

Abstract
Activation of hepatic stellate cells (HSCs) is a pivotal event during hepatic fibrogenesis. Activated HSCs are the main source of collagen and other extracellular matrix (ECM) components, and emerging antifibrotic therapies are aimed at preventing ECM synthesis and deposition. MicroRNAs (miRNAs) have been demonstrated to exert regulatory effects on HSC activation and ECM synthesis. In the present study, the HSC-T6 rat hepatic stellate cell line was transiently transfected with a miRNA (miR)-203 mimic, which is an artificial miRNA that enhances the function of miR-203, with a miR-203 inhibitor or with a scramble miRNA negative control. mRNA and protein expression levels of collagen (COL) 1A1, COL3A1, α-smooth muscle actin (α-SMA) and mothers against decapentaplegic homolog 3 (SMAD3) were assessed using reverse transcription-quantitative polymerase chain reaction and western blot analysis, respectively. The interaction between miR-203 and the 3′-untranslated region (UTR) of SMAD3 mRNA was examined using a dual-luciferase reporter assay. The proliferative capabilities of activated HSCs were measured using an MTT assay. The present results demonstrated that the mRNA and protein expression levels of COL1A1, COL3A1, α-SMA and SMAD3 were significantly upregulated following transfection of HSC-T6 cells with the miR-203 inhibitor. Conversely, COL1A1, COL3A1, α-SMA, and SMAD3 mRNA and protein expression appeared to be downregulated in rat HSCs transfected with miR-203 mimics. Notably, the inhibition of miR-203 expression was revealed to promote HSC proliferation, whereas increased miR-203 expression suppressed the proliferative capabilities of HSC-T6 cells. Furthermore, SMAD3 was revealed to be a direct target of miR-203. The present study suggested that miR-203 may function to prevent the synthesis and deposition of ECM components, including COL1A1, COL3A1 and α-SMA, and to inhibit the proliferation of HSCs through a SMAD3-dependent mechanism. Therefore, it may be hypothesized that miR-203 has potential as a novel target for the development of alternative therapeutic strategies for the treatment of patients with hepatic fibrosis in clinical practice.
Keywords: miR-203, HSCs, collagen, α-SMA, SMAD3
Introduction
Hepatic fibrosis is a continuous wound-healing process that is activated in response to chronic liver injury of various etiologies, including chronic hepatitis B and C infection, alcohol abuse, non-alcoholic steatohepatitis and autoimmune hepatitis (1). Hepatic fibrogenesis is characterized by the aberrant deposition of extracellular matrix (ECM) components in the liver (1,2). Activated hepatic stellate cells (HSCs) are the primary source of excess ECM components, and thus serve a crucial role in the progression of hepatic fibrogenesis (3). During chronic liver injury, HSCs have been reported to transdifferentiate from quiescent vitamin A-storing cells into proliferative myofibroblast-like cells with enhanced secretory capabilities, which causes an imbalance between ECM synthesis and degradation (1). The detailed molecular mechanisms underlying HSC transdifferentiation are complex and have yet to be fully elucidated; various cytokines and signaling pathways have reported to be involved in HSC activation. The transforming growth factor (TGF)-β/mothers against decapentaplegic homolog (SMAD) pathway has been identified as an essential signaling cascade during HSC activation (4). SMAD proteins are intracellular mediators of TGF-β signaling (5) and have been reported to modulate the biogenesis of microRNAs (miRNAs) in several pathophysiological processes (6).
miRNAs are endogenous, short (~22 nucleotides-long), non-coding RNA molecules that are involved in the post-transcriptional regulation of gene expression in plants and animals through binding to the 3′-untranslated regions (UTRs) of target mRNAs, which results in mRNA degradation or the suppression of their translation (7,8). Numerous miRNAs have been identified as pivotal regulators in several physiological processes, including immune responses and cellular differentiation and proliferation (9). Previous studies have reported on the regulatory functions for miRNAs during hepatic fibrogenesis (10,11), and specific miRNAs have been revealed to mediate fibrosis by targeting SMAD3 (12,13). Notably, miR-203 has been demonstrated to negatively regulate hepatic tumorigenesis through various signal transduction pathways (14–16). miR-203 downregulation has been reported in rat and human fibrotic liver tissue, as well as in activated rat HSCs, whereas miR-203 upregulation was demonstrated to prevent HSC activation and proliferation by targeting transient receptor potential vanilloid 4 (TRPV4) channels (17). However, the effects of miR-203 expression on collagen synthesis in HSCs, as well as its interaction with SMAD3, have yet to be elucidated.
