

Abstract
Exposure to chronic stress upregulates blood glucocorticoid levels and impairs cognition via diverse epigenetic mechanisms, such as histone deacetylation. Histone deacetylation can lead to transcriptional silencing of many proteins involved in cognition and may also cause learning and memory dysfunction. Histone deacetylase-2 (HDAC2) has been demonstrated to epigenetically block cognition via a reduction in the histone acetylation level; however, it is unknown whether HDAC2 is involved in the cognitive decline induced by chronic stress. To the best of authors' knowledge, this is the first study to demonstrate that the stress hormone corticosteroid upregulate HDAC2 protein levels in neuro-2a cells and cause cell injuries. HDAC2 knockdown resulted in a significant amelioration of the pathological changes in N2a cells via the upregulation of histone acetylation and modifications in the phosphoinositide 3-kinase/protein kinase B signaling pathway. In addition, the HDAC2 protein levels were upregulated in 12-month-old female C57BL/6J mice under chronic stress in vivo. Taken together, these findings suggested that HDAC2 may be an important negative regulator involved in chronic stress-induced cognitive impairment.
Keywords: histone deacetylation-2, chronic stress, cognition, epigenetics, glucocorticoid
Introduction
Chronic stress exposure induces pathological changes in the central nervous system that affect both emotion and cognition (1). The hypothalamic-pituitary-adrenal (HPA) axis is activated and glucocorticoids (corticosterone in rodents and cortisol in humans) are released at higher levels in response to stress. Glucocorticoid receptors (GRs) are located throughout the brain, including the cortex and hippocampus, which represent regions closely associated with learning and memory (2). Glucocorticoids bind to GRs and inhibit cognitive function during stress (3). Numerous studies have demonstrated that chronic stress and/or release of stress hormones lead to the deterioration of cognition through various mechanisms, including epigenetic regulation (4–6). Histone deacetylation is a typical epigenetic regulatory mechanism involved in stress. Many gene transcription events related to cognition are blocked by histone deacetylation during stress (7,8). Histone deacetylases (HDACs) are vital for the catalysis of histone deacetylation (9,10). The relationship between specific HDAC proteins and chronic stress with regards to cognitive impairment remains unclear.
Histone deacetylase-2 (HDAC2) is a member of the class I HDAC family. Although HDAC2 is diffusely distributed in the cerebrum, the most densely stained neurons are located in the CA1-CA3 regions of the hippocampus (11). A recent study has demonstrated that HDAC2 upregulation exerts an important epigenetic blockade of cognition (12). HDAC2 overexpression exerts negative effects regarding memory because it suppresses spine formation and decreases the spine density of hippocampal neurons (12). HDAC2 activity leads to reduced histone H3 and H4 acetylation levels, which results in memory-related gene silencing, such as brain-derived neurotrophic factor (BDNF), c-Fos, and glutamate receptor 1, in mice (12). Both HDAC inhibitors and HDAC2 knockdown are neuroprotective (13–16). Moreover, HDAC2 is increased in both animal models and patients with neurodegenerative diseases, such as Alzheimer's disease (AD), and may represent a novel drug target (13). Despite the recent attention focused on HDAC2, the mechanism of HDAC2 upregulation in diseases is largely unknown (17,18).
GRs and HDAC2 are both expressed in the hippocampus (19,20). Therefore, there is a possibility that they co-localize and interact. In the present study, it is demonstrated that the administration of the stress hormone corticosteroid or chronic stress increased HDAC2 levels. The pathological changes and potential mechanisms were subsequently investigated, and it was determined that a stress-induced increase in HDAC2 was accompanied by cellular dysfunction and cognitive decline in mice. HDAC2 knockdown partially prevented stress-induced cellular injuries and partially restored the levels of histone H3K9 acetylation and phosphoinositide 3-kinase/protein kinase B pathway phosphorylation. The current findings indicated that HDAC2 is involved in stress-induced cognitive impairment. To the best of the authors' knowledge, this is the first study to demonstrate that HDAC2 is directly involved in stress-related cognitive decline.
Materials and methods
Cell culture and treatments
Mouse neuroblastoma N2a cells (China National Infrastructure of Cell Line Resource, Beijing, China) were used and cultured in minimum essential medium (MEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml streptomycin and 100 U/ml penicillin (Gibco; Thermo Fisher Scientific, Inc.) in a 37°C humidified incubator with 95% air and 5% CO2. The cells were resuspended and plated into fresh dishes prior to treatment. For stress hormone administration, following cell attachment, the medium was replaced by fresh MEM with corticosteroids (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at different final concentrations and incubated for an additional 24–48 h prior to collection. For HDAC2 silencing, N2a cells were transfected with a lentivirus-HDAC2-short hairpin (sh)RNA plasmid (LV-HDAC2-shRNA; Biowit Technologies, Ltd., Shenzhen, China), which encoded shRNA that targeted the mouse HDAC2 transcript. The HDAC2 expression levels were confirmed by western blot analysis.
Cell MTT assay
MTT (Sigma-Aldrich; Merck KGaA) was used to assess cell viability as described by Wang et al (21). N2a cells were seeded into a 96-well plate at 1×104 cells/ml and 200 µl/well. The medium was refreshed following cell attachment, and the cells were exposed to corticosteroids at various concentrations (1–10 µM). Following a 24 h incubation, 20 µl MTT solution (5 mg/ml) was added to each well for an additional 4 h culture at 37°C. Cells without corticosteroid treatment were treated with culture medium under the same culture conditions and were used as the control group. The media were emptied, and 200 µl dimethylsulfoxide were added to each well to dissolve the crystals. The absorbance was assessed at 550 nm to determine the cell viability; four vice holes were used per dose.
