

Abstract
The myofibroblast has been implicated to be an important pathogenic cell in all fibrotic diseases, through synthesis of excess extracellular matrix. Lung fibroblast migration, proliferation and differentiation into a myofibroblast-like cell type are regarded as important steps in the formation of lung fibrosis. In the present study, the effect of maresin 1 (MaR 1), a pro-resolving lipid mediator, on transforming growth factor (TGF)-β1-stimulated lung fibroblasts was investigated, and the underlying molecular mechanisms were examined. The results of the present study demonstrated that MaR 1 inhibited TGF-β1-induced proliferative and migratory ability, assessed using MTT and scratch wound healing assays. The TGF-β1-induced expression of α-smooth muscle actin (α-SMA) and collagen type I, the hallmarks of myofibroblast differentiation, was decreased by MaR 1 at the mRNA and protein levels, determined using the reverse transcription-quantitative polymerase chain reaction and western blot analysis, respectively. Immunofluorescence demonstrated that MaR 1 downregulated the TGF-β1-induced expression of α-SMA. In addition, phosphorylated mothers against decapentaplegic homolog 2/3 (Smad2/3) and extracellular signal-related kinases (ERK) 1/2 were upregulated in TGF-β1-induced lung fibroblasts, and these effects were attenuated by MaR 1 administration. In conclusion, the results of the present study demonstrated that MaR 1 inhibited the TGF-β1-induced proliferation, migration and differentiation of human lung fibroblasts. These observed effects may be mediated in part by decreased phosphorylation of Smad2/3 and ERK1/2 signaling pathways. Therefore, MaR 1 may be a potential therapeutic approach to lung fibrotic diseases.
Keywords: maresin 1, proliferation, migration, differentiation, lung fibroblast, transforming growth factor-β1
Introduction
Pulmonary fibrosis occurs in various clinical conditions, including chronic obstructive pulmonary disease, asthma, idiopathic pulmonary fibrosis, interstitial pneumonia, radiation-induced lung injury and end-stage lung failure (1–3). Pulmonary fibrosis is characterized by destruction of the lung architecture, and the production and excessive accumulation of a collagen-rich extracellular matrix (ECM), eventually leading to respiratory insufficiency. The primary etiology of fibrosis remains poorly understood, and there is a lack of therapeutic interventions and novel therapies for pulmonary fibrosis (4,5).
Previous studies have demonstrated that fibroblast proliferation and activation serve a role in the pathogenic fibrotic process. When stimulated by transforming growth factor (TGF)-β1 or other local cytokines, fibroblasts proliferate, migrate, and differentiate into myofibroblasts, which drive lung fibrogenesis (6). Fibroblasts and myofibroblasts produce numerous cytokines and growth factors in addition to the ECM, which are the hallmarks of fibrotic pulmonary disease (7). A previous study demonstrated that dysregulation of fibroblast activation and differentiation into myofibroblasts may be a therapeutic strategy for pulmonary fibrosis (8).
A family of endogenous specialized pro-resolving mediators, including lipoxins, resolvins, protectins and maresins, serve a role in the regulation of tissue inflammation, organ protection and fibrotic diseases (9,10). Maresin 1 is biosynthesized via lipoxygenase by docosahexaenoic acid to generate 14S-hydroperoxydocosa-4Z, 7Z, 10Z, 12E, 16Z, 19Z-hexaenoic acid, which undergoes further conversion via epoxidation in macrophages and is subsequently converted to 7R, 14S-dihydroxydocosa-4Z, 8Z, 10E, 12Z, 16Z, 19Z-hexaenoic acid (Fig. 1A), known as maresin 1 (MaR 1) (11). Previous studies in vitro and in vivo demonstrated that MaR 1 exhibited homeostatic activity in models, including colitis, tissue generation, liver injury, lung injury and intimal hyperplasia (12–16). A recent study reported that MaR 1 ameliorated bleomycin-induced pulmonary fibrosis in mice and suppressed epithelial-mesenchymal transition in the mouse type II alveolar epithelial cell line MLE-12 (17). Due to the important role of fibroblasts in lung fibrogenesis, it was hypothesized that MaR 1 may directly influence lung fibroblast activation.
Figure 1.