The present study aimed to explore the putative regulatory roles of miR-203 during the development of hepatic fibrosis. Bioinformatics analysis was used to predict SMAD3 as a target gene for miR-203. In addition, the effects of altered miR-203 expression on HSC-T6 proliferation and on the expression of collagen-related genes were investigated. Furthermore, the putative interaction between miR-203 and SMAD3 was confirmed, revealing SMAD3 as a direct target gene for miR-203 in hepatocytes.
Materials and methods
Cell culture and transfection
HSC-T6 (18), rat hepatic stellate cells were purchased from Yingrun Biotechnologies, Inc. (Hunan, China). HSC-T6 cells were seeded (3×105 cells/well) in six-well plates and cultured with high-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.). Cells were maintained at 37°C in a 5% CO2 humidified atmosphere. Following a 24-h incubation, cells were cultured in serum-free DMEM for ~12 h and then transfected with a miR-203 mimic, which is an artificial miRNA that enhances the function of miR-203, a miR-203 inhibitor, which is an antisense oligonucleotide for miR-203, or a negative control scramble miRNA. All miRNAs were purchased from Shanghai GenePharma Co., Ltd., (Shanghai, China). Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) was used as the transfection reagent, according to the manufacturer's protocol. The concentration of miR-203 mimic, miR-203 inhibitor and scramble miRNA that were used for transfection was 0.02 µM, and each step was completed in 3 min at room temperature. The culture medium was replaced 4–6 h post-transfection, the cells were cultured for an additional 48 h and then used for subsequent experiments. The sequences of the miRNAs that were used were as follows: miR-203 mimic, forward 5′-GUGAAAUGUUUAGGACCACUAG-3′, reverse 5′-AGUGGUCCUAAACAUUUCACUU-3′; miR-203 antisense oligonucleotide, 5′-CUAGUGGUCCUAAACAUUUCAC-3′; and scramble miRNA, 5′-CAGUACUUUUGUGUAGUACAA-3′.
Transfection efficiency assessment
The efficiency of HSC-T6 cell transfection was assessed in order to optimize the concentrations of miRNAs and transfection reagent that were used in the present experiments, using transient transfection with scramble miRNAs, including fluorescein amidite (FAM)-labeled and unlabeled miRNAs. FAM-labeled miRNAs emit fluorescence following excitation at 420–485 nm, which can be observed using fluorescence microscopy, whereas unlabeled miRNAs do not. Culture and transfection conditions were similar to the aforementioned.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from HSC-T6 cells (~2×106) using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. miR-203 was reverse transcribed using the following stem-loop RT primer:5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGTTGAA-3′. Thermocycling conditions were as follows: denaturation at 42°C for 60 min, extension at 70°C for 10 min, storage at 4°C. qPCR was performed on cDNA using SYBR Green Realtime PCR Master Mix-Plus (Toyobo Co., Ltd., Osaka, Japan) and miR-203-specific qPCR primers (Table I; Shanghai GenePharma Co., Ltd.). The reaction volume was 20 µl [7.2 µl diethyl pyrocarbonate-treated water, 10 µl SYBR Green mix, 0.4 µl (10 µM) of each primer and 2 µl cDNA]. Thermocycling conditions were as follows: Initial 1 cycle at 95°C for 180 sec, followed by 40 cycles at 95°C for 12 sec and at 62°C for 40 sec.
Table I.
Gene-specific primers for reverse transcription-quantitative polymerase chain reaction.
| Gene | Sequence (5′→3′) |
|---|---|
| β-actin | F: CGTAAAGACCTCTATGCCAACA |
| R: -GGAGGAGCAATGATCTTGATCT | |
| α-SMA | F: GTGCTGTCCCTCTATGCCTCTGG |
| R: GGCACGTTGTGAGTCACACCATC | |
| COL1A1 | F: GTACATCAGCCCAAACCCCAAG |
| R: CGGAACCTTCGCTTCCATACTC | |
| COL3A1 | F: GACTGCCCCAACCCAGAGATC |
| R: TACCATCAGGAATGACAGGAGCAG′ | |
| SMAD3 | F: CGATGTCCCCAGCACACAATAAC |
| R: TAGTAGGAGATGGAGCACCAAAAGG | |
| miR-203 | F: CGATGCTGTGAAATGTTTAGGGAC |
| R: TATGGTTTTGACGACTGTGTGAT | |
| U6 | F: ATTGGAACGATACAGAGAAGATT |
| R: GGAACGCTTCACGAATTTG |
COL, collagen; F, forward; miR, microRNA; R, reverse; SMA, smooth muscle actin; SMAD3, mothers against decapentaplegic homolog 3.