Cell morphological analysis
The N2a cells were treated with a 2 µM solution of corticosteroids (cat. no. C2505; Sigma-Aldrich; Merck KGaA) for 48 h, and the morphology was analyzed and compared with the control cells. An inverted phase-contrast microscope (Olympus Corporation, Tokyo, Japan) and Image-Pro Plus software (version, 6.0; Media Cybernetics, Inc., Rockville, MD, USA) were used to evaluate morphological changes. The cell density and length of the neurite-like outgrowths were measured, and the cell size, including the number of primary dendrites, were determined as previously described (22). Then, >100 cells were counted per group at random at the same magnification (x40), and at least 3 cultures were used per group.
Immunofluorescence staining
Corticosteroid-treated and control cells were fixed with 4% paraformaldehyde for 10 min at room temperature; the frozen brain sections (8 µm) were fixed for 20 min. The slides were subsequently incubated with 0.5% Triton followed by 3% hydrogen peroxide (H2O2) for 10 min each. The slides were then incubated with mouse anti-HDAC2 (1:200; cat. no. ab51832) and rabbit anti-PSD95 primary antibodies (1:200; cat. no. ab18258), which were both obtained from Abcam (Cambridge, MA, USA), overnight at 4°C. The following day, the slides were incubated with goat anti-mouse and goat anti-rabbit fluorescein-conjugated IgG secondary antibodies (1:100; cat. nos. ZB-0312 and ZB-0311, respectively; Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China) for 40 min at room temperature. The slides were examined via a laser scanning confocal microscope (Olympus Corporation) and analyzed using ImageJ 3.1 software (National Institutes of Health, Bethesda, MD, USA).
Protein extraction and western blot analysis
To obtain the total protein, N2a cells maintained in plates or hippocampal tissue were lysed with a mixture of lysis buffer (25 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA and 0.5% Triton X-100) and protease inhibitors on ice for 30 min. The lysates were clarified via centrifugation at 13,000 × g for 30 min at 4°C. The supernatant was discarded prior to protein analysis and western blotting. A total of 40 µg total proteins from each sample were separated by 10% SDS-PAGE, and transferred to a polyvinylidene difluoride membrane (0.45 µm). The membrane was incubated with 5% fat-free milk to block the blots for 1 h at room temperature prior to incubation with primary antibodies [HDAC2, 1:1,000, cat. no. ab51832; PSD-95, 1:1,000, cat. no. ab18258; acetyl-H3K9, 1:500, cat. no. ab10812; H3, 1:500, cat. no. ab1791; acetyl-H4K8, 1:500, cat. no. ab15823; H4, 1:500, cat. no. ab10158; all obtained from Abcam; Akt, 1:1,000, cat. no. 4691; p-Akt (Ser 473), 1:1,000, cat. no. 4058; PI3K, 1:500, cat. no. 4292; and p-PI3K-p85, 1:500, cat. no. 4228; all obtained from Cell Signaling Technology, Inc., Danvers, MA, USA] overnight at 4°C. The blots were probed with horseradish peroxidase-conjugated GAPDH (1:15,000; KC-5G5; Kangcheng Technology Co., Ltd.), and goat anti-mouse and goat anti-rabbit horseradish peroxidase-conjugated IgG secondary antibodies (1:15,000; cat. nos. ZB-2305 and ZB-2301, respectively; Zhongshan Golden Bridge Biotechnology Co., Ltd.) the following day for 1 h at room temperature and the protein expression levels were visualized via X-ray films and quantified using ImageJ 3.1 software. GAPDH, total H3, total H4, total AKT, and total PI3K were used as loading controls for HDAC2 and PSD-95, acetyl-H3K9, acetyl-H4K8, p-Akt and p-PI3K, respectively.
Animals and the chronic restraint stress procedure
In total, 20 female C57BL/6J mice (12-months-old; weight, 20–30 g) were used because older mice are more sensitive to stress (23) and mice were divided into 2 groups randomly (n=10 per group). The mice were maintained in a temperature- and humidity-controlled room on a 12 h light-dark cycle and had free access to food and water. The use of animals was approved by the Animal Care and Use Committee of The Institute of Laboratory Animal Science of Peking Union Medical College (Beijing, China; ILAS-GC-2012-004). Chronic restraint stress was performed and modified according to Yang et al (24). Briefly, the stressed mice were restrained in cylinder tubes to limit their autonomous actions for 7 h/day (from 11:00 p.m. to 6:00 a.m.) for 28 consecutive days in a behavior science laboratory. The control mice remained in their home cages without stress exposure.
Determination of blood corticosteroids
Fresh serum was obtained immediately following euthanasia at 8:30-9:30 a.m., and corticosteroids were measured with a corticosteroid ELISA kit (ADI-900-097; Enzo Life Sciences, Inc., Farmingdale, NY, USA) according to the instruction manual.
Morris water maze (MWM) test
The MWM procedure was similar to previous descriptions (25) and was conducted for 7 days following the completion of the stress protocol. Briefly, the mice were trained twice per day on days 1–6 during the learning period and examined via a probe test on day 7. The platform was hidden starting on day 2 and was absent on day 7. Each swimming cycle was 60 sec. The mice were considered to have located the platform when they remained on it for at least 5 sec. The automatic tracking system Noldus Ethovision XT (Noldus Information Technology BV, Wageningen, Netherlands) was used to monitor and analyze the data.
Statistical analysis
The data were analyzed using SPSS software (version, 13.0; SPSS, Inc., Chicago, IL, USA). The data are expressed as the means ± standard error of the mean, and P<0.05 was considered to indicate a statistically significant difference. T-tests were used to evaluate the differences between two groups, and one-way analyses of variance (ANOVA) followed by Fisher's least significant difference test were performed to evaluate the differences among multiple groups. Repeated measures and multivariate ANOVAs were used for the MWM.
