxygenase that is not physically linked in the cluster, termed carT, converts the torulene precursor to neurosporaxanthin [63]. The hypothesis that Z. tritici may also produce a similar carotenoid is supported by the presence of a carT orthologue in the Z. tritici genome (Mycgr3G72713, Table 2). With regards to biosynthesis of physiologically active apocarotenoids, in F. fujikuroi this cluster is required for rential biosynthesis, containing genes encoding an opsin like protein (carO), and retinal synthesizing enzyme (carX) [64, 65]. Orthologues for both these genes are present in the corresponding Z. tritici cluster (Table 2 and Fig. 2c). Opsins are transmembrane proteins that bind retinal via a conserved lysine residue [65], and light mediated isomerization of retinal enables the opsins to act as light receptors at the fungal membrane. While Z. tritici light responses have not been comprehensively determined, the link between light and secondary metabolism has already been demonstrated, as the production of aerial mycelium in vitro is light dependent, and is regulated by the velvet complex, which also controls melanin production [35]. Taken together, we predict that cluster 34 (Table 1) may be multifunctional, biosynthesizing a torulene-like carotenoid pigment and light responsive opsin/chromophore, and ultimately this locus may co-ordinate light responses and pigment production in Z. tritici.
Putative epipolythiodioxopiperazine biosynthetic gene cluster
Cluster 9 contains a total of 19 predicted genes, including a NRPS, and was highly comparable to epipolythiodioxopiperazine (ETP) biosynthetic clusters from numerous fungi. This included the gliotoxin and sirodesmin PL biosynthetic loci in A. fumigatus and L. maculans, with 8 and 6 conserved genes respectively (Fig. 2d and Additional file 1) [66, 67]. Both gliotoxin and sirodesmin PL are ETPs, which consist of a diketopiperazine core and contain a disulphide bridge [33]. This latter motif is important for ETP toxicity towards both plants and mammals, as it is required for protein-ETP conjugates [68]. For example, the presence of a Z. tritici orthologue for the thioredoxin reductase gliT, which is required for disulphide bridge formation during gliotoxin biosynthesis, suggests that this key moiety is also present on the putative Z. tritici ETP [69]. Interestingly, we did not find any gene encoding a putative toxin efflux pump or transporter in this cluster (Tables 1 and 2). Toxin efflux pumps are present in 15 out of 16 ETP clusters analyzed in filamentous ascomycetes [33]. In A. fumigatus, for example, the transporter is encoded by the gliA gene which is required for efflux of gliotoxin [32]. The absence of an orthologous gene in the Z. tritici cluster, and any putative transporters in the 18 predicted tailoring genes, suggests that the ETP may be intracellular. Consequently, the predicted ETP encoded by this locus in Z. tritici may not primarily function as a phytotoxin. Interestingly, ETPs have been shown to potently reduce H202 [70], one of the major reactive oxygen species encountered by Z. tritici in the host [71], and consequently, this putative ETP may act a defensive molecule during infection. Similar defensive functions related to detoxification have also been postulated for presumed toxins, including the carcinogenic polyketide aflatoxin [72].
With regards to regulation of this cluster, we did not identify a resident transcription factor (Table 1). Out of 16 ETP clusters surveyed amongst ascomycetes, resident transcription factors are only absent in A. terreus [33]. In A. fumigatus, for example, the resident transcription factor GliZ regulates genes expression of the ETP cluster and is essential for toxin biosynthesis [73]. These data suggest that in Z. tritici ETP gene expression does not rely on regulation by a resident transcription factor. Consequently global regulators of secondary metabolite biosynthesis, such as the transcription factor StuA or the velvet complex, may play important roles in regulation of this cluster in Z. tritici [34].
Putative fumonisin biosynthetic gene cluster
The second largest cluster predicted by AntiSMASH analysis was number 27, with 54 putative genes surrounding a PKS (Mycgr3G101493, Table 1). SMURF predicted a considerably smaller cluster of 7 genes containing the same key enzyme. MultiGeneBLAST analysis demonstrated significant similarity of this locus to the fumonisin biosynthetic gene cluster in Fusarium oxysporum (a total of 4 conserved genes, Fig. 2e and Table 2) [74], in addition to predicted fumonisin clusters in various other species, including Neosartorya fischeri, A. fumigatus and Aspergillus niger. Fumonisins are a structurally diverse class of linear, 19–20-carbon backbone mycotoxins which cause significant crop contamination, and the genetics of their biosynthesis have been well characterized in various Fusarium species [75].
Interestingly, the Z. tritici cluster has some deviations from those of Fusarium spp., most notably the absence of genes encoding the oxoamine synthase Fum8 and the P450 monooxygenase Fum6, which are predicted to catalyze the second and third biosynthetic steps respectively [75], and both of which are essential for fumonisin production in F. verticillioides [76]. However we found significant homology between Z. tritici Mycgr3G101493 and PKS fum1 (Table 2), which catalyzes the condensation of two methyl and nine acetate units to produce a linear polyketide in the first step in fumonisin biosynthesis in Fusarium spp. [75]. Additionally, we predict Z. tritici orthologues at this locus for fum2 and fum3, which hydroxylate C-10 and C-5 in the fifth and final steps of fumonisin B1 biosynthesis [77]. Finally, a single Z. tritici gene had significant conservation with two cytochrome p450 encoding genes in the Fusarium cluster (fum15 and cpm1) which presumably function to hydroxylate an as yet unknown carbon [75]. We therefore predict that Z. tritici produces a PKS which might be structurally similar to fungal fumonisins. This is of particular interest for necrotrophic pathogens, as fumonisins can induce plant cell death by depletion of extracellular ATP [78]. It is interesting to speculate that the product of this cluster may contribute to virulence by causing host cell death, a hypothesis supported by transcriptional upregulation of the PKS during necrotrophic phases of infection relative to laboratory culture [6].
