

Abstract
Background:
Hepatocellular carcinomas are frequently nonresponsive to systemically delivered drugs. Local delivery provides an alternative to systemic administration, maximizing the dose delivered to the tumor, achieving sustained elevated concentrations of the drug, while minimizing systemic exposure.
Results:
Ultrasound-guided deposition of doxorubicin (Dox)-eluting in situ forming implants (ISFI) in an orthotopic tumor model significantly lowers systemic drug levels. As much as 60 µg Dox/g tumors were observed 21 days after ISFI injection. Tumors treated with Dox implants also showed a considerable reduction in progression at 21 days.
Conclusion:
Dox-eluting ISFIs provide a promising platform for the treatment of hepatocellular carcinomas by which drug can be delivered directly into the lesion, bypassing distribution and elimination by the circulatory system.
Keywords: : drug delivery, hepatocellular carcinoma, in situ forming implant, phase inversion, ultrasound
Hepatocellular carcinoma (HCC) is the most common primary malignancy that occurs in the liver, with more than 500,000 new cases discovered annually worldwide [1,2]. HCC is the third most common cause of death worldwide [2,3], and the number of reported cases is predicted to increase in developed countries over the course of the next decade [4]. In the USA alone, there has been an 80% increase in the incidence of HCC during the last 20 years, with incidence rates tripling between 1975 and 2005 [2,4,5]. For patients who are diagnosed early, liver transplantation or tumor resection are the most successful treatment options, but because HCC is often asymptomatic, in many cases detection occurs after significant disease progression has already occurred. As a result, it has been reported that as many as 80% of patients diagnosed with HCC are not eligible for resection or transplantation surgeries [6–9], limiting potential treatment options [3]. Furthermore, the low metabolic activity of the liver in many HCC patients leads to enhanced systemic toxicity of chemotherapeutic agents due to increased half-life of the circulating drugs [2–4].
Chemotherapy is ineffective in most patients, with no improvement in overall survival [3]. However, when drugs have been administered through the hepatic artery, a partial response was observed in as many as 70% of the reported cases [3], indicating a potential benefit for locoregional delivery treatment regimens. While no chemotherapeutic regimen has been successful at improving patient survival, doxorubicin (Dox) has been shown to elicit a response to treatment [10]. Dox acts through two mechanisms. The primary method of action is by intercalating into DNA and interfering with DNA repair mechanisms. Additionally, oxidation of Dox facilitates the formation of reactive oxygen species that can damage lipid membranes and DNA leading to apoptosis [11]. While Dox is a particularly effective chemotherapeutic agent, metabolic bi-products that lead to cardiotoxicity limit the cumulative dose of the drug that the patient can receive [11–13]. In the clinical formulation Doxil, Dox is encapsulated in a liposomal delivery system that is pegylated in order to improve circulation time and reduce uptake by the cardiac tissue [14]. An elevated dose of the drug is delivered to the tumor through the enhanced permeation and retention effect [15]. However, even with the improved accumulation of the drug into the tumor through the enhanced permeation and retention effect [16], only a small percentage of the administered payload is accumulated in the tumor [17].
For nonresectable tumors, image-guided therapies provide alternative treatment strategies to surgical resection and systemic chemotherapy. Image-guided techniques for treating HCC include transarterial chemoembolization (TACE), radio frequency ablation (RFA) and image-guided intratumoral drug delivery [18,19]. TACE is a technique which utilizes embolic agents to block the blood flow of the feeding artery of the tumor, and has been successful at reducing the tumor size for patients with nonresectable tumors [20,21]. However, cirrhosis, portal hypertension, limited hepatic reserve and portal vein involvement with the tumor can all limit the effectiveness of TACE [21]. RFA is a minimally invasive technique performed by placing an electrode into the tumor, under image guidance, and applying radio frequency to heat the surrounding tissue [18,19]. Proximity of the ablation zone to a blood supply can reduce effectiveness of the therapy due to the vascularization functioning as a heat sink [22]. Additionally, RFA treatments can result in abscess formation, bile duct injury and nontargeted damage to adjacent organs [18,19]. Alternatively, intratumoral drug delivery provides a promising means by which drugs can be administered directly into the lesion, while minimizing systemic exposure. Local doxorubicin delivery has been achieved through the use of drug-loaded polymer millirods and implants, which can be placed within a lesion in a minimally invasive manner, and have been shown to facilitate elevated drug concentrations throughout the tumor volume [23–26].
In situ forming implants (ISFIs) provide another method for local long-term administration of the therapeutic agent, which can be achieved through a minimally invasive injection. ISFIs typically consist of a biodegradable hydrophobic polymer dissolved in a biocompatible organic solvent, which can be mixed with a therapeutic agent [27–32]. The drug-loaded polymer solution can then be injected directly into the lesion, and as a function of solvent/nonsolvent mass transfer events, the implant solidifies into a drug eluting depot in situ [33–36] through a process known as phase inversion. The release profile of these implants can be controlled by altering a number of factors to include (but not limited to) the polymer molecular weight, polymer concentration, polymer crystallinity or the choice of solvent used. This minimally invasive system can be used to deliver therapeutic agents over period of weeks to months [32,37–41].
The successful development of targeted therapies requires a suitable in vivo model, however only a limited number of animal models exist. Immune deficient mice can be used to make a subcutaneous xenograft, but these models lack the physiological complexity of the liver. The Novikoff hepatoma (N1-S1) cells can be used to create an orthotopic model in Sprague-Dawley rats, which not only more accurately recapitulate the tumor-associated physiology, but the model uses immune competent animals [42]. Physiological features are important to consider with minimally invasive procedures, because the collateral blood supply may reduce treatment efficacy of the system [42]. Therefore, the N1-S1 animal model was used in these studies.