Results
Corticosteroid administration decreased N2a cell viability
Corticosteroids were used as a stressor agent to mimic stress conditions in vitro. To determine a suitable corticosteroid concentration, the MTT assay was used to evaluate N2a cell viability. Cells were incubated with corticosteroids at a series of concentrations (1, 2, 5 and 10 µM) for 24 h. The absorbance at 550 nm was measured following incubation, and the optical density (OD) value in the defined conditions reflected the number of viable cells present. The OD value exhibited a decreasing trend when the corticosteroid concentration was >1 µM, which indicated that the cells were injured. When the concentration increased to 5 µM, the cell viability significantly decreased, which indicated substantial cell death. Taken together, the stress hormone corticosteroid exhibited debilitative effects on N2a cells, and at a higher dose, it was associated with cell lethality (P<0.05; Fig. 1). Therefore, 2 µM was chosen as a suitable dose to induce injuries without excessive cell apoptosis in the present study.
Figure 1.
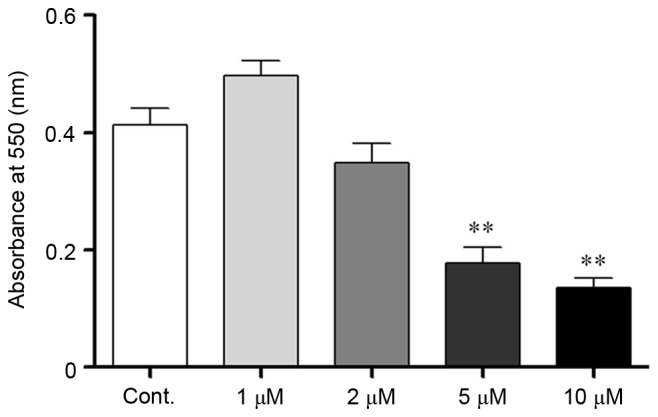
Cell viability assay. Neuro-2a cell viability was determined using a MTT assay following corticosteroid administration at various concentrations (1–10 µM). The absorbance at 550 nm was significantly decreased with increased corticosteroid concentrations >5 µM. Cont, control cells. Data are presented as the mean ± standard error of the mean. **P<0.01 vs. control cells.
Corticosteroid treatment disturbed N2a neurite outgrowth and connections
In the current study, the authors chose a continuous 48 h treatment of 2 µM corticosteroids for the experimental conditions to mimic chronic stress. These conditions induced significant changes in cell morphology and density (P<0.05; Fig. 2A-C). The cell number significantly decreased in these experimental conditions and resulted in a lower cell density, which reflected an effect of corticosteroid growth inhibition on the N2a cells (P<0.05; Fig. 2B). In addition to the reduced cell density, the N2a cells treated with corticosteroid had shorter neurites, and the longest neurite length of the cells significantly decreased after corticosteroid treatment compared with the control cells (P<0.05; Fig. 2C). There were no significant changes in the average neurite number per cell (P>0.05; Fig. 2D). The decreased cell number and neurite outgrowth resulted in a loosened cellular attachment, which reflected a weakened connection and communication among cells. In addition to cell growth inhibition, cell function was also injured after corticosteroid incubation. Postsynaptic density protein 95 (PSD95), a scaffold protein, is a well-established synapse-associated marker that is crucial for synaptic plasticity (26). PSD95 is located in the postsynaptic density domain, and its expression is important for synaptic function. The PSD95 protein was located at the neurite outgrowth of the N2a cells (Fig. 2G). During the experimental conditions, the PSD95 expression level decreased as detected by western blot analysis (P<0.05; Fig. 2E and F). Furthermore, immunofluorescence staining indicated decreased PSD95 expression following corticosteroid incubation (P<0.05; Fig. 2G and H). The downregulated PSD95 verified the functional impairment of the N2a cells.
Figure 2.

Corticosteroid administration induced injury in N2a cells. (A-D) Cell morphological changes following corticosteroid administration at a dose of 2 µM for 48 h. (A and B) Corticosteroid treatment reduced the cell density. The cell number was reduced following treatment compared with the control cells. (A and C) Corticosteroid treatment shortened the neurite outgrowth length of the N2a cells, and the longest neurite length was significantly decreased compared with the control cells. (A and D) There was no significant difference in the average neurite number per cell. (E and F) The expression of synapse-associated protein PSD95 decreased. Western blotting indicated a decrease in PSD95 after 48 h of corticosteroid treatment at a dose of 2 µM. GAPDH was used as the loading control. (G and H) The location and decreased expression of PSD95 were demonstrated by immunofluorescence staining. DAPI was used to stain the nucleus. (G) PSD95 was located at the neurite outgrowth of N2a cells. (G and H) Corticosteroid treatment decreased the fluorescent intensity of PSD95 in N2a cells. For all panels, at least 100 cells in each culture were evaluated, and at least 3 cultures were used per group. The magnification and scale are as follows: (A) magnification, ×40; (G) magnification, ×200; bar, 20 µm. Data are presented as the mean ± standard error of the mean. *P<0.05 vs. control cells; NS, P>0.05 vs. control cells. N2a, neuro-2a; PSD95, postsynaptic density 95; cont, control cells; cort, corticosteroid treated cells.