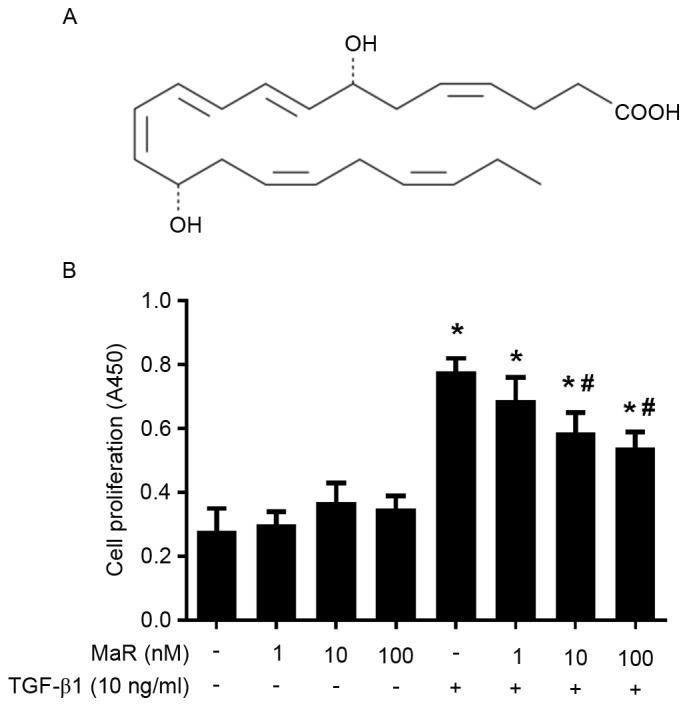
MaR 1 inhibits the TGF-β1-induced proliferation of MRC5 cells. (A) Chemical structure of MaR 1. (B) MRC5 cells were treated with 1, 10 and 100 nM MaR 1. MaR 1 inhibited MRC5 cell proliferation stimulated by TGF-β1, as demonstrated using an MTT assay. Data are presented as the mean ± standard error. n=6. *P<0.05 vs. untreated group. #P<0.05 vs. TGF-β1 treated group. MaR 1, maresin 1; TGF-β1, transforming growth factor β1; A450, absorbance at 450 nm.
The aim of the present study was to evaluate the impact of MaR 1 on TGF-β1-induced human lung fibroblast (MRC-5) proliferation, migration and differentiation in vitro. In order to obtain an increased understanding of the underlying mechanisms, the effects of MaR 1 on mothers against decapentaplegic homolog 2/3 (Smad2/3) and extracellular signal-regulated kinase (ERK) 1/2 phosphorylation signaling pathways were investigated in TGF-β1-treated MRC5 cells.
Materials and methods
Cell culture and stimulation
MRC5 lung fibroblasts (human fetal lung fibroblasts; American Type Culture Collection, Manassas, VA, USA) were grown in Dulbecco's modified Eagle medium (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) with 4.5 g/l glucose, 10% fetal bovine serum (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin/streptomycin solution. Cells were maintained at 37°C in a humidified incubator, in the presence of 5% CO2. Following incubation for 3 days, the cells were cultured to ~80% confluence.
MRC5 cells were pretreated with MaR 1 (Cayman Chemical Company, Ann Arbor, MI, USA) at 1, 10 or 100 nM, or vehicle (0.035% ethanol) for 30 min. Subsequently, TGF-β1 (10 ng/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added and co-incubated for 24 h.
Cell viability assay
Cell viability was assessed using an MTT assay according to the manufacturer's protocol (Cayman Chemical Company). MRC5 cells (2.5×103/100 µl) were seeded into the wells of 96-well culture plates and treated as described above. A total of 24 h subsequent to treatment, cells were incubated with 50 µg MTT solution for 4 h. The supernatants were removed and dimethyl sulfoxide was added to each well. The quantity of aqueous soluble formazan, which was produced by viable cells from tetrazolium salt, was measured using spectrophotometry at absorbance (A) 570 nm, and was equal to the number of living cells. Optical density A570 was measured in six samples of each group.
Cell migration
In order to evaluate the migratory response of MaR 1 pretreated cells following TGF-β1 exposure, a scratch wound healing assay was performed. Cells were seeded at confluent status for 24 h and pre-incubated with MaR 1 (10 nM) for 30 min prior to the addition of TGF-β1 (10 ng/ml) as a chemotactic factor. A cell-free area (900-µm scratch wound) in each well was created using a 200-µl pipette tip. A total of 24 h subsequently, the migratory cells in the gap were counted using a light microscope at ×200 magnification. All of the data were obtained in six independent experiments.
RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Cells were pretreated with or without MaR 1 (10 nM) for 30 min, followed by the addition of TGF-β1 (10 ng/ml) for 24 h. Total cellular RNA was extracted using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.), and 2 µg RNA was reverse transcribed into cDNA using the RevertAid™ First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. The specific primer sequences (Invitrogen; Thermo Fisher Scientific, Inc.) used were as follows (5′-3′): α-SMA forward, GAC AAT GGC TCT GGG CTC TGT AA and reverse, ATG CCA TGT TCT ATC GGG TAC TT; collagen α-1(I) chain (COL1A1) forward, GAG GGC CAA GAC GAA GAC ATC and reverse, CAG ATC ACG TCA TCG CAC AAC; and GAPDH forward, AGT GCC AGC CTC GTC TCA TAG and reverse, CGT TGA ACT TGC CGT GGG TAG. Each specific gene product was amplified by real-time PCR using SYBR-Green Master Mix in the StepOne™ Real-Time PCR system (Thermo Fisher Scientific, Inc.). PCR thermal cycling was conducted at 95°C for 15 sec, followed by denaturing at 95°C for 5 sec, annealing at 60°C for 60 sec and a final extension step at 72°C for 10 sec, for a total of 40 cycles. The reaction was duplicated for each sample. The relative expression levels of each transcript were normalized to GAPDH expression. The quantitative fold changes in gene expression were calculated using the 2−ΔΔCq (18) method following normalization to GAPDH.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 30 min and permeabilized using 0.5% Triton X-100 for 5 min, followed by blocking with 5% bull serum albumin (Dingguo Changsheng Biotechnology Co., Ltd., Beijing, China) at room temperature for 60 min. The fixed cells were incubated with specific primary α-SMA monoclonal antibody (1:200, cat no. AA132; Beyotime Institute of Biotechnology, Haimen, China) for 12 h at room temperature, washed with PBS, and incubated with streptavidin biotin complex-fluorescein isothiocyanate (Boster Biological Technology, Pleasanton, CA, USA) at 37°C for 30 min. The cells were incubated with DAPI (BestBio, Shanghai, China) at room temperature for 1 min, immunofluorescence was observed and images were captured using an Olympus immunofluorescence microscope (Olympus Corporation, Tokyo, Japan) at ×40 magnification.
Western blot analysis
Cells were washed off the plates with PBS at 4°C and total protein was extracted using radioimmunoprecipitation assay lysis buffer (Boster Biological Technology) supplemented with protease inhibitors (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) for 30 min. The lysates were centrifuged at 14,000 × g for 15 min at 4°C. The protein concentration of lysates was measured using a bicinchoninic acid protein assay (Beyotime Institute of Biotechnology). Equal amounts of protein from each sample (40 µg) were separated using 10% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The membranes were blocked with 5% non-fat dry milk in TBS for 1 h at room temperature and incubated overnight at 4°C with the following primary antibodies: Mouse monoclonal collagen type I antibody (1:1,000; cat no. sc-59772; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), mouse anti-rat α-SMA monoclonal antibody (1:1,000; cat no. AA132; Beyotime Institute of Biotechnology), rabbit anti-rat phosphorylated (p)ERK1/2 (1:500; cat no. 4370L) and ERK1/2 (1:500; cat no. 4695S) monoclonal antibodies, rabbit anti-rat p-Smad2 (1:500; cat no. 3108P) and Smad2 (1:500; cat no. 3122S) monoclonal antibodies, rabbit anti-rat p-Smad3 (1:500; cat no. 9520S) and Smad3 (1:500; cat no. 9523P) monoclonal antibodies (all from Cell Signaling Technology, Inc., Danvers, MA, USA), and rabbit anti-rat GAPDH polyclonal antibody (1:1,000; cat no. 25778; Santa Cruz Biotechnology, Inc.), for 12 h at 4°C. Membranes were washed three times and subsequently incubated with horseradish peroxidase-conjugated goat anti-rabbit (1:2,000; cat no. A0208) or goat anti-mouse (1:2,000; cat no. A0216) immunoglobulin G secondary antibodies (Beyotime Institute of Biotechnology) at 37°C for 2 h. The membranes were washed with PBS and were exposed to an enhanced chemiluminescence reagent (Beyotime Institute of Biotechnology). Protein bands were quantified using Quantity One software (version 4.6.3; Bio-Rad Laboratories, Inc.) and normalized against the loading control, GAPDH.
