

Abstract
Alcohol is one of the most socially accepted addictive drugs in modern society. Its abuse affects virtually all organ systems with the central nervous system (CNS) being particularly vulnerable to excessive alcohol exposure. Alcohol exposure also causes profound damage to both the adult and developing brain. Excessive alcohol consumption induces numerous pathophysiological stress responses, one of which is the endoplasmic reticulum (ER) stress response. Potential mechanisms that trigger the alcohol induced ER stress response are either directly or indirectly related to alcohol metabolism, which include toxic levels of acetaldehyde and homocysteine, oxidative stress and abnormal epigenetic modifications. Growing evidence suggests that H2S is the most recently recognized gasotransmitter with tremendous physiological protective functions against oxidative stress induced neurotoxicity. In this review we address the alcohol induced oxidative stress mediated ER stress and the role of H2S in its mitigation in the context of alcohol neurotoxicity. Interruption of ER stress triggers is anticipated to have therapeutic benefits for alcohol mediated diseases and disorders.
Keywords: Endoplasmic reticulum stress, Alcohol, Homocysteine, Unfolded protein response, Reactive oxygen species, Hydrogen sulfide
1. Introduction
Alcohol is one of the most socially accepted addictive drugs in modern society. Alcohol abuse can have devastating effects on individuals’ health, careers and relationships (Cheng, 2012). Excessive alcohol use is the third leading cause of preventable death in the United States and is responsible for 3.8% of deaths worldwide (Gunzerath et al., 2011; Rehm et al., 2009). Alcohol is readily spread throughout the body in the blood stream and crosses biological membranes which, affects virtually all biological processes. Continual alcohol abuse leaves profound effects in virtually all organ systems with the central nervous system (CNS) being particularly vulnerable to excessive alcohol exposure. Alcohol exposure also causes profound damage to both the adult and developing brain (Yang and Luo, 2015). Excessive alcohol consumption induces numerous pathophysiological stress responses part of which, is the endoplasmic reticulum (ER) stress response. ER stress, a condition under which unfolded/misfolded proteins accumulate in the ER, contributes to alcoholic disorders of major organs such as the liver, pancreas, heart and especially the brain. Potential mechanisms that trigger the alcoholic ER stress response are either directly or indirectly related to alcohol metabolism which, include toxic levels of acetaldehyde and homocysteine, oxidative stress and abnormal epigenetic modifications (Cheng, 2012).
The ER is a regulator of posttranslational protein processing and transport. The accumulation of unfolded or misfolded proteins in the ER lumen triggers ER stress inducing the unfolded protein response (UPR) which, is mediated by three transmembrane ER signaling proteins: pancreatic endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6) (Yang and Luo, 2015). UPR is initiated to protect cells from overwhelming ER protein loading and sustained ER stress may result in cell death. ER stress has been implied in various CNS injuries including brain ischemia, traumatic brain injury and aging-associated neurodegeneration such as vascular dementia (VAD), Alzheimer’s disease (AD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD). However; the effects of alcohol on ER stress in the CNS receive less attention (Yang and Luo, 2015). In this review we address the alcohol induced oxidative stress mediated ER stress and the role of H2S in its mitigation in the context of alcohol neurotoxicity. Interruption of ER stress triggers is anticipated to have therapeutic potential to manage alcohol related diseases and disorders.