Putative AM-toxin biosynthetic gene cluster
Our analysis also identified a putative secondary metabolite locus in the subtelomeric region of chromosome six (cluster number 18, Table 1) with significant similarity to the AM-toxin biosynthetic gene cluster from the apple pathotype of A. alternata [14, 79]. Gene architecture between these two loci was highly conserved, with a total of 8 orthologous genes found at Z. tritici cluster and corresponding A. alternata locus (Fig. 2f and Table 2). This included the NRPS encoding gene amt1, which is essential for AM-toxin biosynthesis [79]. Although the functions of all genes within this cluster have not been elucidated in A. alternata, they are transcriptionally co-induced under AM-toxin producing conditions [80], and encode proteins associated with secondary metabolite biosynthesis, including thioesterases, methyltransferases, and dehydrogenases (Table 2). In Z. tritici, the cluster also includes two predicted transporters (Table 1) strongly suggesting that this metabolite is secreted. Interestingly, our analysis revealed poor conservation of this cluster in Dothideomycetes outside the Mycosphaerella genus (Additional file 1). In A. alternata, several toxin biosynthetic gene clusters, including the AM-toxin locus, reside on small (1.1–1.8 Mb) supernumerary chromosomes [80]. The biosynthetic products of these clusters are host specific toxins (HSTs) which are necessary for virulence of the various pathotypes, including apple, pear, strawberry or tangerine, but dispensable for normal development, growth, and cell viability [14]. amt1 null mutants, for example, cannot produce the AM-toxin and are unable to cause disease symptoms on susceptible apple cultivars [79]. It has been suggested that supernumerary chromosomes are horizontally transferred across pathotypes and therefore facilitate host-range expansion [81]. Our data is indicative of horizontal gene transfer of this cluster between Z. tritici and A. alternata, and we postulate that Z. tritici produces a secondary metabolite similar to the A. alternata AM-toxin. With regards to predicting the structure and possible mechanism of action of this molecule, the AM-toxin is a cyclic depsipeptide with two sites of action, firstly, causing invagination and electrolyte loss across host plasma membranes and secondly membrane perturbation in choloroplasts, resulting in reduced chlorophyll content and photosynthesis [14]. It is interesting to speculate that this cluster in Z. tritici may biosynthesize an NRP with a similar structure and/or mechanism of action, which could conceivably be required for virulence and host or cultivar specificity.
Gene expression analysis of predicted secondary metabolite loci
We analyzed an existing RNA seq dataset [6] to determine if predicted secondary metabolite loci from our study demonstrated co-expression during in vitro growth and throughout a virulence model of Z. tritici infection (Table 1). This analysis suggested that 16 putative secondary metabolite loci demonstrate evidence of transcriptional co-regulation (Table 1 and Additional file 3). Several clusters demonstrated stage-specific transcriptional upregulation at key phases of infection (Fig. 3), for example during germination (day 1, cluster 15), asymptomatic growth (day 4, cluster 8), the switch to necrotrophic infection (day 9, clusters 18, 31, 33) and throughout rapid fungal growth and development of reproductive pycnidia (day 14 and 21, cluster 17, 27 and 28). These data indicate that the biosynthetic products of these clusters may play key roles at specific stages of disease, and it is possible use these expression patterns to suggest putative biological functions. For example, during the earliest stages of disease, transcriptionally upregulated metabolic clusters (e.g. cluster 15, Fig. 3) may biosynthesize germination inhibitors in order to spatially or temporally coordinate production of infectious hyphae [15]. Alternatively, initiating phases of infection characterized by slow, symptomless fungal growth may require extracellular metabolites for masking or counteracting host immune surveillance (cluster 15 and 8, Fig. 3). In contrast, the switch from symptomless disease to host tissue necrosis at 9 and 14 days post infection may require generalist or host-selective phytotoxins, an observation supported by the stage specific transcriptional upregulation of genes from clusters 18 and 27, which demonstrate cluster homology to known phytotoxins (Fig. 2). Consequently, it is possible to use transcriptional profiles of co-expressed secondary metabolite loci during disease to aid hypothesis construction regarding their biological function, which can then be validated using genetic and metabolomic approaches.
Fig. 3.