Corticosteroids induced the deacetylation of histone H3K9 and HDAC2
The acetylation of histone residues, including H3K9, H3K14, H4K8 and H4K12, enhances gene expression related to learning and memory (27). In previous studies of the authors, decreased acetylation levels of histone H3K9 and H4K8 were identified in a mouse model of AD, which is characterized by cognitive impairment (unpublished data). Glucocorticoids can also cause cognitive impairments in mice via a reduction in histone acetylation (28). In the present study, the acetylation levels of H3K9 were significantly decreased following corticosteroid treatment (P<0.05; Fig. 3E and F), consistent with abnormal epigenetic regulation involved in stress, even though H4K8 acetylation remained stable (P>0.05; Fig. 3G-I). Histone deacetylation leads to more compact chromosomes and reduces gene transcription, which thus reduces proteins crucial for learning and memory. Histone deacetylation is catalyzed by histone deacetylases (HDACs), and the protein levels indicated both dose-dependent and time-dependent augmentation of HDAC2 (P<0.05; Fig. 3A-D). HDAC2 was prominently increased following 48 h of corticosteroid treatment at the same dose of 2 µM and exhibited a persistent increase with an increase in dose, when compared with the control group. The levels of HDAC2 were determined using immunofluorescence staining. HDAC2 was expressed in the nucleus (Fig. 3G). At 48 h of corticosteroid incubation at a dose of 2 µM, there was an augmentation of HDAC2 compared with the control cells (Fig. 4G and H). This corresponding upregulation of HDAC2 and cell injuries suggested that HDAC2 may be a negative mediator in corticosteroid-induced neural damage.
Figure 3.

Corticosteroid treatment increased HDAC2 expression. (A and B) Time-dependent effects on HDAC2 protein expression in N2a cells. The cells were incubated with corticosteroid at 2 µM for the indicated times from 24–48 h prior to collection. The HDAC2 protein levels were measured by western blot analysis. The HDAC2 protein increased over time; GAPDH was used as the loading control. (C and D) Dose-dependent expression effects on the HDAC2 protein in N2a cells. The cells treated with an increased dose of corticosteroid from 1–2 µM for 48 h exhibited enhanced HDAC2 protein levels as evaluated by western blot analysis. HDAC2 increased with an increase in the corticosteroid dose. GAPDH was used as the loading control. (G and H) Immunofluorescence staining indicated the subcellular location of HDAC2 protein. HDAC2 was located in the nucleus of N2a cells. Differences in fluorescent staining were identified between groups, and an increasing trend was apparent. DAPI was used to stain the nucleus. (E and F, I and J) Acetylated levels of histone residues related to cognition. Corticosteroid incubation (2 µM, 48 h) significantly decreased the acetylated level of H3K9 but not H4K8. (E and F) The histone H3K9 acetylation levels were reduced following corticosteroid treatment compared with the control cells. Total histone H3/H4 was used as the loading control. Data are presented as the mean ± standard error of the mean. *P<0.05, **P<0.01 vs. control cells. At least 3 cultures were used per group. HDAC2, histone deacetylase 2; N2a, neuro-2a; H4K8, histone 4 lysine 8; H3K9, histone 3 lysine 9; cont, control cell; cort, corticosteroid treated cells.
Figure 4.

Specific HDAC2 knockdown partially resisted corticosteroid-induced N2a cell injury. (A) Lentivirus-shRNA transfection targeted the HDAC2 protein of N2a cells. The N2a cell transfection efficiency was confirmed by the presence of green fluorescence in the cytoplasm. (C and D) The HDAC2 protein expression level following transfection was detected by western blotting. The HDAC2 protein significantly decreased 48 h after the transfection efficiency evaluation. The lentivirus-shRNA transfection that targeted HDAC2 protein was resistant to the corticosteroid-induced HDAC2 upregulation in N2a cells. HDAC2 did not increase during corticosteroid administration following transfection. GAPDH was used as the loading control. (B) Morphological recovery of HDAC2 knockdown N2a cells following corticosteroid administration (2 µM, 48 h). The cells exhibited a partially restored density and the longest neurite length following transfection. (E and F) The PSD95 levels exhibited a trend toward recovery following HDAC2 knockdown in N2a cells. Data are presented as the mean ± standard error of the mean. *P<0.05 vs. control cells or between two groups. At least 3 cultures were used per group. HDAC2, histone deacetylase 2; N2a, neuro-2a; shRNA, short hairpin RNA; PSD95, postsynaptic density 95; cont, control cells; cort, corticosteroid treated cells (2 µM, 48 h); shR, transfected cells; scr, scramble shRNA transfected cells; GFP, green fluorescence protein.
HDAC2 acts as a negative regulator involved in corticosteroid-induced injuries via the PI3K/AKT signaling cascade
Based on previous results, the authors hypothesized that HDAC2 mediates cell injuries following incubation with corticosteroids. To test this hypothesis, further studies were performed. To knockdown HDAC2, cells were transfected with LV-shRNA-mHdac2. Lentivirus vectors that expressed Hdac2 shRNA were added to the media, with the multiplicity of infection = 4.6IU/cell. Polybrene was used as the transfection enhancer. The vectors also carried the gene encoding green fluorescent protein, and successfully transfected cells were recognized via green fluorescence. Scrambled shRNA was used as a control. Green fluorescence was present after transfection, and the efficiency was >50% (Fig. 4A). Western blot analysis indicated a significant reduction in HDAC2 expression following transfection (P<0.05; Fig. 4C and D). HDAC2 silencing was maintained during corticosteroid treatment, as presented in Fig. 4C and D. The transfected cells did not exhibit an increase in HDAC2 following corticosteroid treatment compared with the control cells.
Cell morphology was assessed again following transfection. Consistent with the authors' hypothesis, knockdown of HDAC2 restored cell density in the stressed cells (Fig. 4B). In addition, the cell neurite outgrowth length was partially restored (Fig. 4B). These morphological improvements in the cells provided an indicator of improved cell viability. In addition, PSD95 expression was partially restored, which was evaluated by western blot analysis (Fig. 4E and F). HDAC2 knockdown was followed by an improvement of cell function, and the cell recovery mechanism was subsequently explored. As previously discussed, the acetylation of histones, such as histone H3K9 acetylation, was reduced. Following corticosteroid treatment, transfected cells exhibited an augmentation of acetylated H3K9 compared with the control cells, as demonstrated by western blot analysis (P<0.05; Fig. 5A and B), which may explain, in part, the restoration of protein PSD95 expression.
Figure 5.