2. Homocysteine and alcohol mediated neurotoxicity
Homocysteine (Hcy) is a normal intermediate involved in the metabolism of the essential amino acid methionine. Alcohol interferes with the Hcy metabolism in multiple ways (Fig. 1) which leads to increased accumulation of Hcy in plasma, inducing hyperho-mocysteinemia (HHcy) (Gibson et al., 2008). Along with alcohol, dietary methionine drives a transient increase in methionine, S-adenosylmethionine (SAM) and Hcy (Finkelstein and Martin, 1986). HHcy is reportedly associated with insulin resistance (Meigs et al., 2001) and it is a common risk factor in Type 2 Diabetes (Qureshi et al., 2003). HHcy is an independent risk factor in cardiovascular, neuroinflammatory and neurodegenerative diseases, obesity and hepatic steatosis (Seshadri et al., 2002). HHcy is also associated with damage to the vascular system by a mechanism related to oxidative stress resulting in a build-up of damaging free hydrogen radicals (Tyagi et al., 2005). Different investigations have shown that alcohol consumption, particularly in actively drinking alcoholics, is closely associated with elevated plasma Hcy levels. Human studies suggest that Hcy plays a role in brain damage including a decline in cognitive functions and memory. Mild to moderate HHcy is a known risk factor for neurodegenerative and neurovascular diseases. Hcy or folate and vitamin B12 deficiency can cause disturbed methylation and/or redox potentials thereby, promoting calcium influx, amyloid and tau protein accumulation, apoptosis and neuronal death. Hcy promotes neuronal degeneration contributing to psychiatric and age-related neurodegenerative diseases. Hcy metabolism in relation to neurodegenerative diseases arises from the fact that plasma concentrations of Hcy increase with age (Herrmann et al., 1999) and it is considered a risk factor for vascular disease as well as brain atrophy (den Heijer et al., 2003). Total fasting Hcy (tHcy) concentrations greater than 11.9 μmol/L were associated with a higher risk for white matter damage when compared to concentrations below 8.6 μmol/L (Wright et al., 2005). The epidemiological and longitudinal studies of Nurk et al. (2005) suggested a causal link between Hcy and cognitive impairment. This link might be due to cerebrovascular as well as direct neurotoxic mechanisms (Sachdev, 2005). Changes in Hcy levels are also predicted to cause a decline in memory scores in elderly subjects (Nurk et al., 2005). Several follow up studies demonstrated a positive association between the level of Hcy and cognitive functions (Seshadri et al., 2002). Alcohol induced genetic alteration and its consequences lead to decreased expression of Hcy-metabolizing enzymes such as cystathionine β-synthase (CBS), cystathionine-7-lyase (CSE), methylene tetrahy-drofolate reductase (MTHFR) and methionine synthase (MS) thus, inducing HHcy (Dayal et al., 2004). Changes in plasma Hcy concentrations reflect one aspect of the metabolic consequences of methyl group deficiencies. Folic acid supplementation helps to alleviate these deficiencies because betaine works as a methyl donor. Betaine is a significant determinant of plasma Hcy particularly in cases of folate deficiency, methionine overload or alcohol consumption. Betaine supplementation has a lowering effect on methionine load and high Hcy levels. Increasing choline or betaine levels can reduce hypomethylation and lower Hcy levels. Folic acid supplementation also lowers the risk factor for stroke by reducing tHcy levels. Increased levels of Hcy can cause elevated blood pressure and are considered a risk factor for cerebrovascular dysfunction. In particular, Hcy levels are increased in the body when metabolism of cysteine or methionine is impaired. This often occurs due to dietary deficiencies in B vitamins such as vitamin B6, vitamin B12 and folic acid. Chronic alcohol consumption negatively impacts dietary choices and significantly reduces absorption of B vitamins. It has been shown that Hcy is regulated through a series of pathways which, are dependent on B vitamins particularly folate (Halsted et al., 2002). However; it remains obscure whether alcohol dependent patients benefit from Hcy lowering strategies through folate, vitamin B6 or B12 supplementation particularly, in those who have a low folate status. Reports have also suggested that consumption of alcohol along with a folate-deficient diet is associated with increased levels of Hcy (Halsted et al., 2002). Thus, it may be concluded that increased alcohol intake and a low folate diet may lead to HHcy. Previous studies imply, excess accumulation of Hcy in the body causes cell damage and promotes vascular and microvascular disorders that lead to cerebrovascular dysfunction (Dayal et al., 2004; Kamat et al., 2013).
Fig. 1.