Transcriptional analysis reveals stage specific expression of numerous predicted secondary metabolite loci during infection. Numbers in parentheses indicate cluster number from this study (Table 1). Transcriptional values (y-axis) are average fragments per kilobase for each gene per million fragments (FPKM), with the Ensemble gene identifier numbers given. Data were taken from Rudd and colleagues Additional file 3: Table S3 ([6]). In this study, RNA samples were isolated from Czapek-Dox broth (CDB), potato dextrose broth (PDB), and from various days post inoculation in a virulence assay of IP0323 on ‘Riband’ wheat cultivar. We identified co-expressed contiguous loci using the FunGeneClusterS web interface [26]
Conclusions
This study has used in silico approaches to predict, and subsequently analyze, 32 secondary metabolite loci in the genome of the wheat pathogen Z. tritici. We were able to assign putative biosynthetic products to numerous clusters based on their conservation amongst other fungi. These analyses suggest a siderophore, melanin, carotenoid, opsin, ETP, fumonisin-like polyketide and AM-toxin-like NRP are part of the Z. tritici secondary metabolite repertoire. Interestingly, analysis of most secondary metabolite loci did not enable prediction of an experimentally confirmed product, yet several were transcriptionally deployed during infection, and consequently the capacity of these clusters to play as yet undetermined roles in disease or other stages of the Z. tritici lifecycle is significant.
While our analyses have been conducted using isolate IP0323, it is not currently clear if this is a representative or average genome for Z. tritici. Indeed, high rates of sexual reproduction [9] and gene flow [82] result in extreme genomic and phenotypic diversity in Z. tritici populations, which is likely to result in a range of secondary metabolite repertoires amongst field isolates. As noted earlier, genes predicted to encode key biosynthetic enzymes from the IP0323 genome are absent in certain field strains [39]. Future comparative genomic analyses of multiple Z. tritici isolates will enable identification of secondary metabolite loci that are essential for virulence or other phases of the pathogen lifecycle, and those that are either dispensable or required for cultivar specificity.
Clearly, to validate the products of the identified loci and comprehensively determine their biological function, gene functional characterization and metabolomics analyses will be essential, and numerous tools now exist for such experiments in Z. tritici. Finally, from a biotechnological perspective, these loci and cognate products may be promising for the discovery of new bioactive molecules.
Additional files
MultiGeneBLAST analysis of putative secondary metabolite clusters. All encoded amino acid sequences from genes residing in clusters predicted by AntiSMASH are given as FASTA file format. All output data from MultiGeneBLASTs are also provided. (ZIP 42911 kb)
Subtelomeric co-ordinates and genes in the Z. tritici genome. Genome co-ordinates within 300 kb of each chromosome end are given. All genes located in these regions are provided. (XLSX 153 kb)
FungalGeneClusteRs gene co-expression analysis. All co-regulated loci are given (Table 1), secondary metabolite co-regulated loci (Table 2), and FPKM expression values for each co-regulated secondary metabolite gene (Additional file 3: Table S3, [6]). (XLSX 143 kb)
All putative secondary metabolite genes predicted in this study. (XLSX 59 kb)
Genes encoding predicted ABC or MFS transporters located in putative secondary metabolite clusters. (XLSX 13 kb)
Genes encoding predicted transcription factors located in putative secondary metabolite clusters. (XLSX 9 kb)
Genes residing in secondary metabolite loci with at least one paralogue that also resides in a biosynthetic gene cluster. (XLSX 20 kb)
{kind=link}
Schematic representation of putative gene duplication events at exemplar secondary metabolite loci. Predicted gene boundaries are depicted by arrows and paralogous genes assigned Ensemble identifiers. Dashed lines indicate paralogous gene pairs. Ensemble identifiers for key biosynthetic genes (PKS, clusters 17 and 27, and an NRPS, cluster 31) are given in parentheses. (GIF 21 kb)
Acknowledgements
Not applicable.
Funding
TC gratefully acknowledges Postdoctoral funding from Berlin University of Technology. Funding sources played no role in the design of this study or the collection, analysis, and the interpretation of data, or in writing the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- ABC
ATP-binding cassette
- AntiSMASH
Antibiotics and secondary metabolite analysis shell
- ATP
Adenosine triphosphate
- BLAST
Basic local alignment search tool
- CDB
Czapek-dox broth
- CoA
Coenzyme A
- DDL
Duplication, diversification and differential gene loss
- DHN
Dihydroxynaphthalene
- DNA
Deoxyribonucleic acid
- ETP
Epipolythiodioxopiperazine
- FPKM
Fragments per kilobase of transcript per million mapped reads
- HMG-CoA
Hydroxymethylglutaryl-CoA
- HST
Host specific toxins
- MFS
Major facilitator superfamily
- NRP
Non-ribosomal peptide
- NRPS
Non-ribosomal peptide synthetase
- PAMP
Pathogen associated molecular pattern
- PDB
Potato dextrose broth
- PKS
Polyketide synthase
- PRR
Pattern recognition receptor
- RNA
Ribonucleic acid
- SMURF
Secondary metabolite unique regions finder
- TAFC
Triacetyl fusarine C
- THN
Tetrahydroxynapthalene
- UV
Ultraviolet
Authors’ contributions
Conceived of the study- TC, VM; conducted analysis- TC; wrote and reviewed manuscript- TC, VM. All authors read and approved the final manuscript.
Ethics approval and consent to participate
No plant or fungal materials were used directly, and no field work was conducted in this study.
Consent for publication
All authors give their informed consent for the publication of submitted documents and data in their entirety.
Competing interests
The authors declare they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3969-y) contains supplementary material, which is available to authorized users.
Contributor Information
Timothy Cairns, Email: t.cairns@campus.tu-berlin.de.