Statistical analysis
All experimental data are presented as the mean ± standard error of the mean and were analyzed using SPSS software (version 17.0; SPSS Inc., Chicago, IL, USA). Statistical analysis was performed using one-way analysis of variance and Student-Newman-Keuls post hoc analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
MaR 1 inhibits TGF-β1-induced proliferation in MRC5 cells
As presented in Fig. 1B, TGF-β1 significantly increased MRC5 cell viability measured by MTT assay (P<0.05). Incubation with MaR 1 alone exerted no effects on MRC5 cell viability compared with the untreated group (P>0.05). Pretreated with MaR 1, the TGF-β1-induced proliferation of MRC5 cells was significantly decreased at 10 and 100 nM (P<0.05). The lower concentration of MaR 1 (1 nM) exerted no effect on viability. As presented in Fig. 1B, the TGF-β1-induced proliferative ability decreased in a dose-dependent manner. The concentrations of 10 ng/ml and 10 nM were selected to be the working concentration of TGF-β1 and MaR 1, respectively.
MaR 1 attenuates TGF-β1-induced migration in MRC5 cells
The scratch assay was used to measure the effect of MaR 1 on TGF-β1-induced fibroblast migration. TGF-β1 significantly increased the migration of fibroblasts compared with the control and MaR 1-only treated groups (P<0.05). MaR 1 alone exerted no apparent influence on the migration of fibroblasts. However, administration of MaR 1 significantly decreased fibroblast migration and scratch closure induced by TGF-β1 (P<0.05; Fig. 2).
Figure 2.

MaR 1 inhibits MRC5 cell migration induced by TGF-β1. (A) Images were captured 24 h subsequent to the wound being made. (B) Quantification of cell migration was performed by counting the cell numbers within the rectangle in four fields. Original magnification, ×200. Data are presented as the mean ± standard error. n=6. *P<0.05 vs. control group. #P<0.05 vs. TGF-β1-treated group. MaR1, maresin 1; TGF-β1, transforming growth factor β1.
MaR 1 inhibits MRC5 cell differentiation into myofibroblasts, induced by TGF-β1 in vitro
In order to determine whether MaR 1 exerts an inhibitory effect on the TGF-β1-induced differentiation of fibroblasts into myofibroblasts, the expression of α-SMA and collagen type I was examined in MRC5 cells treated with TGF-β1, in the presence or absence of MaR 1. RT-qPCR analysis demonstrated that administration of TGF-β1 resulted in a significant increase in α-SMA and COL1A1 (the gene that encodes the pro-α1 chains of collagen type I) mRNA levels compared with the control and MaR 1-only treated groups. By contrast, treatment with MaR 1 decreased the mRNA level of α-SMA induced by TGF-β1. It was observed that MaR 1 decreased the TGF-β1-induced expression of COL1A1 mRNA, an additional marker of myofibroblast differentiation (P<0.05; Fig. 3). Immunofluorescence staining analysis demonstrated that treatment with TGF-β1 increased the intensity of α-SMA staining in MRC5 cells; whereas, treatment of MRC5 cells with TGF-β1 following MaR 1 significantly inhibited this enhancement (P<0.05). In addition, MaR 1 alone exerted no effect on α-SMA expression (Fig. 4A). Consistent with the results of the RT-qPCR and immunofluorescence staining assays, western blot analysis demonstrated that MaR 1 decreased the TGF-β1-induced expression of α-SMA and collagen type I (P<0.05). However, MaR 1 alone exhibited no effect on protein expression (Fig. 4B and C).
Figure 3.

Reverse transcription-quantitative polymerase chain reaction analysis of (A) α-SMA and (B) COL1A1 mRNA expression levels in MRC5 cells. MaR 1 was observed to suppress the α-SMA and COL1A1 mRNA expression levels. Data are presented as the mean ± standard error. n=6. *P<0.05 vs. control group. #P<0.05 vs. TGF-β1-treated group. α-SMA, α-smooth muscle actin; COL1A1, collagen α-1(I) chain; MaR 1, maresin 1; TGF-β1, transforming growth factor β1.
Figure 4.