To evaluate the efficacy of ISFIs for treating HCC in a rat hepatic tumor model, ISFIs were used to deliver Dox locally over a standard 3-week treatment interval. Ultrasound was used to guide the injection of the ISFI solution into the tumor. Fluorescence imaging was used to determine the biodistribution of drug in the heart, liver, lungs, tumor and kidneys as well as evaluate the spatial distribution of the drug within the tumor. Imaging results were compared with those obtained by extracting drug from tissue homogenate. Tumor growth was monitored using diagnostic ultrasound and compared in animals receiving no treatment, blank implants, intravenous administration of drug or ISFIs using two different polymer solutions.
Materials & methods
Materials
Poly(DL-lactic-co-glycolic acid; PLGA) (50:50 lactic acid [LA]:glycolic acid [GA], 3A, Mw 21,000 Da) was obtained from Evonik Industries (Rellinghauser Strasse, Germany) and used as received. 85:15 LA:GA, PLGA Mw 41,000 Da was obtained from PolySciTech (IN, USA) and used as received. N-methyl-2-pyrrolidinone (NMP) and Dox were used as received from Sigma Aldrich (MO, USA). Water soluble tetrazolium-1 (WST-1) assay, improved minimum essential medium (IMEM) and phosphate-buffered saline (PBS) were used as received from Fischer Scientific (MA, USA). Rat Novikoff hepatoma (N1-S1) cells were obtained from ATCC (VA, USA). Sprague-Dawley rats were purchased from Charles Rivers Laboratories (OH, USA).
Cell culture
N1-S1 cells were cultured in complete IMEM, with 10% fetal bovine serum (FBS) and 1% penicillin streptomycin (Invitrogen) and grown to 90% confluence. They were plated in 96-well round bottomed tissue culture plates for cytotoxicity studies and cultured in T75 tissue culture flasks for the tumor inoculations. Cells were cultured in 5% CO2 under humidified conditions at 37°C. N1-S1 cells are a nonadherent cell line and were maintained at an initial density of 40,000 cells/ml and subcultured after every 3 days.
Doxorubicin toxicity
The toxicity of Dox on N1-S1 cells was determined using a WST-1 assay, following previously established conditions [43]. Briefly, 24 h prior to treatment, cells were plated in 96-well round bottomed tissue culture plates with complete medium and incubated at 37°C and 5% CO2. The cells were then suspended at a concentration of 16,000 cells/ml in serum free medium containing differing concentrations of the drug (0, 0.005, 0.05, 0.1, 0.3, 0.7, 1, 10 and 100 µg/ml). After different exposure times (3, 6, 12, 24, 48 and 96 h), the medium containing drug was removed and replaced with 100 µl of complete medium and allowed to incubate 96 h before colorimetric evaluation with the WST-1 assay (Roche, IN, USA). Here, 100 µl of medium containing 10 µl of WST-1 cell proliferation reagent was added and allowed to incubate for 1 h at 37°C, and protected from light. The absorbance was then read at 450 nm using a microplate reader (Tecan Ltd, Infinite 200 series). The data were then fit to a 4-parameter logistic model to calculate the LC50 values.
Implant formulation
Control polymer implant solutions using 85:15 LA:GA PLGA, with no additives were prepared using a 40:60 mass ratio of PLGA:NMP by dissolving the PLGA in NMP overnight at 37°C on an orbital shaker at 90 rpm. Solutions containing Dox (39:60:1 mass ratio of PLGA:NMP:Dox) were prepared in a similar manner as the control solution using either a 50:50 LA:GA PLGA or an 85:15 LA:GA PLGA. Dox was first dissolved in NMP, and allowed to mix using a magnetic stirrer for 30 min before addition of the polymer. The solutions were then mixed overnight at 37°C in an orbital shaker at 90 rpm. Polymer solutions were stored at 4°C and used within 3 days.
Drug dissolution kinetics
Drug release profiles were evaluated as previously described [41]. Briefly, 50 µl of polymer solution was injected into 10 ml of prewarmed PBS (pH 7.4, 37°C). The sample was then immediately placed in an incubated orbital shaker (37°C at 90 rpm). After 30m, 1 h, 2 h, 4 h and 6 h, a 1 ml sample was taken and replaced with a fresh 1 ml of PBS to maintain sink conditions. After 24 h, the bath solution was sampled then completely replaced by 10 ml of fresh buffer daily for 21 days. After 21 days, implants were removed from the bath solution, lyophilized and then dissolved in 5 ml of NMP. Dox mass was determined by comparing fluorescence with a standard curve of known Dox concentrations using a multimode microplate reader (Tecan Ltd, Infinite 200 series) at excitation/emission wavelengths (Ex/Em) of 470/585 nm. The cumulative drug release was calculated from these measurements and normalized by the total mass of drug in the implant.
Tumor inoculation
Due to the low take rate of the N1-S1 tumor model, inoculation was performed by laparotomy in 34 8-week old male Sprague-Dawley rats following protocols approved by the Case Western Reserve University Institutional Animal Care and Use Committee in accordance with established guidelines for animal use. After anesthetization, using 1% isofluorane at a flowing oxygen rate of 1 l/m (EZ150 Isoflurane Vaporizer, EZ Anesthesias™), the rats were shaved and the incision site was cleaned using isopropanol and betadine. Then an incision was made along the ventral midline in order to expose liver, and 100 µl N1-S1 cells were injected into the liver parenchyma using a 26-gauge syringe at a concentration of 5 × 106 cells/ml in IMEM [42].