Vera Meyer, Email: vera.meyer@tu-berlin.de.
References
- 1.Fones H, Gurr S. The impact of Septoria Tritici blotch disease on wheat: an {EU} perspective. Fungal Genet Biol. 2015;79:3–7. doi: 10.1016/j.fgb.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torriani SF, Melichar JP, Mills C, Pain N, Sierotzki H, Courbot M. Zymoseptoria tritici: a major threat to wheat production, integrated approaches to control. Fungal Genet Biol. 2015;79:8–12. doi: 10.1016/j.fgb.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Vermeulen E, Lagrou K, Verweij PE. Azole resistance in Aspergillus fumigatus: a growing public health concern. Curr Opin Infect Dis. 2013;26:493–500. doi: 10.1097/QCO.0000000000000005. [DOI] [PubMed] [Google Scholar]
- 4.Steinberg G. Cell biology of Zymoseptoria tritici: pathogen cell organization and wheat infection. Fungal Genet Biol. 2015;79:17–23. doi: 10.1016/j.fgb.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keon J, Antoniw J, Carzaniga R, Deller S, Ward JL, Baker JM, Beale MH, Hammond-Kosack K, Rudd JJ. Transcriptional adaptation of Mycosphaerella graminicola to programmed cell death (PCD) of its susceptible wheat host. Mol Plant-Microbe Interact. 2007;20:178–193. doi: 10.1094/MPMI-20-2-0178. [DOI] [PubMed] [Google Scholar]
- 6.Rudd JJ, Kanyuka K, Hassani-Pak K, Derbyshire M, Andongabo A, Devonshire J, Lysenko A, Saqi M, Desai NM, Powers SJ, Hooper J, Ambroso L, Bharti A, Farmer A, Hammond-Kosack KE, Dietrich RA, Courbot M. Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the Hemibiotrophic lifestyle Def. Plant Physiol. 2015;167:1158–1185. doi: 10.1104/pp.114.255927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong Z, Marcel TC, Hartmann FE, Ma X, Plissonneau C, Zala M, Ducasse A, Confais J, Compain J, Lapalu N, Amselem J, McDonald BA, Croll D, Palma-Guerrero J. A small secreted protein in Zymoseptoria tritici is responsible for avirulence on wheat cultivars carrying the Stb6 resistance gene. New Phytol. 2017;214(2):619–31. [DOI] [PubMed]
- 8.Sánchez-Vallet A, McDonald MC, Solomon PS, McDonald BA. Is Zymoseptoria tritici a hemibiotroph? Fungal Genet Biol. 2015;79:29–32. doi: 10.1016/j.fgb.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Goodwin SB, M’Barek SB, Dhillon B, Wittenberg AH, Crane CF, Hane JK, Foster AJ, Van der Lee TA, Grimwood J, Aerts A, Antoniw J, Bailey A, Bluhm B, Bowler J, Bristow J, van der Burgt A, Canto-Canche B, Churchill AC, Conde-Ferraez L, Cools HJ, Coutinho PM, Csukai M, Dehal P, De Wit P, Donzelli B, van de Geest HC, van Ham RC, Hammond-Kosack KE, Henrissat B, Kilian A, et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011;7:e1002070. [DOI] [PMC free article] [PubMed]
- 10.Lee W, Rudd JJ, Hammond-kosack KE, Kanyuka KK. Mycosphaerella graminicola LysM effector-mediated stealth pathogenesis subverts recognition through both CERK1 and CEBiP homologues in wheat. Mol Plant-Microbe Interact. 2013;27:236–243. doi: 10.1094/MPMI-07-13-0201-R. [DOI] [PubMed] [Google Scholar]
- 11.Kettles GJ, Bayon C, Canning G, Rudd JJ, Kanyuka K. Apoplastic recognition of multiple candidate effectors from the wheat pathogen Zymoseptoria tritici in the nonhost plant Nicotiana benthamiana. New Phytol. 2017;213:338–350. doi: 10.1111/nph.14215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poppe S, Dorsheimer L, Happel P, Stukenbrock EH. Rapidly evolving genes are key players in host specialization and virulence of the fungal wheat pathogen Zymoseptoria tritici (Mycosphaerella graminicola) PLoS Pathog. 2015;11:1–21. doi: 10.1371/journal.ppat.1005055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walton JD. HC-toxin. Phytochemistry. 2006;67:1406–1413. doi: 10.1016/j.phytochem.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 14.Tsuge T, Harimoto Y, Akimitsu K, Ohtani K, Kodama M, Akagi Y, Egusa M, Yamamoto M, Otani H. Host-selective toxins produced by the plant pathogenic fungus Alternaria alternata. FEMS Microbiol Rev. 2013;37:44–66. doi: 10.1111/j.1574-6976.2012.00350.x. [DOI] [PubMed] [Google Scholar]
- 15.Pusztahelyi T, Holb IJ, Pócsi I. Secondary metabolites in fungus-plant interactions. Front Plant Sci. 2015;6:573. doi: 10.3389/fpls.2015.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keller NP, Turner G, Bennett JW. Fungal secondary metabolism [mdash] from biochemistry to genomics. Nat Rev Micro. 2005;3:937–947. doi: 10.1038/nrmicro1286. [DOI] [PubMed] [Google Scholar]