MaR 1 inhibits α-SMA and collagen type I protein expression levels. (A) Immunofluorescence staining assay of α-SMA in MRC5 cells. Blue: Cellular nucleus; red: α-SMA. (B) Western blotting and (C) statistical analysis of α-SMA and collagen type I protein expression. Data are presented as the mean ± standard error. n=6. *P<0.05 vs. control group. #P<0.05 vs. TGF-β1-treated group. α-SMA, α-smooth muscle actin; MaR 1, maresin 1; TGF-β1, transforming growth factor β1.
MaR 1 decreases TGF-β1-induced Smad2/3 and ERK1/2 phosphorylation in MRC5 cells
TGF-β1 signals were followed by the activation of cytoplasmic effectors, including Smad and non-Smad mediator proteins. In order to elucidate the possible mechanism involved in the effect of MaR 1 on TGF-β1-stimulated profibrotic activity, the expression of p-Smad2, p-Smad3, and p-ERK1/2 was evaluated. As presented in Fig. 5, treatment with TGF-β1 resulted in significantly increased phosphorylation of Smad2, Smad3 and ERK1/2 compared with the control group. Application of MaR 1 decreased the TGF-β1-induced expression of p-Smad2, p-Smad3 and p-ERK1/2 in MRC5 cells, compared with treatment with TGF-β1 alone. Treatment with MaR 1 alone exhibited no effect on Smad and ERK mediator proteins.
Figure 5.

MaR 1 inhibits TGF-β1-induced Smad and ERK phosphorylation in MRC5 cells. Total protein from whole cell lysates was extracted to analyze (A) p-Smad2/Smad2, (B) p-Smad3/Smad3 and (C) p-ERK/ERK, assessed by western blotting and statistical analysis. Data are presented as the mean ± standard error. n=6. *P<0.05 vs. control group. #P<0.05 vs. TGF-β1-treated group. MaR 1, maresin 1; TGF-β1, transforming growth factor β1; ERK, extracellular signal-related kinase; p, phosphorylated; Smad, mothers against decapentaplegic homolog.
Discussion
In the present study, it was demonstrated that pretreatment with MaR 1 was able to attenuate TGF-β1-dependent lung fibroblast proliferation and migration, TGF-β1-dependent increases in lung fibroblast α-SMA and collagen type I expression and TGF-β1-induced Smad2/3 and ERK1/2 phosphorylation in lung fibroblasts. It was additionally observed that administration of MaR 1 post-TGF-β1 stimulation was able to inhibit human lung fibroblast proliferation, migration and differentiation in vitro (data not presented).
The fibroblast/myofibroblast has been identified to be an important pathogenic cell in all fibrotic diseases through the secretion of excess ECM, cytokines and growth factors, and tissue contraction. Myofibroblasts have three biological origins: Tissue resident fibroblasts; epithelial to mesenchymal transition; and bone marrow-derived mesenchymal precursors (19). Previous evidence has suggested that myofibroblasts may be a target for anti-fibrotic therapy (6–8). Lung fibroblast migration, proliferation and differentiation into a myofibroblast-like cell type are regarded important steps in lung fibrogenesis (5). It is well-known that TGF-β1 is the most potent stimulator of the activation and the differentiation of fibroblasts into myofibroblasts (20). Preventing myofibroblast differentiation in migratory and proliferative fibroblasts has been demonstrated to attenuate lung fibrosis (21).
Recently discovered pro-resolving lipid mediators, including lipoxins, protectins, resolvins and maresins, have been reported to exhibit potent anti-inflammatory and pro-resolving effects in multiple disease models, by acting on neutrophils, macrophages and other inflammatory cells (9,10). Previous research demonstrated that pro-resolving lipid mediators exhibit anti-fibrotic and anti-proliferative abilities. Lipoxin A4 was demonstrated to inhibit the proliferation of human lung fibroblasts evoked by connective tissue growth factor, and suppressed the tumor necrosis factor-α-induced proliferation of rat mesangial cells (22,23). Aspirin-triggered-15R-lipoxin A4 and lipoxin A4 inhibited endothelial cell proliferation stimulated with vascular endothelial growth factor or leukotriene D4 (24). Systemic administration of resolvin 2 and MaR 1 attenuated neointimal hyperplasia and vessel remodeling in a mouse model of arterial injury, by reducing early proliferation in the vessel wall and inhibiting vascular smooth muscle cell migration mediated by platelet-derived growth factor (16). As demonstrated by the MTT and scratch wound healing assays, the present study demonstrated that MaR 1 is able to inhibit fibroblast proliferation and migration stimulated by TGF-β1.