Tumor treatment evaluation
10 days after inoculation, animals were anesthetized to monitor tumor growth using diagnostic ultrasound. The tumor volumes were calculated using the formula:
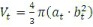 |
where at is the longest tumor diameter and bt is the shortest tumor diameter [44]. Treatments were given after the tumors reached 500 mm3. Implant volumes were adjusted based on animal mass such that 3.3 mg/kg Dox were injected via ultrasound guidance for animals evaluated after 4 h of treatment (n = 3, for each group). Ultrasound images were acquired using an AplioXG (Toshiba Medical Systems), with a 12 MHz transducer. To ensure uniform implant administration for animals treated for 3 weeks (n = 5 50:50 PLGA implant, n = 6 85:15 PLGA implant, n = 3 bolus injection, n = 7 saline control and n = 3 empty vehicle control), the implants were administered via laparotomy. Intravenous administration of 3.3 mg/kg Dox was given once through a tail vein injection, with control animals given isotonic saline. After drug administration, tumor size was monitored twice weekly for the duration of the study. Treatment was halted for multilobular tumors.
Measurement of Dox biodistribution & spatial distribution
A 4-h safety and feasibility study was performed using bolus treatment of Dox, a 50:50 PLGA implant, and a control that received only saline. After the 4-h study, a long-term 21 day study was performed which had expanded experimental groups. At 4 h and 21 days after drug administration, animals were euthanized by CO2 inhalation. The lungs, liver, tumor, kidneys and heart were then dissected from the animal for fluorescence imaging. Each organ was first washed with PBS, and then the organ was imaged using a fluorescence imaging system (CRI Maestro, Caliper Life Sciences, MA, USA). A blue excitation filter (435–480 nm) and a green emission filter (560–750 nm) were used, with an 800 ms exposure time. Then the organ was isolated, and the average signal intensity was measured and recorded. The implants were then removed and a second set of images were taken. Spatial distribution was determined by isolating the implant, then measuring the average signal intensity as a function of distance from the surface of the implant using a custom MatLab program (Mathworks, Inc., MA, USA).
After fluorescence images were obtained, Dox was extracted from the tissue by using an acidified ethanol solution [45]. First, tissue sections were weighed, and then homogenized in a 20-fold volume of 0.3 N HCl in 50% ethanol. After homogenization, the tissues were kept at 4°C for 24 h, and then the homogenate was centrifuged at 20,000 × g for 20 min at 4°C. The supernatant was then collected and analyzed [45]. For each tissue type, a standard curve was made by adding a known mass of Dox into the extracted solution obtained from the animals that did not receive treatment. Then the concentration of Dox in the study samples was determined by comparing the fluorescence intensity of the extracted drug from each tissue with the standard curve for that tissue, using a multimode microplate reader at ex/em wavelengths of 470/585 nm. The mass of extracted Dox was then normalized by the mass of homogenized tissue [45].
Statistical analysis
One way analysis of variance (ANOVA, p < 0.05) was used to determine statistical significance. A difference between groups was determined using a Tukey multicomparison test. All statistical analysis was performed using Minitab (Minitab, Inc., PA, USA). All data are reported as mean ± standard deviation unless otherwise noted.
Results
Doxorubicin toxicity in vitro
Cells treated with Dox were affected by both the dose of the drug as well as the length of exposure time. Exposure time was paramount in eliciting a toxic effect, with 24 h of drug exposure resulting in the greatest response to the drug. The LC50 for a 24 h exposure to Dox was 0.063 μg/ml (48.3 ± 2.25%), with a greater dose required for shorter exposure times (Figure 1). The viability of cells treated with Dox for 24 h decreased significantly with an increasing drug concentration (p < 0.05), until reaching a concentration of 10 μg/ml (2.7 ± 0.9%), after which no significant difference was observed (Figure 1). Cells exposed to Dox for 12 h required a concentration >0.05 μg/ml in order to initiate a response, with 100 μg/ml eliciting the greatest response (2.6 ± 1.6%). A statistically significant dose response (p < 0.05) was observed in cells exposed to Dox for 6 h for a drug dose exceeding 0.05 μg/ml, but the response was lower than that observed when the cells were treated for 12 or 24 h. When cells were exposed to Dox for only 3 h, a dose exceeding 0.1 ug/ml was required to reduce cell viability, and a statistically greater percent of the cell population was viable at a concentration of 100 μg/ml (11.0 ± 5.6%) relative to the other exposure times.
Figure 1. . Dose response of N1-S1 cells to (A) doxorubicin and (B) effect of exposure time on the LC50.

*p < 0.05.
Drug dissolution kinetics
A burst release was observed for both implant formulations, with the 85:15 (LA:GA) polymer exhibiting the greatest burst, 33.9 ± 0.9% after 24 h (Figure 2). The derivative of release was evaluated to detect the onset of degradation facilitated release, but no statistical increase in release occurred for the 85:15 polymer implants. A slight, but significant increase in the rate of release was observed after 5 days in the 50:50 PLGA implants (p < 0.05). The average daily release of Dox was 6.8 ± 2.8 µg/day for 50:50 PLGA implants, and 4.4 ± 2.9 µg/day for 85:15 PLGA implants.
Figure 2. . Cumulative release of doxorubicin over the course of 21 days.

*p < 0.05.