- 17.Schwelm A, Bradshaw RE. Genetics of Dothistromin biosynthesis of Dothistroma septosporum: an update. Toxins (Basel) 2010;2:2680–2698. doi: 10.3390/toxins2112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lendenmann MH, Croll D, Stewart EL, McDonald BA. Quantitative Trait Locus Mapping of Melanization in the Plant Pathogenic Fungus Zymoseptoria tritici. G3 Genes|Genomes|Genetics. 2014;4:2519–2533. doi: 10.1534/g3.114.015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Medema MH, Takano E, Breitling R. Detecting sequence homology at the gene cluster level with multigeneblast. Mol Biol Evol. 2013;30:1218–1223. doi: 10.1093/molbev/mst025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bignell E, Cairns TC, Throckmorton K, Nierman WC, Keller NP. Secondary metabolite arsenal of an opportunistic pathogenic fungus. Philos Trans R Soc B Biol Sci. 2016;371 [DOI] [PMC free article] [PubMed]
- 21.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, Breitling R, Takano E, Medema MH. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2016;44(Database issue):D67–D72. doi: 10.1093/nar/gkv1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T, Howe K, Kähäri A, Kokocinski F, Martin FJ, Murphy DN, Nag R, Ruffier M, Schuster M, Tang YA, Vogel J-H, White S, Zadissa A, Flicek P, Searle SMJ: The Ensembl gene annotation system. Database 2016, 2016:baw093. [DOI] [PMC free article] [PubMed]
- 25.McDonagh A, Fedorova ND, Crabtree J, Yu Y, Kim S, Chen D, Loss O, Cairns T, Goldman G, Armstrong-James D, Haynes K, Haas H, Schrettl M, May G, Nierman WC, Bignell E. Sub-telomere directed gene expression during initiation of invasive aspergillosis. PLoS Pathog. 2008;4 doi: 10.1371/journal.ppat.1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vesth TC, Brandl J, Andersen MR. FunGeneClusterS: predicting fungal gene clusters from genome and transcriptome data. Synth Syst Biotechnol. 2016;1:122–129. doi: 10.1016/j.synbio.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inglis DO, Binkley J, Skrzypek MS, Arnaud MB, Cerqueira GC, Shah P, Wymore F, Wortman JR, Sherlock G. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 2013;13:91. doi: 10.1186/1471-2180-13-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiemann P, Guo C-J, Palmer JM, Sekonyela R, Wang CCC, Keller NP. Prototype of an intertwined secondary-metabolite supercluster. Proc Natl Acad Sci U S A. 2013;110:17065–17070. doi: 10.1073/pnas.1313258110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coyle CM, Panaccione DG. An ergot alkaloid biosynthesis gene and Clustered hypothetical genes from Aspergillus fumigatus. Appl Environ Microbiol. 2005;71:3112–3118. doi: 10.1128/AEM.71.6.3112-3118.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman JJ, Mylonakis E. Efflux in Fungi: La Pièce de Résistance. PLoS Pathog. 2009;5:1–7. doi: 10.1371/journal.ppat.1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D-N, Toyotome T, Muraosa Y, Watanabe A, Wuren T, Bunsupa S, Aoyagi K, Yamazaki M, Takino M, Kamei K. GliA in Aspergillus fumigatus is required for its tolerance to gliotoxin and affects the amount of extracellular and intracellular gliotoxin. Med Mycol. 2014;52:506. doi: 10.1093/mmy/myu007. [DOI] [PubMed] [Google Scholar]
- 33.Patron NJ, Waller RF, Cozijnsen AJ, Straney DC, Gardiner DM, Nierman WC, Howlett BJ. Origin and distribution of epipolythiodioxopiperazine (ETP) gene clusters in filamentous ascomycetes. BMC Evol Biol. 2007;7:174. doi: 10.1186/1471-2148-7-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brakhage AA. Regulation of fungal secondary metabolism. Nat Rev Micro. 2013;11:21–32. doi: 10.1038/nrmicro2916. [DOI] [PubMed] [Google Scholar]
- 35.Choi Y-E, Goodwin SB. MVE1, encoding the velvet gene product homolog in Mycosphaerella graminicola, is associated with aerial mycelium formation, melanin biosynthesis, hyphal swelling, and light signaling. Appl Environ Microbiol. 2011;77:942–953. doi: 10.1128/AEM.01830-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farman ML. Telomeres in the rice blast fungus Magnaporthe oryzae: the world of the end as we know it. FEMS Microbiol Lett. 2007;273:125–132. doi: 10.1111/j.1574-6968.2007.00812.x. [DOI] [PubMed] [Google Scholar]
- 37.Cairns T, Minuzzi F, Bignell E. The host-infecting fungal transcriptome. FEMS Microbiol Lett. 2010;307:1–11. doi: 10.1111/j.1574-6968.2010.01961.x. [DOI] [PubMed] [Google Scholar]
- 38.Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, Angiuoli S, Bussey H, Bowyer P, Cotty PJ, Dyer PS, Egan A, Galens K, Fraser-Liggett CM, Haas BJ, Inman JM, Kent R, Lemieux S, Malavazi I, Orvis J, Roemer T, Ronning CM, Sundaram JP, Sutton G, Turner G, Venter JC, et al. Genomic Islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palma-Guerrero J, Ma X, Torriani SFF, Zala M, Francisco CS, Hartmann FE. Croll D. McDonald BA: Comparative transcriptome analyses in Zymoseptoria tritici reveal significant differences in gene expression among strains during plant infection. Mol Plant-Microbe Interact; 2017. [DOI] [PubMed]
- 40.Winterberg B, Uhlmann S, Linne U, Lessing F, Marahiel MA, Eichhorn H, Kahmann R, Schirawski J. Elucidation of the complete ferrichrome a biosynthetic pathway in Ustilago maydis. Mol Microbiol. 2010;75:1260–1271. doi: 10.1111/j.1365-2958.2010.07048.x. [DOI] [PubMed] [Google Scholar]