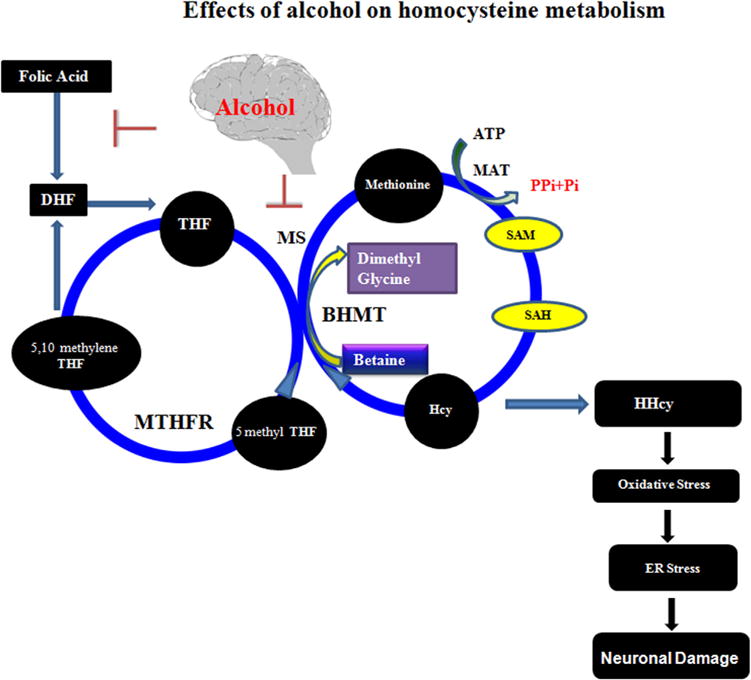
Interaction of alcohol in Hcy metabolism and ER stress: Alcohol interacts with methionine synthase (MS), which is responsible for the conversion of Hcy to methionine. One of the possible reasons for HHcy during alcohol exposure may be due to inhibition of the enzyme MS by alcohol, which disallows Hcy to be converted into methionine thus leading to the condition of HHcy. On the other hand, other pathways such as transmethylation may also be affected by the inhibition of MS and may cause abrupt disturbance of S-Adenosyl methionine (SAM) and S-Adenosyl-L-homocysteine (SAH). SAM to SAH conversion is controlled by DNA methyltransferase (DNMT). If the pathway becomes disturbed, the transmethylation process also becomes disturbed. On the other hand, high Hcy also causes the low expression of cystathionine β-synthase (CBS), an antioxidant enzyme in the brain which maintains the redox system. Alcohol also interacts with folic acid pathways. Folic acid is converted into tetrahydrofolate (THF) by the enzyme dihydrofolate reductase (DHFR). THF is converted into methylene tetrahydrofolate (MTHF) by the cofactor vitamin B6. Again MTHF is transformed to THF by the key enzyme, MTHF reductase (MTHFR). In this diagram, the author speculates that alcohol may inhibit folic acid to DHF conversion and methionine synthase activity. In this mechanism, alcohol can trigger HHcy and thus affect the redox system, leading to ER stress resulting in neuronal damage that ends in impaired brain function. H2S inhibits free-radical reactions and oxidative stress thereby, alleviating ER stress, providing neuronal protection.
3. ER stress and alcohol mediated neurotoxicity
The efficient functioning of the ER is indispensable for most cellular activities and survival, as it involves the synthesis, folding, modification and transport of newly synthesized transmembrane and secretory proteins. The accumulation of unfolded or misfolded proteins in the ER lumen induces UPR which, triggers ER stress that is mediated by three transmembrane ER signaling proteins: PERK, IRE1 and ATF6. UPR is initiated to protect cells from overwhelming the ER with protein loading. However; sustained ER stress may result in cell death. ER stress has been implied in various CNS injuries, including brain ischemia, traumatic brain injury and aging-associated neurodegenerative and neurovascular disorders. However; the effects of alcohol on ER stress in the CNS receive less attention.