Image guided implantation & Dox biodistribution
Implants were injected into the HCC lesions under image guidance (Figure 3). After 4 h, the animal was euthanized and the placement of the implant in the lesion was validated. All ISFIs injected under image guidance were found within the tumor. The biodistribution of drug was determined using fluorescence imaging (Figure 4). The systemically delivered Dox via a bolus tail vein injection resulted in a statistically higher mass of drug in the heart (146.8 ± 39.3% normalized to negative control) and lungs (69.0 ± 17.0%) relative to 50:50 PLGA implants (levels were below the detectable limit). No observable difference in Dox concentration was found in the kidneys using fluorescence imaging. In the tumor, the Dox concentration was the highest in the 50:50 PLGA group (123.9 ± 27.4%), followed by the systemically delivered dose (48.7 ± 6.6%; Figure 5). Extraction of Dox from the tissues, demonstrated a statistically higher dose of the drug in the heart (13.1 ± 4.4 µg/g), liver (6.9 ± 3.8 μg/g) and kidneys (8.0 ± 1.6 μg/g) after a bolus tail vein administration of the drug when compared with the polymer implants. No statistical difference was observed after 4 h in the mass of Dox extracted from the tumor or lungs. Twenty-one days after treatment, no statistical difference was found in the tissues using fluorescence imaging. A significantly higher mass of Dox was extracted from the tumor with animals receiving 50:50 PLGA implants (60.3 ± 48.5 μg/g) than animals treated using the 85:15 PLGA implants (5.3 ± 7.5 μg/g) or a bolus injection of Dox (p < 0.05).
Figure 3. . Time-lapse image showing the image-guided injection of the implants into the tumor.

The tumor is circled in white, the needle and injection solution have been identified with arrows, and the time stamp is indicated in the lower left of each panel.
Figure 4. . Representative fluorescence images of whole organs evaluated for biodistribution of doxorubicin.

Column 1 is the saline control, Column 2 is the bolus injection of doxorubicin after 4 h, Column 3 is the 50:50 PLGA implant after 4 h, Column 4 is the 50:50 PLGA implant after 21 days, and Column 5 is the 85:15 PLGA implant after 21 days.
Figure 5. . Biodistribution of doxorubicin.

Extracted mass of Dox in the tissue after (A) 4 h and (B) 21 days. Quantitative evaluation of the fluorescence signal to evaluate biodistribution of Dox in the tissue after (C) 4 h and (D) 21 days.
*p < 0.05.
Dox: Doxorubicin.
Spatial distribution of Dox in tumors
After 21 days, no significant differences were observed between the two implant formulations at the implant/tissue interface, however a more rapid loss in the Dox fluorescent signal was observed for the 85:15 PLGA implants relative to the 50:50 PLGA implants. A low fluorescence signal was observed 1 mm away from the surface of the 85:15 PLGA implants, while the 50:50 PLGA implants had detectable fluorescence signal 3 mm away from the surface of the drug-eluting depot. Overall, a significantly greater treatment area (p < 0.05) was observed with the 50:50 PLGA implants relative to 85:15 PLGA implants (Figure 6).
Figure 6. . Spatial distribution of doxorubicin from the implants into the adjacent tissue.

Treatment efficacy in vivo
No statistical differences were observed in tumor sizes between groups until 14 days after administration of the drug, at which time, the tumors treated with 50:50 PLGA implants (2.5 ± 2.0-fold) were significantly smaller than the negative controls (9.1 ± 2.3-fold; p < 0.05). The tumors treated using 85:15 PLGA implants became significantly smaller than the negative controls within 17 days of treatment (Figure 7). The growth rate of the tumors continued to increase for the untreated control animals, for 17 days, and then no significant increase in volume occurred. For tumors treated with an ISFI implant that did not contain Dox, tumor growth rate increased for 10 days, after which the increase in volume continued at a slower rate. The tumor growth rate for all three treatment groups was equivalent for the first 3 days after treatment. Tumor growth stopped after 7 days for the 50:50 PLGA implants, followed by a loss of tumor volume after 17 d. The 85:15 implants initiated a decrease in tumor volume after 7 days, but the initial decrease in tumor volume was followed by a brief period of tumor growth, then a subsequent decrease in tumor volume after 14 days which lasted the duration of the study. Animals treated with a bolus injection of Dox continued to grow until after 17 days, and did not significantly change volume for the remainder of the study (Figure 6). After 21 days, 50:50 PLGA implants had the smallest mean normalized tumor volume, followed by the 85:15 PLGA implants, with the bolus injection having the largest mean normalized tumor volume among the animals that were treated (Figure 7).
Figure 7. . In vivo treatment efficacy.

Representative ultrasound image used to track changes in tumor volume over time (A). Change in normalized tumor volume over the course of 21 days (B). Normalized tumor volume of treated groups after 21 days (C).
*p < 0.05.
Discussion
Injection of a drug-bearing implant directly into a tumor provides a means of delivering elevated concentrations of the active agent through a minimally invasive injection, subsequently reducing systemic involvement [46,47]. This is of particular importance when using drugs with high toxicity, in that limiting systemic exposure can significantly reduce off target effects, ultimately improving the quality of life for a patient [20]. Since the implants are placed directly into the lesion, the exposure time of cancer cells to the drug is prolonged, enhancing the drug efficacy. On average implants released between 4.4 and 6.8 μg/day of doxorubicin in vitro, which was greater than the 1 μg/ml needed to reduce cell viability by 95%. Furthermore, the ability to inject an implant into an orthotopic tumor under image guidance is not only faster, but also reduces complications that arise from placement through laparotomy such as bleeding and infection [42].