To detect the mRNA expression of collagen type 1, α1 (COL1A1), COL3A1, α-smooth muscle actin (α-SMA) and SMAD3, total RNA was reverse transcribed into cDNA using the RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. qPCR was performed on the cDNA using specific primers (Table I; Shanghai GenePharma Co., Ltd.) and SYBR Green Realtime PCR Master Mix-Plus (Toyobo Co., Ltd.). The reaction volume was 20 µl [7.2 µl diethyl pyrocarbonate-treated water, 10 µl SYBR Green mix, 0.4 µl (10 µM) of each primer and 2 µl cDNA]. Thermocycling conditions were as follows: Initial 1 cycle at 95°C for 180 sec, followed by 40 cycles at 95°C for 12 sec and at 62°C for 40 sec. Relative gene expression was quantified using the 2−ΔΔCq method (19); miR-203 expression was normalized to the expression of U6, whereas COL1A1, COL3A1, α-SMA and SMAD3 expression was normalized to β-actin. Experiments were performed in triplicate. GraphPad Prism software version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA) was used for analysis.
Western blot analysis
HSC-T6 cells (~2×106) were lysed using RIPA lysis buffer (Beyotime Institute of Biotechnology, Haimen, China) at 4°C for 30 min and total protein concentration was measured using a bicinchoninic acid protein assay kit (Beyotime Institute of Biotechnology). Protein samples were denatured at 100°C for 5 min, and equal amounts of extracted protein samples (20 µg) were separated by 8–10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Following blocking against non-specific protein binding using 5% bovine serum albumin (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 60 min at room temperature, membranes were incubated with the following primary antibodies at 4°C overnight: rabbit monoclonal anti-α-SMA (1:1,000; cat no. ab32575; Abcam, Cambridge, MA, USA), rabbit monoclonal anti-Smad3 (1:1,000; cat no. C67H9; Cell Signaling Technology, Inc., Danvers, MA, USA), mouse monoclonal anti-COL1A1 (1:500; cat no. ab6308; Abcam) and anti-COL3A1 (1:500; cat no. ab6310; Abcam), and rabbit polyclonal anti-GAPDH (1:500; cat no. ab8245; Abcam). Membranes were then washed with 1X TBS containing 0.1% Tween-20 (TBST) 3 times for 10 min and incubated with the following horseradish peroxidase-conjugated AffiniPure secondary antibodies: Goat anti-rabbit (1:8,000; cat no. 111-035-003; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) and goat anti-mouse (1:2,000; cat no. 115-035-003; Jackson ImmunoResearch Laboratories, Inc.) for 1 h at room temperature. The membranes were washed with TBST 3 times for 10 min and the protein bands were visualized by enhanced chemiluminescence (ECL) using SuperSignal™ West Femto Maximum Sensitivity Substrate for ECL (Thermo Fisher Scientific, Inc.). GAPDH was used as the loading control. Blots were semi-quantified by densitometric analysis using the Image Lab software version 4.1 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cellular proliferation assay
HSC-T6 cells were seeded (~1×104 cells/well) in 96-well plates and transfected with miR-203 mimic, miR-203 inhibitor or scramble miRNA for 48 h, using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). Cellular proliferation was measured using an MTT assay (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). Following transfection, 0.5% MTT (20 µl) was added in each well and cells were incubated at 37°C for 4 h. Subsequently, 150 µmol DMSO were added to each well to dissolve the formazan crystals. The absorbance was measured at 490 nm and the optical density (OD) of the samples was calculated. Results were averaged from at least three independent experiments.