- 41.Haas H. Fungal siderophore metabolism with a focus on Aspergillus fumigatus. Nat Prod Rep. 2014;31:1266–1276. doi: 10.1039/C4NP00071D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eichhorn H, Lessing F, Winterberg B, Schirawski J, Kämper J, Müller P, Kahmann R. A Ferroxidation/permeation iron uptake system is required for virulence in Ustilago maydis. Plant Cell. 2006;18:3332–3345. doi: 10.1105/tpc.106.043588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ardon O, Nudelman R, Caris C, Libman J, Shanzer A, Chen Y, Hadar Y. Iron uptake in Ustilago maydis: tracking the iron path. J Bacteriol. 1998;180:2021–2026. doi: 10.1128/jb.180.8.2021-2026.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oide S, Krasnoff SB, Gibson DM, Turgeon BG. Intracellular Siderophores are essential for ascomycete sexual development in heterothallic Cochliobolus heterostrophus and homothallic Gibberella zeae. Eukaryot Cell. 2007;6:1339–1353. doi: 10.1128/EC.00111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oide S, Berthiller F, Wiesenberger G, Adam G, Turgeon BG. Individual and combined roles of malonichrome, ferricrocin, and TAFC siderophores in Fusarium graminearum pathogenic and sexual development. Front Microbiol. 2014;5:759. doi: 10.3389/fmicb.2014.00759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, Hesse CN, Kosti I, LaButti K, Lindquist EA, Lucas S, Salamov AA, Bradshaw RE, Ciuffetti L, Hamelin RC, Kema GHJ, Lawrence C, Scott JA, Spatafora JW, Turgeon BG, de Wit PJGM, Zhong S, Goodwin SB, Grigoriev IV. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes Fungi. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1003037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dellagi A, Rigault M, Segond D, Roux C, Kraepiel Y, Cellier F, Briat J-F, Gaymard F, Expert D. Siderophore-mediated upregulation of Arabidopsis ferritin expression in response to Erwinia chrysanthemi infection. Plant J. 2005;43:262–272. doi: 10.1111/j.1365-313X.2005.02451.x. [DOI] [PubMed] [Google Scholar]
- 48.Albarouki E, Schafferer L, Ye F, von Wirén N, Haas H, Deising HB. Biotrophy-specific downregulation of siderophore biosynthesis in Colletotrichum graminicola is required for modulation of immune responses of maize. Mol Microbiol. 2014;92:338–355. doi: 10.1111/mmi.12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franck WL, Gokce E, Oh Y, Muddiman DC, Dean RA. Temporal analysis of the Magnaporthe oryzae proteome during conidial germination and cyclic AMP (cAMP)-mediated Appressorium formation. Mol Cell Proteomics. 2013;12:2249–2265. doi: 10.1074/mcp.M112.025874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson JE, Fahnestock S, Farrall L, Liao D-I, Valent B, Jordan DB. The second Naphthol reductase of fungal melanin biosynthesis inMagnaporthe grisea : Tetrahydroxynaphthalene reductase. J Biol Chem. 2000;275:34867–34872. doi: 10.1074/jbc.M006659200. [DOI] [PubMed] [Google Scholar]
- 51.Tsuji G, Kenmochi Y, Takano Y, Sweigard J, Farrall L, Furusawa I, Horino O, Kubo Y. Novel fungal transcriptional activators, Cmr1p of Colletotrichum lagenarium and Pig1p of Magnaporthe grisea, contain Cys2His2 zinc finger and Zn(II)2Cys6 binuclear cluster DNA-binding motifs and regulate transcription of melanin biosynthesis genes in a developmentally specific manner. Mol Microbiol. 2000;38:940–954. doi: 10.1046/j.1365-2958.2000.02181.x. [DOI] [PubMed] [Google Scholar]
- 52.Schmaler-Ripcke J, Sugareva V, Gebhardt P, Winkler R, Kniemeyer O, Heinekamp T, Brakhage AA. Production of Pyomelanin, a second type of melanin, via the tyrosine degradation pathway in Aspergillus fumigatus. Appl Environ Microbiol. 2009;75:493–503. doi: 10.1128/AEM.02077-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beltrán-García MJ, Prado FM, Oliveira MS, Ortiz-Mendoza D, Scalfo AC, Pessoa A, Jr, Medeiros MHG, White JF, Di Mascio P. Singlet molecular oxygen generation by light-activated DHN-melanin of the fungal pathogen Mycosphaerella Fijiensis in black Sigatoka disease of bananas. PLoS One. 2014;9:1–15. doi: 10.1371/journal.pone.0091616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paolo WF, Dadachova E, Mandal P, Casadevall A, Szaniszlo PJ, Nosanchuk JD. Effects of disrupting the polyketide synthase gene WdPKS1 in Wangiella [Exophiala] dermatitidis on melanin production and resistance to killing by antifungal compounds, enzymatic degradation, and extremes in temperature. BMC Microbiol. 2006;6:55. doi: 10.1186/1471-2180-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, Casadevall A. Decreased susceptibility of melanized Cryptococcus neoformans to UV light. Appl Environ Microbiol. 1994;60:3864–3866. doi: 10.1128/aem.60.10.3864-3866.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goncalves R. De CR, Pombeiro-Sponchiado SR: antioxidant activity of the melanin pigment extracted from Aspergillus nidulans. Biol Pharm Bull. 2005;28:1129–1131. doi: 10.1248/bpb.28.1129. [DOI] [PubMed] [Google Scholar]