In vitro, implants demonstrated a sharp burst with a subsequent slower release over the course of 21 days. Elevated daily release was observed with the 50:50 PLGA implants, relative to the 85:15 PLGA implants. This was anticipated due to the lower crystallinity of the polymer and faster rate of degradation [48–52]. A sharp rise in Dox release occurred after 6 days, but the flux returned to predegradation rates after 9 days, which we hypothesize to be a function of electrostatic interactions forming between the anionic degradation biproducts and the cationic doxorubicin [41]. Since Dox can form crystalline domains at elevated concentrations, we hypothesize that it forms a polyionic complex with the degrading matrix, reducing erosion, and stabilizing the implant microstructure [41,53–55]. These release kinetics provide a means for rapidly achieving a therapeutic dose, followed by lower daily maintenance doses released by the implant [21,23,56]. Polymer millirods, which follow a similar release profile, have shown promise as a local delivery system for treating ectopic lesions [24,25].
Local delivery has been demonstrated to reduce the systemic exposure, while maintaining elevated drug doses [33,34,36]. It was observed through fluorescent imaging that off-target accumulation of drug was significantly reduced when using ISFI when compared with systemic delivery of the drug, which was then validated by extraction of the drug from the tissue (Figures 3 & 4). It has been reported that after 24 h, Dox is not readily detected in tissues when delivered systemically [45], therefore the levels of Dox in the heart, liver, kidneys, lungs and tumor were measured 4 h after drug administration. When given a bolus dose, elevated Dox concentrations were found in the heart 4 h after injection when compared with Dox administered using an ISFI. This is particularly important in the case of Dox delivery, due to the potential for cardiotoxicity of the drug [11–13,57]. After 21 days, no detectable mass of Dox was found in any of the tissues tested, for animals treated with a bolus injection of the drug. In contrast, elevated doses were still found in the tumor at the end of one treatment cycle with the animals treated using ISFIs. The residual drug extracted from the tumors after 21 days was higher than what was extracted 4 h after treatment. Overall, Dox administered using the 50:50 PLGA formulation resulted in a higher dose of drug extracted from the tissue when compared with the 85:15 implants, which we hypothesize to be a function of elevated rate of polymer degradation for the 50:50 PLGA implants leading to a greater accumulation of the drug in the tumor.
Furthermore, when comparing the distribution of Dox within the tumor, it was observed that the 50:50 PLGA implants had a significantly greater distribution of drug relative to the 85:15 PLGA implants (Figure 5). We hypothesize that the difference in drug distribution was primarily a result of elevated levels of free drug available due to the more rapid polymer degradation of the 50:50 PLGA implants. Additionally, the injection site has been reported to alter the release profile of ISFIs due to reactive forces from the tissue that occur as a result of implant swelling [35]. Therefore, the difference in the polymer swelling between the amorphous 50:50 PLGA and the more crystalline 85:15 PLGA may have attributed to the different drug distribution patterns observed between the two implant formulations.
It was anticipated that local delivery would be more effective at reducing the tumor volume, but no statistical differences were observed among any of the treatment groups (Figure 6). While not statistically different, within 7 days after injection of the polymer solution, tumors injected with implants formulated using 50:50 PLGA were an average of 28.6% smaller than the animals receiving a bolus injection of drug. Over the course of the final 2 weeks of the study, these same animals averaged tumors that were 36% smaller than the animals receiving a bolus injection. Similar results were observed with animals receiving injections of the 85:15 PLGA implants, but the tumors did not become smaller until 10 days after treatment, with an average tumor volume 15.3% smaller than animals receiving a bolus injection of drug. One animal treated with a 50:50 PLGA implant had no observable tumor at the end of the 3-week treatment cycle. Interestingly, even though the implants were injected into the center of the tumors, after 21 days the implants were all discovered near the tumor boundary, which may have reduced the implants efficacy. It was not clear if the change in implant location was a result of tumor cells growing more rapidly outside of the treatment volume, or if the implant solution moved along the needle track after the injection as a function of polymer swelling [40]. While local therapy was not more effective at reducing tumor volume, it did effectively reduce systemic exposure. In this study a single implant was injected into the center of the lesion, while a potentially more effective approach would be to uniformly inject small implants throughout the tumor volume to ensure homogenous distribution of the drug throughout the entire tumor volume [25].
While this study was performed using the drug Dox primarily due to the ease of detection through fluorescence imaging, as well as a track record of use to treat HCC [10], ISFIs can be formulated using a variety of different drugs or drug combinations. For example, the drug sorafenib, which has shown promise as a new treatment option for HCC by blocking tumor angiogenesis and inhibiting different aspects of the Raf signaling pathway [58] could easily be loaded alone or with Dox and delivered locally using ISFIs [59]. Additionally, due to the minimally invasive nature of this system, these implants could also be used to provide adjuvant therapy after RFA [24,34,35,60] or also be placed within the tumor to provide additional therapy after TACE [61].
Conclusion
Phase sensitive ISFI implants provide a promising means by which drugs can be administered locally in a minimally invasive manner. Implants were shown to slow the rate of tumor growth, and demonstrated the potential for reducing tumor volume. Additionally, implants reduced the mass of drug found in off target locations, such as the lungs and heart. The reduction in systemic exposure lowers the potential for unwanted affects such a myelosuppression or cardiotoxicity. These findings are of particular importance for hepatocellular carcinoma, where patients are often not eligible for more aggressive therapies, and typically only show a modest response to systemically delivered chemotherapy. While promising, improved distribution of the polymer solution throughout the lesion may further improve the therapeutic outcome.
Future perspective
Intratumoral image-guided drug delivery provides a platform through which drugs can be administered to malignant tissues that cannot be surgically resected or managed effectively through systemic chemotherapy. Building upon existing, clinically utilized interventional radiology tools, image-guided access to the tumor site, and accurate, on demand deposition of the drug-loaded polymer can be achieved. Drugs delivered in this way result it 100% of the injected dose residing immediately within the tumor itself, while minimizing systemic involvement and reducing the typical dose-limiting side effects associated with chemotherapy. Complementary imaging techniques can be developed to monitor implant performance and therapeutic efficacy, creating the framework for the development of more rational, personalized drug delivery systems. Ultimately image-guided local drug delivery techniques can streamline and advance patient care.