Dual-luciferase reporter assay
Bioinformatics analysis was used to predict potential target genes for miR-203; TargetScan (www.targetscan.org/vert_71/) (20), PicTar (pictar.mdc-berlin.de/) (21) and miRanda (www.microrna.org/microrna/home.do) (22) software were used. Based on the results of the bioinformatics analysis, oligonucleotides (62 bp) of wild-type and mutant SMAD3 3′-untranslated region (UTR), containing the putative miR-203 binding sites, were synthesized by Shanghai GenePharma Co., Ltd. and cloned into the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega Corporation, Madison, WI, USA) by double digestion with SacI/XhoI restriction enzymes by Shanghai GenePharma Co., Ltd. The dual-luciferase reporter assay was performed in 293T human embryonic kidney cells obtained from the Central Laboratory of the First Affiliated Hospital of Wenzhou Medical University (Wenzhou, China). The 293T cell line is characterized by high transfection efficiency, with no influence on the expression of target genes; therefore, 293T cells were used for this experiment. Briefly, 293T cells were seeded in 24-well plates at a density of 3×104 cells/well, and incubated for 24 h at 37°C in a 5% CO2 humidified atmosphere. Subsequently, cells were co-transfected with either miR-203 mimics (20 pmol) or scramble miRNA negative control (20 pmol) and a reporter plasmid (50 ng) containing either wild-type or mutant SMAD3-3′UTR sequences, or a blank reporter plasmid, using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) as the transfection reagent. Following 48 h transfection at 37°C in a 5% CO2 humidified atmosphere, luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega Corporation), and the signal was recorded using the Smart Line TL Tube Luminometer (Berthold Detection Systems GmbH, Pforzheim, Germany). Renilla luciferase was used as the internal control.
Statistical analysis
The statistical significance of the differences between groups was assessed using a Student's t-test for pair-wise comparisons and a one-way analysis of variance followed by a post hoc Dunnett's test for multiple comparisons. Data are expressed as the mean ± standard deviation of at least three independent experiments. P<0.05 was considered to indicate a statistically significant difference. Statistical analysis was performed using SPSS software version 19.0 (IBM Corp., Armonk, NY, USA).
Results
miR-203 suppresses COL1A1, COL3A1 and α-SMA expression in HSCs
To investigate the putative roles of miR-203 in the development of hepatic fibrosis, the expression levels of genes associated with hepatic fibrogenesis were assessed. During chronic liver injury, activated stellate cells have been reported to remodel the ECM and enrich the fibril-forming collagen contents, particularly collagen types I and III (23), whereas α-SMA has been identified as a marker of hepatic fibrogenesis (24). In the present study, HSC-T6 cells were transfected with a miR-203 mimic, a miR-203 inhibitor or with scramble miRNA. Transfection efficiency was assessed using FAM-labeled miRNAs (Fig. 1A-D). RT-qPCR was used to confirm that miR-203 expression was significantly increased in HSC-T6 cells following miR-203 mimic transfection, whereas it was significantly suppressed following transfection with the miR-203 inhibitor (P<0.01 vs. scramble miRNA; Fig. 1E).
Figure 1.

Expression of miR-203 in rat HSCs. Rat HSC-T6 cells were transfected with a miR-203 mimic, a miR-203 inhibitor or scramble miRNA. Transfection efficiency was assessed following transient transfection with (A and B) FAM-labeled NC miRNAs and (C and D) unlabeled NC miRNAs. Photomicrographs were captured under (A and C) bright field and (B and D) fluorescence microscopy. Scale bar, 100 µm. (E) Reverse transcription-quantitative polymerase chain reaction was used to assess miR-203 expression levels in HSC-T6 cells transfected with a miR-203 mimic, a miR-203 inhibitor or scramble miRNA. Data are expressed as the mean ± standard deviation of at least three independent experiments. **P<0.01. FAM, fluorescein amidite; HSC, hepatic stellate cell; miR, microRNA; NC, negative control.
RT-qPCR and western blot analyses were used to investigate mRNA and protein expression levels of COL1A1, COL3A1 and α-SMA (Fig. 2). Notably, mRNA expression levels of the fibrosis-associated genes COL1A1, COL3A1 and α-SMA were significantly upregulated in HSC-T6 cells transfected with the miR-203 inhibitor, whereas they were significantly downregulated in miR-203 mimic-transfected cells compared with the scramble group (P<0.01; Fig. 2A and B). Similarly, COL1A1, COL3A1 and α-SMA protein expression levels were increased following transfection with the miR-203 inhibitor and decreased following transfection with mimics, compared with the scramble miRNA control group (P<0.01 and P<0.01, respectively; Fig. 2C and D).