Differentiated myofibroblasts are characterized by increased expression of collagen and α-SMA, the most commonly used molecular markers (6). The results of the present study demonstrated that MaR 1 prevents TGF-β1-induced myofibroblast differentiation in cultured lung fibroblasts, as exhibited by the underexpression of collagen type I and α-SMA at the mRNA and protein levels. It was hypothesized that, in addition to inhibiting epithelial-mesenchymal transition in lung alveolar epithelial cells (17), MaR 1 may inhibit the effects of TGF-β1 on fibroblasts, demonstrating a second, synergistic mechanism whereby MaR 1 exerts its anti-fibrotic effects. To the best of our knowledge, the present study is the first to directly demonstrate that MaR 1 inhibits TGF-β1-induced myofibroblast differentiation from lung fibroblasts.
TGF-β1 exhibits numerous biological functions associated with airway remodeling, fibrosis and myofibroblast differentiation, including enhanced cellular migration, proliferation and ECM synthesis (25,26). TGF-β1 elicits its biological activities by signaling through a receptor complex of serine/threonine kinase type I and type II receptors, which induces signal transduction through receptor-mediated Smad signaling pathway in addition to non-Smad signaling pathways (27). In order to investigate the possible signaling cascade by which MaR 1 may suppress human lung fibroblast cell activation, the effects of MaR 1 on the Smad2/3 and ERK1/2 signaling pathways in TGF-β1-stimulated fibroblasts were investigated. It has been demonstrated that the TGF-β1/Smad pathway is activated in myofibroblast differentiation, and inhibiting its phosphorylation may attenuate myofibroblast differentiation and the fibrogenic response. Dong et al (28) demonstrated that interleukin-27 inhibited the proliferation, differentiation and collagen synthesis of lung fibroblasts by reducing the activation of the TGF-β1/Smad signaling pathways. In cystic fibrosis lungs, TGF-β1 signaling was markedly increased, as observed by p-Smad2 expression, increased myofibroblast differentiation and tissue fibrosis (29). Tumelty et al (30) reported that aortic carboxypeptidase-like protein increased α-SMA and collagen protein expression by inducing Smad2/3 phosphorylation in primary lung fibroblasts and promoted myofibroblast differentiation. As expected, the results of the present study demonstrated that p-Smad2 and p-Smad3 were highly expressed in TGF-β1 treated cells compared with controls, and that this effect was inhibited by treatment with MaR 1. The results of the present study suggested that MaR 1 inhibited myofibroblast differentiation, in part through the inhibition of Smad2 and Smad3 phosphorylation.
In addition to Smad signaling pathways, previous studies have demonstrated that non-Smad pathways, including ERK1/2, may serve a role in TGF-β1-mediated biological activities. The ratio of p-ERK to ERK indicates the degree of activation of the ERK pathway, which serves a role in a number of fibroblast/myofibroblast functions, including proliferation, migration and differentiation. A previous study demonstrated that MaR 1 may protect against liver injury induced by carbon tetrachloride by inactivating mitogen-activated protein kinase/ERK1/2 signaling pathways (15). An additional previous study demonstrated that the downregulation of ERK1/2 inhibited high mobility group protein B1-induced myofibroblast differentiation and migration, using the human lung fibroblast cell line WI-38 (31). In addition, early reports indicated that IM-412 inhibited TGF-β1 induced expression of the fibrotic markers α-SMA and fibronectin, and collagen accumulation in human lung fibroblasts by decreasing Smad2 and Smad3 phosphorylation, in addition to ERK activity (32). Chung et al (33) reported that resistin like α induced myofibroblast differentiation by increasing the rapid phosphorylation of the ERK signaling pathway. Consistent with this report, the results of the present study demonstrated that the effects of MaR 1 were partly mediated by decreasing ERK1/2 phosphorylation in TGF-β1-treated human lung fibroblasts. A number of growth factors stimulate ERK1/2, and it has been reported that activated ERK phosphorylates Smad proteins within their linker regions, resulting in the maintenance of Smad-mediated biological activities (34). However, the precise mechanisms through which MaR 1 attenuates the TGF-β1-induced proliferation, migration and differentiation of human lung fibroblasts requires further clarification.