Executive summary.
The LC50 value of doxorubicin for the N1-S1 cell line was 0.06 μg/ml and required at least 12 h of exposure to effectively treat cells.
Elevated polymer crystallinity resulted in an increased burst release, but a decrease in diffusion facilitated release from phase sensitive in situ forming implants within the standard treatment window of 3 weeks.
In situ forming implants can successfully be administered under ultrasound guidance, and the implants can be placed within the lesion in <2 min.
Bolus injection of doxorubicin resulted in the systemic delivery of drug, to include the heart, however the dose remained localized if administered using a controlled release system.
The more crystalline polymer resulted in a decreased spatial distribution of drug within the hepatocellular carcinoma tumor.
Both amorphous and crystalline implants were effective treatment options for the N1-S1 orthotopic tumor model.
Footnotes
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Financial & competing interests disclosure
Research reported in this manuscript was supported by the National Cancer Institute of the National Institutes of Health under award number R01CA118399, R01EB016960 to AA Exner and the Department of Defense Breast Cancer Research Fellowship under award number W81XWH-10–1–0582 to L Solorio. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Department of Defense.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.McMasters KM, editor. Hepatocellular Carcinoma Targeted Therapy and Multidisciplinary Care. Volume 1. Springer; NY, USA: 2011. [Google Scholar]
- 2.Rahman R, Hammoud GM, Almashhrawi AA, Ahmed KT, Ibdah JA. Primary hepatocellular carcinoma and metabolic syndrome: an update. World J. Gastrointest. Oncol. 2013;5(9):186–194. doi: 10.4251/wjgo.v5.i9.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carr BI, editor. Hepatocellular Carcinoma Diagnosis and Treatment. Volume 2. Humana Press; NY, USA: 2010. [Google Scholar]
- 4.Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat. Rev. Gastroenterol. Hepatol. 2010;7(8):448–458. doi: 10.1038/nrgastro.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Serag HB, Davila JA, Petersen NJ, Mcglynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann. Intern. Med. 2003;139(10):817–823. doi: 10.7326/0003-4819-139-10-200311180-00009. [DOI] [PubMed] [Google Scholar]
- 6.Gervais DA, Goldberg SN, Brown DB, Soulen MC, Millward SF, Rajan DK. Society of Interventional Radiology position statement on percutaneous radiofrequency ablation for the treatment of liver tumors. J. Vasc. Interv. Radiol. 2009;20(7 Suppl.):S342–347. doi: 10.1016/j.jvir.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 7.Massarweh NN, Park JO, Farjah F, et al. Trends in the utilization and impact of radiofrequency ablation for hepatocellular carcinoma. J. Am. Coll. Surg. 2010;210(4):441–448. doi: 10.1016/j.jamcollsurg.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spangenberg HC, Thimme R, Blum HE. Targeted therapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2009;6(7):423–432. doi: 10.1038/nrgastro.2009.86. [DOI] [PubMed] [Google Scholar]
- 9.Wong SL, Mangu PB, Choti MA, et al. American Society of Clinical Oncology 2009 clinical evidence review on radiofrequency ablation of hepatic metastases from colorectal cancer. J. Clin. Oncol. 2010;28(3):493–508. doi: 10.1200/JCO.2009.23.4450. [DOI] [PubMed] [Google Scholar]
- 10.Llovet JM, Real MI, Montana X, et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet. 2002;359(9319):1734–1739. doi: 10.1016/S0140-6736(02)08649-X. [DOI] [PubMed] [Google Scholar]
- 11.Thorn CF, Oshiro C, Marsh S, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet. Genomics. 2011;21(7):440–446. doi: 10.1097/FPC.0b013e32833ffb56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris L, Batist G, Belt R, et al. Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first-line therapy of metastatic breast carcinoma. Cancer. 2002;94(1):25–36. doi: 10.1002/cncr.10201. [DOI] [PubMed] [Google Scholar]
- 13.Yildirim Y, Gultekin E, Avci ME, Inal MM, Yunus S, Tinar S. Cardiac safety profile of pegylated liposomal doxorubicin reaching or exceeding lifetime cumulative doses of 550 mg/m2 in patients with recurrent ovarian and peritoneal cancer. Int. J. Gynecol. Cancer. 2008;18(2):223–227. doi: 10.1111/j.1525-1438.2007.00992.x. [DOI] [PubMed] [Google Scholar]
- 14.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin. Pharmacokinet. 2003;42(5):419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 15.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. [PubMed] [Google Scholar]; •• First description of the enhanced permeation and retention effect.