Figure 2.

Effects of miR-203 on COL1A1, COL3A1 and α-SMA expression in rat HSCs. Rat HSC-T6 cells were transfected with a miR-203 mimic, a miR-203 inhibitor or scramble miRNA. mRNA expression levels for (A) α-SMA, (B) COL1A1 and COL3A1 were assessed using reverse transcription-quantitative polymerase chain reaction. (C) α-SMA, (D) COL1A1 and COL3A1 protein expression levels were assessed by western blot analysis. Data are expressed as the mean ± standard deviation of at least three independent experiments. **P<0.01. COL, collagen; HSC, hepatic stellate cell; miR, microRNA; SMA, smooth muscle actin; inh, miR-203 inhibitor; scr, scramble miRNA; mim, miR-203 mimic.
miR-203 inhibits HSC proliferation
To investigate whether miR-203 may be involved in the regulation of HSC proliferation, the MTT assay was used to assess the effects of miR-203 inhibition and upregulation on the proliferative capabilities of rat HSCs. The results demonstrated that HSC-T6 cell proliferation was significantly increased following transfection with the miR-203 inhibitor (P<0.01), whereas it was significantly suppressed in cells transfected with the miR-203 mimic (P<0.01) compared with the scramble group (Fig. 3). These results suggested that miR-203 may serve a role as a modulator of HSC proliferation.
Figure 3.
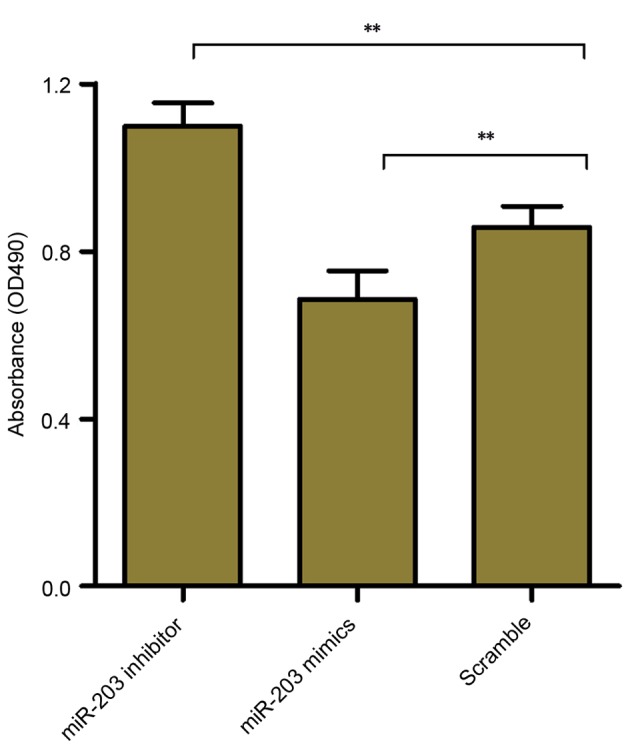
Effects of miR-203 on rat HSC proliferation. Rat HSC-T6 cells were transfected with a miR-203 mimic, a miR-203 inhibitor or scramble miRNA, and cellular proliferation was evaluated using the MTT assay and measurement of the absorbance at 490 nm. Data are expressed as the mean ± standard deviation of at least three independent experiments. **P<0.01. HSC, hepatic stellate cell; miR, microRNA; OD, optical density.
SMAD3 is a target of miR-203
To investigate the potential mechanisms underlying the implication of miR-203 in HSC activation and proliferation, bioinformatics analysis was performed to identify potential target genes of miR-203. TRPV4 has previously been reported as a direct target gene of miR-203 during HSC activation (17), and the TGF-β/Smad signaling pathway has been demonstrated to serve an important role in hepatic fibrosis (25). Therefore, the present study investigated whether SMAD3 may be a direct target gene for miR-203. RT-qPCR and western blot analyses were used to evaluate SMAD3 mRNA and protein expression levels, and revealed that SMAD3 mRNA and protein expressions in miR-203 mimic-transfected cells were significantly reduced compared with the scramble group (Fig. 4A and B). Conversely, SMAD3 mRNA and protein expression levels were significantly increased in HSC-T6 cells transfected with the miR-203 inhibitor (P<0.01; Fig. 4A and B).