LV-shRNA-HDAC2 knockdown partially restored the reduced acetylation level of histone H3K9 and increased PI3K-AKT signaling pathway phosphorylation. (A and B) Western blot analysis indicated restored acetylation of H3K9 following lentivirus-shRNA-HDAC2 transfection. Corticosteroid treatment (2 µM, 48 h) significantly decreased the histone acetylation levels of H3K9 in neuro-2a (N2a) cells. HDAC2-shRNA transfected cells exhibited increased histone H3K9 acetylation levels during corticosteroid treatment compared with the untransfected cells in the same condition. Total H3 was used as the loading control. (C-F) HDAC2 knockdown increased PI3K/AKT phosphorylation. (C and D) In the same treatment conditions, the phosphorylation of PI3K p85 in HDAC2-shRNA transfected cells was substantially increased compared with the untransfected cells. Total PI3K was used as the loading control. (E and F) Following corticosteroid incubation, AKT (ser473) phosphorylation in HDAC2-shRNA transfected cells was partially restored compared with the untransfected cells. Total AKT was used as the loading control. Data are presented as the mean ± standard error of the mean. *P<0.05, **P<0.01 between two groups. At least 3 cultures were used per group. shRNA, short hairpin RNA; HDAC1, histone deacetylase 2; H3K9 histone 3, lysine 9; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; n2a, neuro-2a; cont, control cells; cort, corticosteroid treated cells; scr, scramble shRNA transfected cells; shR, transfected cells.
Next, the phosphorylation status of the PI3K/AKT signaling cascade was assessed. HDAC2 knockdown improved the protective phosphorylation of this signaling pathway (P<0.05; Fig. 5C-F). The phosphorylation of the PI3K subunit p85 is an activated form that phosphorylates downstream AKT, and the activated PI3K/AKT cascade was beneficial for neurons (29). The phosphorylation levels of p85 and serine 473 of AKT decreased dramatically during corticosteroid incubation; however, this trend could be partially reversed by HDAC2 knockdown.
Chronic restraint stress impaired cognition in mice and up-regulated HDAC2
The chronic restraint procedure is a reliable model of chronic stress and has been widely used (30,31). Following stress, the HPA axis is activated, and the main stress hormone glucocorticoid is overexpressed, which exerts negative effects on cognition. On average, an increased blood corticosteroid level was identified in the stressed mice compared with the control mice (P<0.05; Fig. 6B), which indicates the involvement of stress hormones during stress and is consistent with previous studies (32). The stressed mice simultaneously exhibited lower body weights (P<0.05; Fig. 6A), which indicates disturbed living states. The MWM test is a classical behavior detection method that was used to evaluate cognitive performance and identify cognitive impairment in the chronic stressed mice (Fig. 6C and D). In the learning period, there was a significant difference in the escape latency between groups starting on day 3 (P<0.05; Fig. 6C). Specifically, the stressed mice took a longer time to find the platform compared with the wild-type control mice starting on day 3 in the learning period. The weakened learning abilities of the stressed mice lasted for four days to the end of this test period. During the exploratory period, the frequency of crossing the platform zone in the stressed group was also significantly less than the control group (P<0.05; Fig. 6D), and the stressed mice exhibited a deteriorated ability to remember the exact platform position. These results confirmed that chronic stress could cause cognitive decline via glucocorticoid upregulation, which is consistent with previous reports (33,34).
Figure 6.

Chronic restraint stress caused cognitive impairment in C57BL/6J mice that corresponded to increased hippocampal HDAC2 protein levels. (A) Chronic restraint stress significantly decreased body weight in the stressed mice compared with the wild-type control mice. (B) Chronic restraint stress triggered glucocorticoid release and increased blood glucocorticoid levels; the stressed mice exhibited increased blood glucocorticoid levels on average compared with the wild-type control mice. (C and D) The Morris Water Maze test indicated that chronic restraint stress caused cognitive impairment in mice. (C) Behavioral changes in the learning period. The escape latency was significantly longer in the stressed mice compared with the wide-type mice starting on day 3. (D) Frequency of finding the platform during the exploration period. After stress, the mice less frequently arrived at the platform zone and had a decreased ability to find the platform. (E-H) Changes in HDAC2 brain protein level following stress. (E and F) The hippocampal HDAC2 protein was significantly upregulated following stress compared with the control mice as demonstrated by western blotting. GAPDH was used as the loading control. (G and H) The cortical HDAC2 protein level remained the same after stress. GAPDH was used as the loading control. Data are presented as the mean ± standard error of the mean. *P<0.05 compared with the wild-type group. NS, P>0.05 vs. wild-type group. HDAC2, histone deacetylase 2; wt, wild-type control group; crs, chronic restraint stress group.
Following the MWM trials, both the stressed and control mice were euthanized. Proteins from the hippocampus and cortex related to cognition were independently extracted. Following the identification of cognitive decline during stress, HDAC2 was assessed via western blotting for its specific role in blocking cognition. Interestingly, there was a corresponding change in HDAC2 during chronic stress. The hippocampal HDAC2 protein levels were significantly increased compared with the wild-type control mice (P<0.05; Fig. 6E and F); however, cortical HDAC2 was unchanged (P>0.05, Fig. 6G and H). The hippocampus is the predominate region related to cognition, and the upregulation of hippocampal HDAC2 following stress confirmed that HDAC2 is involved in chronic stress-induced cognitive impairment in vivo.
Discussion
The current findings indicated that both stress hormones and chronic restraint stress induce neural injuries and increase HDAC2. HDAC2 knockdown may partially resist these pathological changes via an upregulation of histone H3K9 acetylation and PI3K/AKT pathway phosphorylation in N2a cells.