- 16.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release. 2000;65(1–2):271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 17.Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 18.Willatt J, Hannawa KK, Ruma JA, Frankel TL, Owen days, Barman PM. Image-guided therapies in the treatment of hepatocellular carcinoma: a multidisciplinary perspective. World J. Hepatol. 2015;7(2):235–244. doi: 10.4254/wjh.v7.i2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunzli BM, Abitabile P, Maurer CA. Radiofrequency ablation of liver tumors: actual limitations and potential solutions in the future. World J. Hepatol. 2011;3(1):8–14. doi: 10.4254/wjh.v3.i1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Exner AA, Saidel GM. Drug-eluting polymer implants in cancer therapy. Expert Opin. Drug Deliv. 2008;5(7):775–788. doi: 10.1517/17425247.5.7.775. [DOI] [PubMed] [Google Scholar]
- 21.Solorio L, Patel RB, Wu H, Krupka T, Exner AA. Advances in image-guided intratumoral drug delivery techniques. Ther. Deliv. 2010;1(2):307–322. doi: 10.4155/tde.10.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horkan C, Ahmed M, Liu Z, et al. Radiofrequency ablation: effect of pharmacologic modulation of hepatic and renal blood flow on coagulation diameter in a VX2 tumor model. J. Vasc. Interv. Radiol. 2004;15(3):269–274. doi: 10.1097/01.rvi.0000109396.74740.c4. [DOI] [PubMed] [Google Scholar]
- 23.Qian F, Saidel GM, Sutton DM, Exner A, Gao J. Combined modeling and experimental approach for the development of dual-release polymer millirods. J. Control. Rel. 2002;83(3):427–435. doi: 10.1016/s0168-3659(02)00217-1. [DOI] [PubMed] [Google Scholar]
- 24.Weinberg BD, Patel RB, Exner AA, Saidel GM, Gao J. Modeling doxorubicin transport to improve intratumoral drug delivery to RF ablated tumors. J. Control. Rel. 2007;124(1–2):11–19. doi: 10.1016/j.jconrel.2007.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinberg BD, Patel RB, Wu H, et al. Model simulation and experimental validation of intratumoral chemotherapy using multiple polymer implants. Med. Biol. Eng. Comput. 2008;46(10):1039–1049. doi: 10.1007/s11517-008-0354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Krupka T, Yao J, et al. Liquid-solid phase-inversion PLGA implant for the treatment of residual tumor tissue after HIFU ablation. PLoS One. 2015;10(2):e0117358. doi: 10.1371/journal.pone.0117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brodbeck KJ, Desnoyer JR, Mchugh AJ. Phase inversion dynamics of PLGA solutions related to drug delivery. Part II. The role of solution thermodynamics and bath-side mass transfer. J. Control. Rel. 1999;62(3):333–344. doi: 10.1016/s0168-3659(99)00159-5. [DOI] [PubMed] [Google Scholar]; • Part of a series that was the first to describe the role of polymer phase inversion on drug release.
- 28.Dunn RL, English JP, Cowsar DR. Vanderbilt DP: biodegradable in situ forming implants and methods of producing the same. 1990. Patent US5990194A.; •• First description of the use of phase-inverting systems for drug delivery.
- 29.Dunn RL, Tipton AJ. Polymeric compositions useful as controlled release implants. 1997. Patent US5702716A.
- 30.Graham PD, Brodbeck KJ, Mchugh AJ. Phase inversion dynamics of PLGA solutions related to drug delivery. J. Control. Rel. 1999;58(2):233–245. doi: 10.1016/s0168-3659(98)00158-8. [DOI] [PubMed] [Google Scholar]; • Part of a series that was the first to describe the role of polymer phase inversion on drug release.
- 31.Mchugh AJ. The role of polymer membrane formation in sustained release drug delivery systems. J. Control. Rel. 2005;109(1–3):211–221. doi: 10.1016/j.jconrel.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 32.Ravivarapu HB, Moyer KL, Dunn RL. Sustained activity and release of leuprolide acetate from an in situ forming polymeric implant. AAPS PharmSciTech. 2000;1(1):E1. doi: 10.1208/pt010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen FA, Kuriakose MA, Zhou MX, Delacure MD, Dunn RL. Biodegradable polymer-mediated intratumoral delivery of cisplatin for treatment of human head and neck squamous cell carcinoma in a chimeric mouse model. Head Neck. 2003;25(7):554–560. doi: 10.1002/hed.10241. [DOI] [PubMed] [Google Scholar]; • Description of in situ forming implants used for local delivery for the treatment of head and neck cancer.
- 34.Krupka TM, Weinberg BD, Ziats NP, Haaga JR, Exner AA. Injectable polymer depot combined with radiofrequency ablation for treatment of experimental carcinoma in rat. Invest. Radiol. 2006;41(12):890–897. doi: 10.1097/01.rli.0000246102.56801.2f. [DOI] [PubMed] [Google Scholar]
- 35.Patel RB, Solorio L, Wu HP, Krupka T, Exner AA. Effect of injection site on in situ implant formation and drug release in vivo . J. Control. Rel. 2010;147(3):350–358. doi: 10.1016/j.jconrel.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• First description of the role that stiffness of the injection site plays on in situ forming implants drug release and phase-transition behavior.