- 57.Jermy A. Fungal pathogenesis: Magnaporthe puts a ring on it. Nat Rev Micro. 2012;10:521. doi: 10.1038/nrmicro2840. [DOI] [PubMed] [Google Scholar]
- 58.Heinekamp T, Thywißen A, Macheleidt J, Keller S, Valiante V, Brakhage AA. Aspergillus fumigatus melanins: Interference with the host endocytosis pathway and impact on virulence. Frontiers in Microbiology. 2012(JAN). [DOI] [PMC free article] [PubMed]
- 59.Bayry J, Beaussart A, YF Dufrêne, Sharma M, Bansal K, Kniemeyer O, Aimanianda V, Brakhage AA, Kaveri SV, Kwon-Chung KJ, Latgé JP, Beauvais A. Surface structure characterization of Aspergillus fumigatus conidia mutated in the melanin synthesis pathway and their human cellular immune response. Infect Immun. 2014;82:3141–3153. doi: 10.1128/IAI.01726-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Avalos J, Carmen Limón M. Biological roles of fungal carotenoids. Curr Genet. 2015;61:309–324. doi: 10.1007/s00294-014-0454-x. [DOI] [PubMed] [Google Scholar]
- 61.Linnemannstöns P, Prado M, Fernández-Martín R, Tudzynski B, Avalos J. A carotenoid biosynthesis gene cluster in Fusarium fujikuroi: the genes carB and carRA. Mol Gen Genomics. 2002;267:593–602. doi: 10.1007/s00438-002-0690-5. [DOI] [PubMed] [Google Scholar]
- 62.Jin J-M, Lee J, Lee Y-W. Characterization of carotenoid biosynthetic genes in the ascomycete Gibberella zeae. FEMS Microbiol Lett. 2010;302:197–202. doi: 10.1111/j.1574-6968.2009.01854.x. [DOI] [PubMed] [Google Scholar]
- 63.Prado-Cabrero A, Estrada AF, Al-Babili S, Avalos J. Identification and biochemical characterization of a novel carotenoid oxygenase: elucidation of the cleavage step in the Fusarium carotenoid pathway. Mol Microbiol. 2007;64:448–60. [DOI] [PubMed]
- 64.Prado MM, Prado-Cabrero A, Fernández-Martín R, Avalos J. A gene of the opsin family in the carotenoid gene cluster of Fusarium fujikuroi. Curr Genet. 2004;46:47–58. doi: 10.1007/s00294-004-0508-6. [DOI] [PubMed] [Google Scholar]
- 65.Prado-Cabrero A, Scherzinger D, Avalos J, Al-Babili S. Retinal biosynthesis in fungi: characterization of the carotenoid oxygenase CarX from Fusarium fujikuroi. Eukaryot Cell. 2007;6:650–657. doi: 10.1128/EC.00392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elliott CE, Gardiner DM, Thomas G, Cozijnsen A, VAN DE Wouw A, Howlett BJ. Production of the toxin sirodesmin PL by Leptosphaeria Maculans during infection of Brassica Napus. Mol Plant Pathol. 2007;8:791–802. doi: 10.1111/j.1364-3703.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- 67.Gardiner DM, Howlett BJ. Bioinformatic and expression analysis of the putative gliotoxin biosynthetic gene cluster of Aspergillus fumigatus. FEMS Microbiol Lett. 2005;248:241–8. doi: 10.1016/j.femsle.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 68.Gardiner DM, Waring P, Howlett BJ. The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]
- 69.Scharf DH, Brakhage AA, Mukherjee PK. Gliotoxin – bane or boon? Environ Microbiol. 2016;18:1096–1109. doi: 10.1111/1462-2920.13080. [DOI] [PubMed] [Google Scholar]
- 70.Choi HS, Shim JS, Kim J-A, Kang SW, Kwon HJ. Discovery of gliotoxin as a new small molecule targeting thioredoxin redox system. Biochem Biophys Res Commun. 2007;359:523–528. doi: 10.1016/j.bbrc.2007.05.139. [DOI] [PubMed] [Google Scholar]
- 71.Shetty NP, Kristensen BK, Newman M-A, Møller K, Gregersen PL, Jørgensen HJL. Association of hydrogen peroxide with restriction of Septoria Tritici in resistant wheat. Physiol Mol Plant Pathol. 2003;62:333–346. doi: 10.1016/S0885-5765(03)00079-1. [DOI] [Google Scholar]
- 72.Roze VL, Laivenieks M, Hong S-Y, Wee J, Wong S-S, Vanos B, Awad D, Ehrlich CK, Linz EJ: Aflatoxin biosynthesis is a novel source of reactive oxygen species—a potential redox signal to initiate resistance to oxidative stress? Toxins 2015. [DOI] [PMC free article] [PubMed]
- 73.Bok JW, Chung D, Balajee SA, Marr KA, Andes D, Nielsen KF, Frisvad JC, Kirby KA, Keller NP. GliZ, a transcriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect Immun. 2006;74:6761–6768. doi: 10.1128/IAI.00780-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Proctor RH, Busman M, Seo J-A, Lee YW, Plattner RD. A fumonisin biosynthetic gene cluster in Fusarium oxysporum strain O-1890 and the genetic basis for B versus C fumonisin production. Fungal Genet Biol. 2008;45:1016–1026. doi: 10.1016/j.fgb.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 75.Alexander NJ, Proctor RH, McCormick SP. Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxin Rev. 2009;28:198–215.