Figure 4.

SMAD3 is a direct target gene of miR-203. Rat HSC-T6 cells were transfected with a miR-203 mimic, a miR-203 inhibitor or scramble miRNA. (A) Western blot analysis was used to assess SMAD3 protein expression levels in HSC-T6 cells. (B) Reverse transcription-quantitative polymerase chain reaction was used to assess SMAD3 mRNA expression levels. (C) Bioinformatics analysis predicted the SMAD3 3′-UTR as a potential miR-203 target. The SMAD3 3′-UTR target region for miR-203 and the mutant sequence are presented in red. (D) A dual-luciferase reporter assay was performed on 293T cells co-transfected with reporter constructs containing either wild-type or mutant SMAD3 3′-UTR or a blank reporter plasmid, and either miR-203 mimics or scramble miRNA. Renilla luciferase was used as a control for the normalization of luciferase activity. Data are expressed as the mean ± standard deviation of at least three independent experiments. **P<0.01. HSC, hepatic stellate cell; miR, microRNA; mut, mutant; SMAD3, mothers against decapentaplegic homolog 3; UTR, untranslated region; Rno, Rattus norvegicus.
To investigate whether SMAD3 may be a target of miR-203, the 3′-UTR of the SMAD3 mRNA was cloned into a pmirGLO vector (Fig. 4C). A dual-luciferase reporter assay demonstrated that cells co-transfected with the plasmid containing the wild-type SMAD3 3′-UTR and the miR-203 mimic exhibited the lowest luciferase activity (P<0.01 vs. mutant SMAD3; Fig. 4D). These results suggested that SMAD3 may be a direct target gene of miR-203.
Discussion
Hepatic fibrosis is a common pathophysiological process that underlies chronic liver disease, regardless of etiology. Hepatic fibrogenesis may progress to liver cirrhosis, hepatic failure or even hepatocellular carcinoma (1). Currently, no effective antifibrotic therapeutic strategies are available for clinical use; therefore, the elucidation of the pathophysiological mechanisms underlying fibrogenesis and the development of novel therapeutic strategies are imperative for the treatment of patients with hepatic fibrosis (26). HSC activation has been identified among the primary critical events in the development of hepatic fibrosis, and activated HSCs are the major source of ECM components (3,27). Altered ECM composition has been demonstrated in the fibrotic liver, with increased prevalence of fibrillar types of collagen, such as type I and type III. Furthermore, activated HSCs have been reported to express myogenic markers, including α-SMA (28). Previous studies have suggested that the prevention of the synthesis and deposition of ECM components may have potential as an effective antifibrotic therapeutic strategy (28,29). Song et al (17) reported decreased miR-203 expression levels in human and rodent fibrotic liver tissue, and in TGF-β1-treated HSC-T6 cells. However, the roles of miR-203 in collagen synthesis and HSC proliferation have yet to be elucidated. The present study aimed to investigate the roles of miR-203 in collagen synthesis and HSC proliferation, as well as to explore the molecular mechanisms underlying its actions. Following transfection of the HSC-T6 rat hepatic stellate cell line with a miR-203 inhibitor, the mRNA and protein expression levels of the fibrosis-related proteins COL1A1, COL3A1 and α-SMA were significantly upregulated; whereas transfection with a miR-203 mimic resulted in a significant decrease in the mRNA and protein expression levels. In addition, HSC proliferation was significantly enhanced following miR-203 inhibition, and suppressed following miR-203 potentiation.