The ER also has important roles in the storage of intracellular Ca2+ and regulation of Ca2+ homeostasis. The integrity of the Ca2+ homeostasis in the ER lumen is vital for proper folding of proteins. A downregulation of ER Ca2+ could result in an increase of unfolded or misfolded proteins and ER stress. The ER stress response constitutes a cellular process that is triggered by a variety of conditions that disturb the folding of proteins in the ER. It has been observed that activated ER stress pathways contribute to the pathogenesis of important diseases including diabetes and cancer (Wang and Kaufman, 2012). Altered regulation of ER stress has also been revealed in various forms of CNS injury including brain ischemia, traumatic brain injury, spinal cord injury, epilepticus as well as neurodegeneration encompassing PD, ALS, AD and HD (Placido et al., 2015). The expression of ER stress markers, such as GPR78, PERK, EIF2 and IRE1 are altered in AD patient samples (Hoozemans et al., 2009). The accumulation of ER stress-associated proteins such as, Hrd1p/Der3p (HRD1) that enhance ubiquitination, have been observed within neurons of brain tissue in PD patients (Nakashima et al., 2012). Enhanced ER stress stimulates production of reactive oxygen species (ROS) in multiple pathways. This interruption of ER stress triggers is anticipated to have immense therapeutic benefits and clinical potential for alcohol related disorders.
4. Oxidative stress and alcohol mediated neurotoxicity
Cells have basal levels of ROS for signaling and normal functioning. In contrast, ROS levels increase upon stress or during enzymatic reactions e.g., mitochondrial respiratory chain reactions, arachidonic acid pathway, cytochrome P450 family and those involving glucose oxidase, amino acid oxidase, xanthine oxidase, NADP/NADPH oxidase, NO synthases (Malhotra and Kaufman, 2007), protein disulfide isomerases, endoplasmic reticulum oxidoreductin-1(ERO-1) and NADPH oxidase (Santos et al., 2009). Both oxidative stress and ER stress increase the leakage of Ca2+ from the ER lumen (Berridge et al., 2003). Increases in cytosolic Ca2+ can stimulate mitochondrial ROS production. Oxidative stress has been proposed as a major contributor to alcohol induced multiorgan damage, including CNS pathogenesis (Comporti et al., 2010).
ROS is generated during alcohol metabolism (Fig. 2) or Hcy can undergo autoxidation thus, causing the disruption of redox homeostasis and effecting redox signaling pathways (Zou and Banerjee, 2005). Hcy has also been found to induce neurological dysfunction via oxidative stress (Ho et al., 2001). Antioxidant treatment restores several of the toxic effects Hcy has on neurological function (Jara-Prado et al., 2003). The role of oxidative stress in neurodegeneration has previously been intensively studied. Oxidative stress was one important mechanism for Hcy toxicity in neuronal cells (Ho et al., 2003). Hcy directly increased the neurotoxicity of amyloid-β peptide (Aβ) by inducing oxidative stress (Ho et al., 2001). The cytotoxicity of Hcy was mitigated by antioxidants like N-acetyl cysteine, vitamin E or vitamin C (Ho et al., 2001). Antioxidants (vitamin E or vitamin C) also prevented memory dysfunction and ATPase activity caused by HHcy in rats. Other studies by Ho et al. (2003) showed the effect of folate deficiency on the CNS (Kruman et al., 2005). Folate deprivation induced a marked increase in Hcy, ROS, and Aβ-induced apoptosis, while folate supplementation prevented the generation of ROS by Aβ (Ho et al., 2003). Treatment with the S-adenosyl hydrolase inhibitor, 3-deaza adenosine, provided neuroprotection in normal and apolipoprotein E-deficient mice, as well as in cultured neuronal cells deprived of folate and vitamin E subjected to oxidative challenge (Tchantchou et al., 2004). Thus, it is evident that conscious intervention to reduce ROS mitigates ER stress and consequently neuronal cell survival.
Fig. 2.