Chronic stress is harmful for cognition and is a risk factor for the onset of neurodegenerative diseases. The injury mechanisms during stress have been well established (3). The HPA axis is activated during stress, which is followed by the over-release of glucocorticoids (corticosterone in rodents and cortisol in humans). In the present study, the blood glucocorticoid levels of the stressed mice were significantly upregulated as expected. Epigenetic mechanisms, especially histone acetylation/deacetylation, have been demonstrated to be involved in cognitive regulation and indicate novel regulation patterns associated with stress. Chronic stress can reduce global histone acetylation levels (35). The glucocorticoid receptor, which comprises the key stress hormone receptor, can directly regulate histone acetylation levels, including histone H3K14 (36) and H4K12 (37). Many histone residues, such as H2BK5, H3K9, H3K14, H4K5 and H4K12, are important for learning, memory and synaptic plasticity (38). Histone acetylation makes the transcription of cognition-related genes substantially easier; these genes include PSD95 and BDNF, which facilitate learning and memory (39,40).
N2a cells are broadly utilized in neuroscience research, as well as in stress related fields. Stress hormones, such as dexamethasone and corticosteroid, have also been used to mimic stress conditions (41). In general, a 48 h or 72 h administration is sufficient to induce long lasting observations according to the growth curve of N2a cells. In the present study, a 48 h corticosteroid treatment was sufficient to induce pathological and molecular changes. Corticosteroid caused neural impairments and reduced the histone H3K9 acetylation level. At the same time, PSD95 was downregulated, which supported epigenetic dysregulation during stress. Histone deacetylases are necessary to catalyze histone deacetylation. Glucocorticoid receptors can interact with transcription factors, such as histone acetylases and histone deacetylases (42,43), which indicate that chronic stress may exert epigenetic regulation via HDACs. HDAC2, a member of the class I HDAC family, is a key negative regulator involved in cognition (12,16,44,45). HDAC2 and glucocorticoid receptors are both located in the hippocampus (11,19), and a glucocorticoid receptor recognition element has been identified in the HDAC2 proximal promoter region (13). These findings suggested that HDAC2 may be involved in stress-related cognitive decline, a hypothesis that was preliminary demonstrated in the present study. Furthermore, increased HDAC2 protein levels were induced during chronic stress conditions both in vitro and in vivo, in parallel with injuries. Following HDAC2 knockdown, both the histone H3K9 acetylation and PSD95 protein levels in N2a cells could be partially restored. Taken together, HDAC2 is a negative regulator of cognition during stress via histone deacetylation.
The PI3K/AKT signaling pathway has been explored in depth in the field of learning and memory. AKT (ser-473) phosphorylation induces its active form. The active form of the PI3K/AKT signaling pathway can inhibit neural apoptosis and improve neural morphology, which enhances neuronal communication and synaptic plasticity (46). In addition, PI3K/AKT signaling pathway activation mediates the protective functions of several nerve growth factors, such as insulin growth factor-1 and BDNF. The expression of several dendritic spine density and presynaptic markers in hippocampal neurons is dependent on PI3K/AKT signaling pathway activation (47). Glucocorticoids may damage this signal transduction (48). Furthermore, a decrease in PI3K/AKT signaling pathway phosphorylation may account, in part, for the reduced PSD95.
HDAC2 can regulate AKT activity. HDAC2 has been demonstrated to provide critical support regarding malignant progression via AKT activation in hepatocellular carcinoma (49). In a previous study of the authors, it was demonstrated that a HDAC2 conditional knockout significantly improved cognition via the upregulation of AKT phosphorylation in a mouse model of AD (unpublished data). Insulin resistance has been identified as a risk factor for cognition decline and is involved in the progression of AD. The PI3/AKT pathway is one of the most important pathways in insulin signaling and may exert an important role in cognition. HDAC2 inhibition has been demonstrated to improve insulin signaling transduction (50). As previously discussed, the present study assessed the phosphorylation modifications in the PI3/AKT signaling pathway before and after HDAC2 knockdown. A significant upregulation of protectable p-PI3K and p-AKT following HDAC2 silencing was identified during stress, which indicates that HDAC2 could exhibit a role in impairment via PI3/AKT signaling pathway modification.
Despite these interesting findings, several limitations of the current study should be considered in the interpretation of these findings. The present study used female mice to conduct these experiments because chronic stress is a risk factor of AD with HDAC2 upregulation, and females are more susceptible to this disease. However, one potential issue is that estrogen can affect cognition. As age increases, both estrogen levels and cognition decline. Furthermore, the female mice in the current study were housed in the absence of male mice; thus, the effect of the estrous cycle was likely minimal. Additional studies in male mice should be conducted to examine sex-dependent differences in these findings.
In conclusion, HDAC2 is stress-related and acts as a negative mediator via histone deacetylation and the modification of PI3K/AKT pathway phosphorylation. These findings contribute novel insights into the understanding of the epigenetic mechanisms affected by chronic stress. However, in some neurodegenerative diseases, such as AD, HDAC2 is significantly increased with limited knowledge regarding the mechanisms. Thus, chronic stress-induced increases in HDAC2 protein levels may represent a new research direction, as well as a novel treatment target.