Proinflammatory cytokines, including platelet-derived growth factor, TGF-β, connective tissue growth factor and endothelin-1, have been implicated in the processes of HSC activation (4,30–32). Among these cytokines, the TGF-β signaling pathway has been revealed as an important regulator of HSC activation (4). SMAD proteins, which are intracellular mediators belonging to the TGF-β family, are classified into three groups: receptor-regulated Smads (R-Smads), common mediator Smads (Co-Smads) and inhibitor Smads (30). R-Smads (SMADs 1–3, 5 and 8) are phosphorylated and activated through the TGF-β receptor I kinase, and form heteromeric complexes with Co-Smads (Smad4). These complexes can translocate into the nucleus and associate with other co-mediators to regulate the expression of target genes (33). Smads have previously been identified as miRNA targets in various diseases: miR-203 has been reported to inhibit heat-denatured fibroblast proliferation and migration through the regulation of SMAD3, whereas miR-454 has been demonstrated to inhibit HSC activation by directly targeting SMAD4 (34). Furthermore, SMAD3 has been demonstrated to modulate E-cadherin expression through a miR-200-dependent mechanism (12). These results suggested that SMAD proteins may act as miRNA targets during the modulation of gene expression. In the present study, miR-203 target genes were predicted using bioinformatics analysis, and SMAD3 was identified among ~937 other targets. SMAD3 mRNA and protein expression levels were demonstrated to be significantly upregulated in HSCs transfected with a miR-203 inhibitor, whereas they were downregulated in cells transfected with a miR-203 mimic. In addition, a dual-luciferase reporter assay validated SMAD3 as a direct target gene of miR-203. These results suggested that miR-203 may silence the expression of SMAD3 and thus prevent the activation of the TGF-β/Smad signaling pathway and the activation of HSCs, therefore suppressing the synthesis and secretion of ECM components, including, COL1A1, COL3A1 and α-SMA. However, further studies are required to investigate the specific molecular mechanisms underlying the regulatory effects of miR-203 on the synthesis and deposition of ECM components, and on HSC proliferation.
In conclusion, the results of the present study suggested that miR-203 may inhibit HSC proliferation and suppress the expression of collagen-related genes. Furthermore, SMAD3 was identified as a direct target gene of miR-203, thus suggesting that miR-203-mediated modulation of SMAD3 signaling may be implicated in HSC activation. Further studies are required in order to elucidate the specific roles of miR-203 in the complex molecular mechanisms underlying hepatic fibrogenesis.
Acknowledgements
The present study was supported by The Natural Science Foundation of Zhejiang Province (grant no. LY14H030010).
Glossary
Abbreviations
- HSC
hepatic stellate cell
- α-SMA
α-smooth muscle actin
- SMAD3
mothers against decapentaplegic homolog 3
- ECM
extracellular matrix
- COL1A1
collagen 1A1
- COL3A1
collagen 3A1
- RT-qPCR
reverse transcription-quantitative polymerase chain reaction
- UTR
untranslated region
- miRNA
microRNA
- TGF
transforming growth factor
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- MTT
methyl thiazolyl trazolium
References
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL. Molecular mechanisms of hepatic fibrosis and principles of therapy. J Gastroenterol. 1997;32:424–430. doi: 10.1007/BF02934504. [DOI] [PubMed] [Google Scholar]
- 3.Li D, Friedman SL. Liver fibrogenesis and the role of hepatic stellate cells: New insights and prospects for therapy. J Gastroenterol Hepatol. 1999;14:618–633. doi: 10.1046/j.1440-1746.1999.01928.x. [DOI] [PubMed] [Google Scholar]
- 4.Bissell DM, Roulot D, George J. Transforming growth factor beta and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 5.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 6.Blahna MT, Hata A. Smad-mediated regulation of microRNA biosynthesis. FEBS Lett. 2012;586:1906–1912. doi: 10.1016/j.febslet.2012.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006;20:515–524. doi: 10.1101/gad.1399806. [DOI] [PubMed] [Google Scholar]
- 9.Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: Key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene. 2008;27:5959–5974. doi: 10.1038/onc.2008.274. [DOI] [PubMed] [Google Scholar]