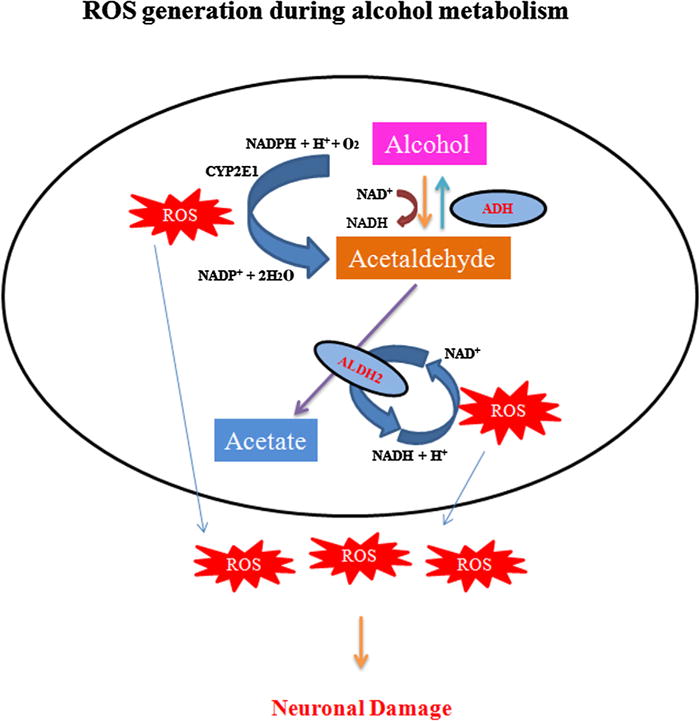
Induction of oxidative pathway response during alcohol metabolism. Upon exposure to alcohol, the cellular alcohol dehydrogenase enzyme (ADH) is activated in the cytosol, which metabolizes to produce acetaldehyde. During long term exposure or chronic alcohol consumption, an enzyme in the endoplasmic reticulum, cytochrome P450 IIE1 (CYP2E1), becomes activated and alcohol gets metabolized to acetaldehyde. The derived acetaldehyde enters into the mitochondria and is further metabolized into acetate mainly by aldehyde dehydrogenase 2 (ALDH2). Thereby, it results in the formation of reactive oxygen species (ROS), which leads to neurotoxicity and damage.
5. Hydrogen sulfide protection against ethanol mediated neuronal damage
Production of gaseous transmitters by mammalian cells has attracted much attention in recent years. The accumulating evidence suggests that despite it being previously seen as a noxious gas, H2S is rapidly emerging as a gaseous transmitter in addition to nitric oxide (NO) and carbon monoxide (CO) (Hosoki et al., 1997). The latter two molecules are endogenously produced gaseous transmitters that reportedly carry out a tremendous physiological role against free radical mediated oxidative damage in neurodegenerative diseases (Rosselli et al., 1998; Wu and Wang, 2005; Uttara et al., 2009). The third gaseous transmitter, H2S contributes to the regulation of cardiac function, systemic and pulmonary blood pressure and vasomotor activity, inflammation and angiogenesis (Kohn et al., 2012). All three gasotransmitters CO, NO H2S have attracted attention because they exert fine modulatory control over cellular functions by influencing an array of intracellular signaling processes (Sen, 2016). H2S was found to be produced endogenously in various parts of the body such as the heart, blood and CNS (Zhao et al., 2001) by two pyridoxal-5′-phosphate-dependent enzymes. These enzymes are responsible for metabolism of l-cysteine which is a by-product of l-methionine, Hcy and cystathionine. CBS, CSE and a newly identified enzyme 3-mercaptopyruvate sulfurtrans-ferase (3MST) (Sen et al., 2012) are involved in the generation of H2S. CBS is the major H2S producing enzyme in the brain (Abe and Kimura, 1996). H2S can easily penetrate the plasma membrane thus, inducing a wide spectrum of signaling cascades in target cells. Until recently, H2S was the least appreciated among the three gasotransmitters nevertheless; growing evidences suggests that it may emerge as the most important one. Particularly due to its ability