- 76.Bojja RS, Cerny RL, Proctor RH, Du L. Determining the biosynthetic sequence in the early steps of the Fumonisin pathway by use of three gene-disruption mutants of Fusarium verticillioides. J Agric Food Chem. 2004;52:2855–60. [DOI] [PubMed]
- 77.Desjardins AE, Plattner RD, Proctor RH. Linkage among genes responsible for fumonisin biosynthesis in Gibberella fujikuroi mating population a. Appl Environ Microbiol. 1996;62:2571–2576. doi: 10.1128/aem.62.7.2571-2576.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chivasa S, Ndimba BK, Simon WJ, Lindsey K, Slabas AR. Extracellular ATP functions as an endogenous external metabolite regulating plant cell viability. Plant Cell. 2005;17:3019–3034. doi: 10.1105/tpc.105.036806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Johnson RD, Johnson L, Itoh Y, Kodama M, Otani H, Kohmoto K. Cloning and characterization of a cyclic peptide Synthetase gene from Alternaria alternata apple Pathotype whose product is involved in AM-toxin synthesis and pathogenicity. Mol Plant-Microbe Interact. 2000;13:742–753. doi: 10.1094/MPMI.2000.13.7.742. [DOI] [PubMed] [Google Scholar]
- 80.Harimoto Y, Hatta R, Kodama M, Yamamoto M, Otani H, Tsuge T. Expression profiles of genes encoded by the supernumerary chromosome controlling AM-toxin biosynthesis and pathogenicity in the apple Pathotype of Alternaria alternata. Mol Plant-Microbe Interact. 2007;20:1463–1476. doi: 10.1094/MPMI-20-12-1463. [DOI] [PubMed] [Google Scholar]
- 81.Mehrabi R, Bahkali AH, Abd-Elsalam KA, Moslem M, Ben M’Barek S, Gohari AM, Jashni MK, Stergiopoulos I, Kema GHJ, de Wit PJGM. Horizontal gene and chromosome transfer in plant pathogenic fungi affecting host range. FEMS Microbiol Rev. 2011;35:542–554. doi: 10.1111/j.1574-6976.2010.00263.x. [DOI] [PubMed] [Google Scholar]
- 82.Zhan J, Pettway RE, McDonald BA. The global genetic structure of the wheat pathogen Mycosphaerella graminicola is characterized by high nuclear diversity, low mitochondrial diversity, regular recombination, and gene flow. Fungal Genet Biol. 2003;38:286–297. doi: 10.1016/S1087-1845(02)00538-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MultiGeneBLAST analysis of putative secondary metabolite clusters. All encoded amino acid sequences from genes residing in clusters predicted by AntiSMASH are given as FASTA file format. All output data from MultiGeneBLASTs are also provided. (ZIP 42911 kb)
Subtelomeric co-ordinates and genes in the Z. tritici genome. Genome co-ordinates within 300 kb of each chromosome end are given. All genes located in these regions are provided. (XLSX 153 kb)
FungalGeneClusteRs gene co-expression analysis. All co-regulated loci are given (Table 1), secondary metabolite co-regulated loci (Table 2), and FPKM expression values for each co-regulated secondary metabolite gene (Additional file 3: Table S3, [6]). (XLSX 143 kb)
All putative secondary metabolite genes predicted in this study. (XLSX 59 kb)
Genes encoding predicted ABC or MFS transporters located in putative secondary metabolite clusters. (XLSX 13 kb)
Genes encoding predicted transcription factors located in putative secondary metabolite clusters. (XLSX 9 kb)
Genes residing in secondary metabolite loci with at least one paralogue that also resides in a biosynthetic gene cluster. (XLSX 20 kb)
Schematic representation of putative gene duplication events at exemplar secondary metabolite loci. Predicted gene boundaries are depicted by arrows and paralogous genes assigned Ensemble identifiers. Dashed lines indicate paralogous gene pairs. Ensemble identifiers for key biosynthetic genes (PKS, clusters 17 and 27, and an NRPS, cluster 31) are given in parentheses. (GIF 21 kb)
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.