- 36.Shikanov A, Shikanov S, Vaisman B, Golenser J, Domb AJ. Cisplatin tumor biodistribution and efficacy after intratumoral injection of a biodegradable extended release implant. Chemother. Res. Pract. 2011;2011:1–9. doi: 10.1155/2011/175054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kranz H, Bodmeier R. Structure formation and characterization of injectable drug loaded biodegradable devices: in situ implants versus in situ microparticles. Eur. J. Pharm. Sci. 2008;34(2–3):164–172. doi: 10.1016/j.ejps.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Luan X, Bodmeier R. In situ forming microparticle system for controlled delivery of leuprolide acetate: influence of the formulation and processing parameters. Eur. J. Pharm. Sci. 2006;27(2–3):143–149. doi: 10.1016/j.ejps.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Ravivarapu HB, Moyer KL, Dunn RL. Sustained suppression of pituitary-gonadal axis with an injectable, in situ forming implant of leuprolide acetate. J. Pharm. Sci. 2000;89(6):732–741. doi: 10.1002/(SICI)1520-6017(200006)89:6<732::AID-JPS4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 40.Solorio L, Olear AM, Hamilton JI, et al. Noninvasive characterization of the effect of varying PLGA molecular weight blends on in situ forming implant behavior using ultrasound imaging. Theranostics. 2012;2(11):1064–1077. doi: 10.7150/thno.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Solorio L, Olear AM, Zhou H, Beiswenger AC, Exner AA. Effect of cargo properties on in situ forming implant behavior determined by noninvasive ultrasound imaging. Drug Deliv. Transl. Res. 2012;2(1):45–55. doi: 10.1007/s13346-011-0054-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan HH, Chu TH, Chien HF, et al. Rapid induction of orthotopic hepatocellular carcinoma in immune-competent rats by non-invasive ultrasound-guided cells implantation. BMC Gastroenterol. 2010;10:83. doi: 10.1186/1471-230X-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eliaz RE, Nir S, Marty C, Jr, Szoka FC. Determination and modeling of kinetics of cancer cell killing by doxorubicin and doxorubicin encapsulated in targeted liposomes. Cancer Res. 2004;64(2):711–718. doi: 10.1158/0008-5472.can-03-0654. [DOI] [PubMed] [Google Scholar]
- 44.Euhus DM, Hudd C, Laregina MC, Johnson FE. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986;31(4):229–234. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- 45.Bachur NR, Moore AL, Bernstein JG, Liu A. Tissue distribution and disposition of daunomycin (NCS-82151) in mice: fluorometric and isotopic methods. Cancer Chemother. Rep. 1970;54(2):89–94. [PubMed] [Google Scholar]
- 46.Langer R. Polymer implants for drug delivery in the brain. J. Control. Rel. 1991;16(1–2):53–59. [Google Scholar]
- 47.Saltzman WM, Fung LK. Polymeric implants for cancer chemotherapy. Adv. Drug Deliv. Rev. 1997;26(2–3):209–230. doi: 10.1016/s0169-409x(97)00036-7. [DOI] [PubMed] [Google Scholar]
- 48.Desnoyer JR, Mchugh AJ. Role of crystallization in the phase inversion dynamics and protein release kinetics of injectable drug delivery systems. J. Control. Rel. 2001;70(3):285–294. doi: 10.1016/s0168-3659(00)00354-0. [DOI] [PubMed] [Google Scholar]
- 49.Hurrell S, Cameron RE. The effect of initial polymer morphology on the degradation and drug release from polyglycolide. Biomaterials. 2002;23(11):2401–2409. doi: 10.1016/s0142-9612(01)00376-3. [DOI] [PubMed] [Google Scholar]
- 50.Tsuji H, Mizuno A, Ikada Y. Properties and morphology of poly(L-lactide). III. Effects of initial crystallinity on long-term in vitro hydrolysis of high molecular weight poly(L-lactide) film in phosphate-buffered solution. J. Appl. Polym. Sci. 2000;77(7):1452–1464. [Google Scholar]
- 51.Alexis F. Factors affecting the degradation and drug-release mechanism of poly(lactic acid) and poly[(lactic acid)-co-(glycolic acid)] Polymer International. 2005;54(1):34–46. [Google Scholar]
- 52.Mikos AG, Thorsen AJ, Czerwonka LA, et al. Preparation and characterizatio nof poly(L-lactic acid) foams. Polymer. 1994;35(5):1068–1077. [Google Scholar]
- 53.Abraham SA, Edwards K, Karlsson G, et al. Formation of transition metall-doxorubicin complexes inside liposomes. Biochim. Biophys. Acta. 2002;1565(1):41–54. doi: 10.1016/s0005-2736(02)00507-2. [DOI] [PubMed] [Google Scholar]
- 54.Deng W, Li JA, Yao P, He F, Huang C. green preparation process, characterization and antitumor effects of doxorubicin-bsa-dextran nanoparticles. Macromol. Biosci. 2010;10(10):1224–1234. doi: 10.1002/mabi.201000125. [DOI] [PubMed] [Google Scholar]
- 55.Manocha B, Margaritis A. Controlled release of doxorubicin from doxorubicin/gamma-polyglutamic acid ionic complex. J. Nanometer. 2010;2010:1–9. [Google Scholar]
- 56.Qian F, Szymanski A, Gao JM. Fabrication and characterization of controlled release poly(D, L-lactide-co-glycolide) millirods. J. Biomed. Mater. Res. 2001;55(4):512–522. doi: 10.1002/1097-4636(20010615)55:4<512::aid-jbm1044>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 57.Zhang HT, Li F, Yi J, et al. Folate-decorated maleilated pullulan-doxorubicin conjugate for active tumor-targeted drug delivery. Eur. J. Pharm. Sci. 2011;43(5):409–409. doi: 10.1016/j.ejps.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 58.Cheng JW, Lv Y. New progress of non-surgical treatments for hepatocellular carcinoma. Med. Oncol. 2013;30(1):381. doi: 10.1007/s12032-012-0381-y. [DOI] [PubMed] [Google Scholar]
- 59.Abou-Alfa GK, Johnson P, Knox JJ, et al. Doxorubicin plus sorafenib vs doxorubicin alone in patients with advanced hepatocellular carcinoma: a randomized trial. JAMA. 2010;304(19):2154–2160. doi: 10.1001/jama.2010.1672. [DOI] [PubMed] [Google Scholar]
- 60.Weinberg BD, Blanco E, Lempka SF, Anderson JM, Exner AA, Gao J. Combined radiofrequency ablation and doxorubicin-eluting polymer implants for liver cancer treatment. J. Biomed. Mater. Res. 2007;81(1):205–213. doi: 10.1002/jbm.a.30926. [DOI] [PubMed] [Google Scholar]
- 61.Lencioni R, Crocetti L. Combination therapies in the treatment of primary liver cancers. In: Dupuy DE, editor. Image-Guided Cancer Therapy. Springer; NY, USA: 2013. pp. 339–343. [Google Scholar]