in signaling activity by sulfhydrating target proteins (Sen, 2016). Some studies in cellular and animal models have suggested several mechanisms to explain the protection associated with H2S including promoting anti-inflammatory responses (Calvert et al., 2010), antiapoptotic effects (Yang et al., 2007), improving mitochondrial action (Kimura et al., 2010), cardiac systolic function, sensory transduction, vasodilation and neuroprotection (Wang, 2012). Endogenous H2S acts as a potent regulator of various biological processes mainly related to vasomotor function. H2S regulates intracellular calcium concentrations via L-type calcium channels, T-type calcium channels, sodium/calcium exchangers, transient receptor potential channels, β-adrenergic receptors and NMDA in various cells (Zhang et al., 2015). Physiological concentrations of H2S enhance the NMDA receptor-mediated responses and modify long-term potentiation (LTP) (Abe and Kimura, 1996). H2S is able to regulate the activity of several sirtuins and enhance AP-1 binding activity with the SIRT3 promoter, reducing oxidant-provoked vascular endothelial dysfunction (Xie et al., 2016). H2S and NO signaling pathways have been described to offer protection against AD associated amyloid vasculopathy and neurodegeneration. There are reports of H2S induced endothelial proliferation and migration as well as enhanced VEGF gene expression. H2S normalizes intracellular ethanol mediated reduced levels of GSH, the major intracellular antioxidant in the biological system (Kimura et al., 2006; Kimura and Kimura, 2004; Chen et al., 2016), alleviating ER stress thus, promoting neuronal protection and improving brain function (Fig. 3). H2S also plays a role in ER stress mitigation by sulfhydration (Krishnan et al., 2011) or scavenging the reactive carbonyl group (Koike and Ogasawara, 2016). There is a growing amount of evidence demonstrating that H2S is a potential therapeutic molecule that carries tremendous physiological functions.
Fig. 3.
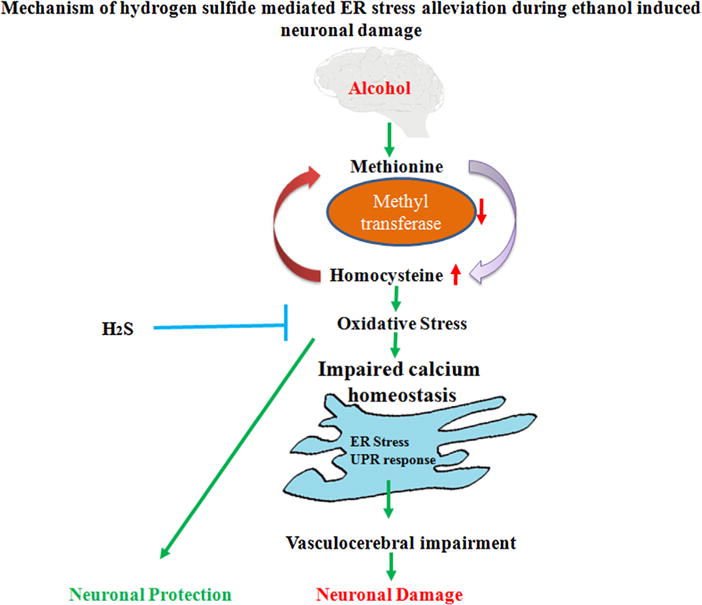
Mechanism of hydrogen sulfide mediated ER stress alleviation during ethanol induced neuronal damage. Ethanol causes deregulation of homocysteine metabolism leading to increased cellular Hcy levels. This further accelerates the oxidative pathways. Ethanol induced oxidative stress alters ER redox status and is manifested by activation of the stress response, which leads to cerebrovascular impairment or neuronal damage. H2S can potentially mitigate the cerebrovascular damage by alleviating the ER stress and thus provides neuroprotection.