In conclusion, the results of the present study demonstrated that MaR 1 inhibited TGF-β1-dependent profibrotic activity in lung fibroblasts when administered prior to or following TGF-β1 stimulation, and that the protective effect of MaR 1 may be associated with inactivation of the Smad2/3 and ERK1/2 signaling pathways in lung fibroblasts. The present study provided evidence for the potential role of MaR 1 in fibrotic lung disease. In addition to lung fibrosis, it is hypothesized that MaR 1 may act on other types of fibroblasts, representing a potentially useful therapeutic strategy in other fibrotic diseases.
References
- 1.Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:434–439. doi: 10.1513/pats.200601-006AW. [DOI] [PubMed] [Google Scholar]
- 2.Ding NH, Li JJ, Sun LQ. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr Drug Targets. 2013;14:1347–1356. doi: 10.2174/13894501113149990198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez AM, Shen Z, Ritzenthaler JD, Roman J. Myofibroblast transdifferentiation in obliterative bronchiolitis: Tgf-beta signaling through smad3-dependent and -independent pathways. Am J Transplant. 2006;6:2080–2088. doi: 10.1111/j.1600-6143.2006.01430.x. [DOI] [PubMed] [Google Scholar]
- 4.Wilson MS, Wynn TA. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: Patterns and perpetrators. J Clin Invest. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maharaj S, Shimbori C, Kolb M. Fibrocytes in pulmonary fibrosis: A brief synopsis. Eur Respir Rev. 2013;22:552–557. doi: 10.1183/09059180.00007713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sivakumar P, Ntolios P, Jenkins G, Laurent G. Into the matrix: Targeting fibroblasts in pulmonary fibrosis. Curr Opin Pulm Med. 2012;18:462–469. doi: 10.1097/MCP.0b013e328356800f. [DOI] [PubMed] [Google Scholar]
- 8.Phan SH. The myofibroblast in pulmonary fibrosis. Chest. 2002;122(Suppl 6):S286–S289. doi: 10.1378/chest.122.6_suppl.286S. [DOI] [PubMed] [Google Scholar]
- 9.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serhan CN, Chiang N. Resolution phase lipid mediators of inflammation: Agonists of resolution. Curr Opin Pharmacol. 2013;13:632–640. doi: 10.1016/j.coph.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dalli J, Zhu M, Vlasenko NA, Deng B, Haeggström JZ, Petasis NA, Serhan CN. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1), inhibits leukotriene A4 hydrolase (LTA4H), and shifts macrophage phenotype. FASEB J. 2013;27:2573–2583. doi: 10.1096/fj.13-227728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marcon R, Bento AF, Dutra RC, Bicca MA, Leite DF, Calixto JB. Maresin 1, a proresolving lipid mediator derived from omega-3 polyunsaturated fatty acids, exerts protective actions in murine models of colitis. J Immunol. 2013;191:4288–4298. doi: 10.4049/jimmunol.1202743. [DOI] [PubMed] [Google Scholar]
- 13.Nordgren TM, Heires AJ, Wyatt TA, Poole JA, LeVan TD, Cerutis DR, Romberger DJ. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respir Res. 2013;14:51. doi: 10.1186/1465-9921-14-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong J, Wu ZY, Qi H, Chen L, Li HB, Li B, Yao CY, Wang YX, Wu J, Yuan SY, et al. Maresin 1 mitigates LPS-induced acute lung injury in mice. Br J Pharmacol. 2014;171:3539–3550. doi: 10.1111/bph.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li R, Wang Y, Zhao E, Wu K, Li W, Shi L, Wang D, Xie G, Yin Y, Deng M, et al. Maresin 1, a proresolving lipid mediator, mitigates carbon tetrachloride-induced liver injury in mice. Oxid Med Cell Longev. 2016;2016:9203716. doi: 10.1155/2016/9203716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akagi D, Chen M, Toy R, Chatterjee A, Conte MS. Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J. 2015;29:2504–2513. doi: 10.1096/fj.14-265363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Li R, Chen L, Tan W, Sun Z, Xia H, Li B, Yu Y, Gong J, Tang M, et al. Maresin 1 inhibits epithelial-to-mesenchymal transition in vitro and attenuates bleomycin induced lung fibrosis in vivo. Shock. 2015;44:496–502. doi: 10.1097/SHK.0000000000000446. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: One function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santana A, Saxena B, Noble NA, Gold LI, Marshall BC. Increased expression of transforming growth factor beta isoforms (beta 1, beta 2, beta 3) in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 1995;13:34–44. doi: 10.1165/ajrcmb.13.1.7541221. [DOI] [PubMed] [Google Scholar]