Glossary
Abbreviations
- HDAC2
histone deacetylase-2
- PI3K
phosphoinositide 3-kinase
- AKT
protein kinase B
- HPA
hypothalamic-pituitary-adrenal
- GRs
glucocorticoid receptors
References
- 1.Nicolaides NC, Kyratzi E, Lamprokostopoulou A, Chrousos GP, Charmandari E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation. 2015;22:6–19. doi: 10.1159/000362736. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, van Heerikhuize J, Aronica E, Kawata M, Seress L, Joels M, Swaab DF, Lucassen PJ. Glucocorticoid receptor protein expression in human hippocampus; stability with age. Neurobiol Aging. 2013;34:1662–1673. doi: 10.1016/j.neurobiolaging.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 3.Lucassen PJ, Pruessner J, Sousa N, Almeida OF, van Dam AM, Rajkowska G, Swaab DF, Czéh B. Neuropathology of stress. Acta Neuropathol. 2014;127:109–135. doi: 10.1007/s00401-013-1223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferland CL, Schrader LA. Regulation of histone acetylation in the hippocampus of chronically stressed rats: A potential role of sirtuins. Neuroscience. 2011;174:104–114. doi: 10.1016/j.neuroscience.2010.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenworthy CA, Sengupta A, Luz SM, Ver Hoeve ES, Meda K, Bhatnagar S, Abel T. Social defeat induces changes in histone acetylation and expression of histone modifying enzymes in the ventral hippocampus, prefrontal cortex, and dorsal raphe nucleus. Neuroscience. 2014;264:88–98. doi: 10.1016/j.neuroscience.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 6.Ferland CL, Harris EP, Lam M, Schrader LA. Facilitation of the HPA axis to a novel acute stress following chronic stress exposure modulates histone acetylation and the ERK/MAPK pathway in the dentate gyrus of male rats. Endocrinology. 2014;155:2942–2952. doi: 10.1210/en.2013-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radley JJ, Kabbaj M, Jacobson L, Heydendael W, Yehuda R, Herman JP. Stress risk factors and stress-related pathology: Neuroplasticity, epigenetics and endophenotypes. Stress. 2011;14:481–497. doi: 10.3109/10253890.2011.604751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnsson A, Durand-Dubief M, Xue-Franzen Y, Rönnerblad M, Ekwall K, Wright A. HAT-HDAC interplay modulates global histone H3K14 acetylation in gene-coding regions during stress. EMBO Rep. 2009;10:1009–1014. doi: 10.1038/embor.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hildmann C, Riester D, Schwienhorst A. Histone deacetylases-an important class of cellular regulators with a variety of functions. Appl Microbiol Biotechnol. 2007;75:487–497. doi: 10.1007/s00253-007-0911-2. [DOI] [PubMed] [Google Scholar]
- 10.Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: A dynamic perspective. Mol Cell. 2006;23:289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 11.Yao ZG, Liu Y, Zhang L, Huang L, Ma CM, Xu YF, Zhu H, Qin C. Co-location of HDAC2 and insulin signaling components in the adult mouse hippocampus. Cell Mol Neurobiol. 2012;32:1337–1342. doi: 10.1007/s10571-012-9859-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gräff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xuan A, Long D, Li J, Ji W, Hong L, Zhang M, Zhang W. Neuroprotective effects of valproic acid following transient global ischemia in rats. Life Sci. 2012;90:463–468. doi: 10.1016/j.lfs.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Hommet C, Mondon K, de Toffol B, Constans T. Reversible cognitive and neurological symptoms during valproic acid therapy. J Am Geriatr Soc. 2007;55:628. doi: 10.1111/j.1532-5415.2007.01117.x. [DOI] [PubMed] [Google Scholar]
- 16.Morris MJ, Mahgoub M, Na ES, Pranav H, Monteggia LM. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J Neurosci. 2013;33:6401–6411. doi: 10.1523/JNEUROSCI.1001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim HS, Chang YG, Bae HJ, Eun JW, Shen Q, Park SJ, Shin WC, Lee EK, Park S, Ahn YM, et al. Oncogenic potential of CK2α and its regulatory role in EGF-induced HDAC2 expression in human liver cancer. FEBS J. 2014;281:851–861. doi: 10.1111/febs.12652. [DOI] [PubMed] [Google Scholar]
- 18.Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- 19.Saaltink DJ, Vreugdenhil E. Stress, glucocorticoid receptors, and adult neurogenesis: A balance between excitation and inhibition? Cell Mol Life Sci. 2014;71:2499–2515. doi: 10.1007/s00018-014-1568-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolber BJ, Muglia LJ. Defining brain region-specific glucocorticoid action during stress by conditional gene disruption in mice. Brain Res. 2009;1293:85–90. doi: 10.1016/j.brainres.2009.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang P, Jiang S, Cui Y, Yue Z, Su C, Sun J, Sheng S, Tian J. The n-terminal 5-MER peptide analogue P165 of amyloid precursor protein exerts protective effects on SH-SY5Y cells and rat hippocampus neuronal synapses. Neuroscience. 2011;173:169–178. doi: 10.1016/j.neuroscience.2010.10.069. [DOI] [PubMed] [Google Scholar]
- 22.Scott HJ, Stebbing MJ, Walters CE, McLenachan S, Ransome MI, Nichols NR, Turnley AM. Differential effects of SOCS2 on neuronal differentiation and morphology. Brain Res. 2006;1067:138–145. doi: 10.1016/j.brainres.2005.10.032. [DOI] [PubMed] [Google Scholar]
- 23.Baumann VR. Stress sensitivity and adaptation. Z Gesamte Inn Med. 1975;30:15–16. passim. (In German) [PubMed] [Google Scholar]
- 24.Yang X, Han ZP, Zhang SS, Zhu PX, Hao C, Fan TT, Yang Y, Li L, Shi YF, Wei LX. Chronic restraint stress decreases the repair potential from mesenchymal stem cells on liver injury by inhibiting TGF-β1 generation. Cell Death Dis. 2014;5:e1308. doi: 10.1038/cddis.2014.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, He Z, Guo L, Huang L, Wang J, He W. Behavioral alterations associated with a down regulation of HCN1 mRNA in hippocampal cornus ammon 1 region and neocortex after chronic incomplete global cerebral ischemia in rats. Neuroscience. 2010;165:654–661. doi: 10.1016/j.neuroscience.2009.10.053. [DOI] [PubMed] [Google Scholar]