- 10.Wang XW, Heegaard NH, Orum H. MicroRNAs in liver disease. Gastroenterology. 2012;142:1431–1443. doi: 10.1053/j.gastro.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu F, Guo Y, Chen B, Dong P, Zheng J. MicroRNA-17-5p activates hepatic stellate cells through targeting of Smad7. Lab Invest. 2015;95:781–789. doi: 10.1038/labinvest.2015.58. [DOI] [PubMed] [Google Scholar]
- 12.Ahn SM, Cha JY, Kim J, Kim D, Trang HT, Kim YM, Cho YH, Park D, Hong S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene. 2012;31:3051–3059. doi: 10.1038/onc.2011.484. [DOI] [PubMed] [Google Scholar]
- 13.Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol. 2011;22:1668–1681. doi: 10.1681/ASN.2010111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang F, Yang Z, Cao M, Xu Y, Li J, Chen X, Gao Z, Xin J, Zhou S, Zhou Z, et al. MiR-203 suppresses tumor growth and invasion and down-regulates MiR-21 expression through repressing Ran in esophageal cancer. Cancer Lett. 2014;342:121–129. doi: 10.1016/j.canlet.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, Wang X, Liang H, Wang T, Yan X, Cao M, Wang N, Zhang S, Zen K, Zhang C, Chen X. miR-203 inhibits cell proliferation and migration of lung cancer cells by targeting PKCα. PLoS One. 2013;8:e73985. doi: 10.1371/journal.pone.0073985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y, Ren F, Rong M, Luo Y, Dang Y, Chen G. Association between underexpression of microrna-203 and clinicopathological significance in hepatocellular carcinoma tissues. Cancer Cell Int. 2015;15:62. doi: 10.1186/s12935-015-0214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song Y, Zhan L, Yu M, Huang C, Meng X, Ma T, Zhang L, Li J. TRPV4 channel inhibits TGF-β1-induced proliferation of hepatic stellate cells. PLoS One. 2014;9:e101179. doi: 10.1371/journal.pone.0101179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel S, Piantedosi R, Frank J, Lalazar A, Rockey DC, Friedman SL, Blaner WS. An immortalized rat liver stellate cell line (HSC-T6): A new cell model for the study of retinoid metabolism in vitro. J Lipid Res. 2000;41:882–893. [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Chen K, Rajewsky N. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet. 2006;38:1452–1456. doi: 10.1038/ng1910. [DOI] [PubMed] [Google Scholar]
- 22.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gressner AM. Cytokines and cellular crosstalk involved in the activation of fat-storing cells. J Hepatol. 1995;22(Suppl 2):S28–S36. [PubMed] [Google Scholar]
- 24.Nouchi T, Tanaka Y, Tsukada T, Sato C, Marumo F. Appearance of alpha-smooth-muscle-actin-positive cells in hepatic fibrosis. Liver. 1991;11:100–105. doi: 10.1111/j.1600-0676.1991.tb00499.x. [DOI] [PubMed] [Google Scholar]
- 25.Wells RG. Fibrogenesis. V. TGF-beta signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2000;279:G845–G850. doi: 10.1152/ajpgi.2000.279.5.G845. [DOI] [PubMed] [Google Scholar]
- 26.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 27.Safadi R, Friedman SL. Hepatic fibrosis-role of hepatic stellate cell activation. Med Gen Med. 2002;4:27. [PubMed] [Google Scholar]
- 28.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI200524282C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghiassi-Nejad Z, Friedman SL. Advances in antifibrotic therapy. Expert Rev Gastroenterol Hepatol. 2008;2:803–816. doi: 10.1586/17474124.2.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borkham-Kamphorst E, van Roeyen CR, Ostendorf T, Floege J, Gressner AM, Weiskirchen R. Pro-fibrogenic potential of PDGF-D in liver fibrosis. J Hepatol. 2007;46:1064–1074. doi: 10.1016/j.jhep.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 31.Paradis V, Dargere D, Vidaud M, de Gouville AC, Huet S, Martinez V, Gauthier JM, Ba N, Sobesky R, Ratziu V, Bedossa P. Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology. 1999;30:968–976. doi: 10.1002/hep.510300425. [DOI] [PubMed] [Google Scholar]
- 32.Rockey DC, Fouassier L, Chung JJ, Carayon A, Vallee P, Rey C, Housset C. Cellular localization of endothelin-1 and increased production in liver injury in the rat: Potential for autocrine and paracrine effects on stellate cells. Hepatology. 1998;27:472–480. doi: 10.1002/hep.510270222. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/S0092-8674(03)00432-X. [DOI] [PubMed] [Google Scholar]
- 34.Zhu D, He X, Duan Y, Chen J, Wang J, Sun X, Qian H, Feng J, Sun W, Xu F, Zhang L. Expression of microRNA-454 in TGF-β1-stimulated hepatic stellate cells and in mouse livers infected with Schistosoma japonicum. Parasit Vectors. 2014;7:148. doi: 10.1186/1756-3305-7-148. [DOI] [PMC free article] [PubMed] [Google Scholar]