- 21.Yang S, Cui H, Xie N, Icyuz M, Banerjee S, Antony VB, Abraham E, Thannickal VJ, Liu G. miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB J. 2013;27:2382–2391. doi: 10.1096/fj.12-219493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu SH, Wu XH, Lu C, Dong L, Chen ZQ. Lipoxin A4 inhibits proliferation of human lung fibroblasts induced by connective tissue growth factor. Am J Respir Cell Mol Biol. 2006;34:65–72. doi: 10.1165/rcmb.2005-0184OC. [DOI] [PubMed] [Google Scholar]
- 23.Wu SH, Wu XH, Lu C, Dong L, Zhou GP, Chen ZQ. Lipoxin A4 inhibits connective tissue growth factor-induced production of chemokines in rat mesangial cells. Kidney Int. 2006;69:248–256. doi: 10.1038/sj.ki.5000025. [DOI] [PubMed] [Google Scholar]
- 24.Fierro IM, Kutok JL, Serhan CN. Novel lipid mediator regulators of endothelial cell proliferation and migration: Aspirin-triggered-15R-lipoxin A(4) and lipoxin A(4) J Pharmacol Exp Ther. 2002;300:385–392. doi: 10.1124/jpet.300.2.385. [DOI] [PubMed] [Google Scholar]
- 25.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 26.Camoretti-Mercado B, Solway J. Transforming growth factor-beta1 and disorders of the lung. Cell Biochem Biophys. 2005;43:131–148. doi: 10.1385/CBB:43:1:131. [DOI] [PubMed] [Google Scholar]
- 27.Samarakoon R, Overstreet JM, Higgins PJ. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell Signal. 2013;25:264–268. doi: 10.1016/j.cellsig.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong Z, Zhao X, Tai W, Lei W, Wang Y, Li Z, Zhang T. IL-27 attenuates the TGF-β1-induced proliferation, differentiation and collagen synthesis in lung fibroblasts. Life Sci. 2016;146:24–33. doi: 10.1016/j.lfs.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 29.Harris WT, Kelly DR, Zhou Y, Wang D, MacEwen M, Hagood JS, Clancy JP, Ambalavanan N, Sorscher EJ. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS One. 2013;8:e70196. doi: 10.1371/annotation/d0132de3-56ca-4258-9119-bdab0ceb2cff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tumelty KE, Smith BD, Nugent MA, Layne MD. Aortic carboxypeptidase-like protein (ACLP) enhances lung myofibroblast differentiation through transforming growth factor β receptor-dependent and -independent pathways. J Biol Chem. 2014;289:2526–2536. doi: 10.1074/jbc.M113.502617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee CC, Wang CN, Lee YL, Tsai YR, Liu JJ. High mobility group box 1 induced human lung myofibroblasts differentiation and enhanced migration by activation of MMP-9. PLoS One. 2015;10:e0116393. doi: 10.1371/journal.pone.0116393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park S, Ahn JY, Lim MJ, Kim MH, Lee SL, Yun YS, Jeong G, Song JY. IM-412 inhibits transforming growth factor beta-induced fibroblast differentiation in human lung fibroblast cells. Biochem Biophys Res Commun. 2010;399:268–273. doi: 10.1016/j.bbrc.2010.07.067. [DOI] [PubMed] [Google Scholar]
- 33.Chung MJ, Liu T, Ullenbruch M, Phan SH. Antiapoptotic effect of found in inflammatory zone (FIZZ)1 on mouse lung fibroblasts. J Pathol. 2007;212:180–187. doi: 10.1002/path.2161. [DOI] [PubMed] [Google Scholar]
- 34.Hough C, Radu M, Doré JJ. Tgf-beta induced Erk phosphorylation of smad linker region regulates smad signaling. PLoS One. 2012;7:e42513. doi: 10.1371/journal.pone.0042513. [DOI] [PMC free article] [PubMed] [Google Scholar]