- 26.Ricobaraza A, Cuadrado-Tejedor M, Pérez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 27.Gräff J, Kim D, Dobbin MM, Tsai LH. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91:603–649. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- 28.Stankiewicz AM, Swiergiel AH, Lisowski P. Epigenetics of stress adaptations in the brain. Brain Res Bull. 2013;98:76–92. doi: 10.1016/j.brainresbull.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Buttrick GJ, Wakefield JG. PI3-K and GSK-3: Akting together with microtubules. Cell Cycle. 2008;7:2621–2625. doi: 10.4161/cc.7.17.6514. [DOI] [PubMed] [Google Scholar]
- 30.Liu N, Wang LH, Guo LL, Wang GQ, Zhou XP, Jiang Y, Shang J, Murao K, Chen JW, Fu WQ, Zhang GX. Chronic restraint stress inhibits hair growth via substance P mediated by reactive oxygen species in mice. PLoS One. 2013;8:e61574. doi: 10.1371/journal.pone.0061574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeong JY, Lee DH, Kang SS. Effects of chronic restraint stress on body weight, food intake, and hypothalamic gene expressions in mice. Endocrinol Metab (Seoul) 2013;28:288–296. doi: 10.3803/EnM.2013.28.4.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mustafa T, Jiang SZ, Eiden AM, Weihe E, Thistlethwaite I, Eiden LE. Impact of PACAP and PAC1 receptor deficiency on the neurochemical and behavioral effects of acute and chronic restraint stress in male C57BL/6 mice. Stress. 2015;18:408–418. doi: 10.3109/10253890.2015.1025044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuadrado-Tejedor M, Ricobaraza A, Del Rio J, Frechilla D, Franco R, Pérez-Mediavilla A, Garcia-Osta A. Chronic mild stress in mice promotes cognitive impairment and CDK5-dependent tau hyperphosphorylation. Behav Brain Res. 2011;220:338–343. doi: 10.1016/j.bbr.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Finsterwald C, Alberini CM. Stress and glucocorticoid receptor-dependent mechanisms in long-term memory: From adaptive responses to psychopathologies. Neurobiol Learn Mem. 2014;112:17–29. doi: 10.1016/j.nlm.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chakravarty S, Pathak SS, Maitra S, Khandelwal N, Chandra KB, Kumar A. Epigenetic regulatory mechanisms in stress-induced behavior. Int Rev Neurobiol. 2014;115:117–154. doi: 10.1016/B978-0-12-801311-3.00004-4. [DOI] [PubMed] [Google Scholar]
- 36.Gutièrrez-Mecinas M, Trollope AF, Collins A, Morfett H, Hesketh SA, Kersanté F, Reul JM. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc Natl Acad Sci USA. 2011;108:13806–13811. doi: 10.1073/pnas.1104383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dombrowsky H, Uhlig S. Steroids and histone deacetylase in ventilation-induced gene transcription. Eur Respir J. 2007;30:865–877. doi: 10.1183/09031936.00134006. [DOI] [PubMed] [Google Scholar]
- 38.Brownell JE, Allis CD. Special HATs for special occasions: Linking histone acetylation to chromatin assembly and gene activation. Curr Opin Genet Dev. 1996;6:176–184. doi: 10.1016/S0959-437X(96)80048-7. [DOI] [PubMed] [Google Scholar]
- 39.Shibasaki M, Mizuno K, Kurokawa K, Ohkuma S. Enhancement of histone acetylation in midbrain of mice with ethanol physical dependence and its withdrawal. Synapse. 2011;65:1244–1250. doi: 10.1002/syn.20947. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–928. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Kloet ER, Karst H, Joëls M. Corticosteroid hormones in the central stress response: Quick-and-slow. Front Neuroendocrinol. 2008;29:268–272. doi: 10.1016/j.yfrne.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Beato M, Sánchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17:587–609. doi: 10.1210/edrv-17-6-587. [DOI] [PubMed] [Google Scholar]
- 43.Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009;1179:167–178. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- 44.Chen Y, Li J, Dunn S, Xiong S, Chen W, Zhao Y, Chen BB, Mallampalli RK, Zou C. Histone deacetylase 2 (HDAC2) protein-dependent deacetylation of mortality factor 4-like 1 (MORF4L1) protein enhances its homodimerization. J Biol Chem. 2014;289:7092–7098. doi: 10.1074/jbc.A113.527507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hou N, Gong M, Bi Y, Zhang Y, Tan B, Liu Y, Wei X, Chen J, Li T. Spatiotemporal expression of HDAC2 during the postnatal development of the rat hippocampus. Int J Med Sci. 2014;11:788–795. doi: 10.7150/ijms.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spencer JP. The impact of fruit flavonoids on memory and cognition. Br J Nutr. 2010;104(Suppl 3):40–47. doi: 10.1017/S0007114510003934. [DOI] [PubMed] [Google Scholar]
- 47.Lee CC, Huang CC, Hsu KS. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology. 2011;61:867–879. doi: 10.1016/j.neuropharm.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Hu J, Price SR. Inhibition of PI3-kinase signaling by glucocorticoids results in increased branched-chain amino acid degradation in renal epithelial cells. Am J Physiol Cell Physiol. 2007;292:C1874–C1879. doi: 10.1152/ajpcell.00617.2006. [DOI] [PubMed] [Google Scholar]
- 49.Noh JH, Bae HJ, Eun JW, Shen Q, Park SJ, Kim HS, Nam B, Shin WC, Lee EK, Lee K, et al. HDAC2 provides a critical support to malignant progression of hepatocellular carcinoma through feedback control of mTORC1 and AKT. Cancer Res. 2014;74:1728–1738. doi: 10.1158/0008-5472.CAN-13-2109. [DOI] [PubMed] [Google Scholar]
- 50.Sun C, Zhou J. Trichostatin A improves insulin stimulated glucose utilization and insulin signaling transduction through the repression of HDAC2. Biochem Pharmacol. 2008;76:120–127. doi: 10.1016/j.bcp.2008.04.004. [DOI] [PubMed] [Google Scholar]