6. Discussion and future prospective
Unhealthy alcohol consumption remains a principal problem for the public health of individuals and is responsible for high morbidity rates by affecting various organs and organ systems, especially the brain. The mammalian cell has evolved a complex and intertwined set of signaling pathways to respond to both physiological and pathological ER stress. Although these pathways are not yet fully characterized, it is becoming clear that ER stress induced ROS generation and UPR are intimately involved in neuronal damage and various neuronal diseases. Interruption of ER stress triggers is anticipated to have therapeutic benefits for controlling alcohol associated neuronal damage. Studies show that H2S has remarkable potential as a therapeutic tool by exploiting ways to increase the endogenous level of H2S. Extensive research on the mechanisms of H2S and its role in modulation of cellular signaling will provide new insights into the physiological functions of H2S.
Acknowledgments
This work was supported by National Institutes of Health grants: HL-107640-NT and AR-067667-NT.
Abbreviations
- CNS
central nervous system
- ER
endoplasmic reticulum
- Hcy
homocysteine
- tHcy
total fasting homocysteine
- HHcy
hyperhomocysteinemia
- CBS
cystathionine β-synthase
- BHMT
betaine methyltransferase
- UPR
unfolded protein response
- MTHFR
methylene tetrahydrofolate reductase
- PERK
pancreatic endoplasmic reticulum kinase
- HD
Huntington’s disease
- ALS
amyotrophic lateral sclerosis
- IRE1
inositol-requiring enzyme 1
- ATF6
activating transcription factor 6
- GPR 78
G-protein coupled receptor 78
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- VAD
vascular dementia
- PD
Parkinson’s disease
- DNMTs
DNA methyltransferases
- MetS
methionine synthase
- CSE
cystathionine-γ-lyase
- PERK
pancreatic endoplasmic reticulum kinase
- IRE1
inositol-requiring enzyme
- EIF2
eukaryotic initiation factor 2
- ROS
reactive oxygen species
- NADP
nicotinamide adenine dinucleotide phosphate
- NMDA
N-methyl-D-aspartate receptor
- LTP
long-term potentiation
- GSH
glutathione
- H2S
hydrogen sulfide
- AD
Alzheimer’s disease
References
- Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signaling: dynamics, homeostasis and remodeling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Elston M, Nicholson CK, Gundewar S, Jha S, Elrod JW, Ramachandran A, Lefer DJ. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation. 2010;122(1):11–19. doi: 10.1161/CIRCULATIONAHA.109.920991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Ji. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int. 2012;2012:216450. doi: 10.1155/2012/216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Chen Q, Cheng YF, Jin HH, Kong DS, Zhang F, Wu L, Shao JJ, Zheng SZ. Diallyl trisulfide attenuates ethanol-induced hepatic steatosis by inhibiting oxidative stress and apoptosis. Biomed Pharmacother. 2016;79:35–43. doi: 10.1016/j.biopha.2016.01.009. [DOI] [PubMed] [Google Scholar]
- Comporti M, Signorini C, Leoncini S, Gardi C, Ciccoli L, Giardini A, Vecchio D, Arezzini B. Alcohol-induced oxidative stress: basic knowledge. Genes Nutr. 2010;5(2):101–109. doi: 10.1007/s12263-009-0159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal S, Arning E, Bottiglieri T, Boger RH, Sigmund CD, Faraci FM, Lentz SR. Cerebral vascular dysfunction mediated by superoxide in hyperhomocysteinemic mice. Stroke. 2004;35:1957–1962. doi: 10.1161/01.STR.0000131749.81508.18. [DOI] [PubMed] [Google Scholar]
- Finkelstein JD, Martin JJ. Methionine metabolism in mammals: adaptation to methionine excess. J Biol Chem. 1986;261:1582–1587. [PubMed] [Google Scholar]
- Gibson A, Woodside JV, Young IS, Sharpe PC, Mercer C, Patterson CC, Mckinley MC, Kluijtmans LAJ, Whitehead AS, Evans A. Alcohol increases homocysteine and reduces B vitamin concentration in healthy male volunteers—a randomized, crossover intervention study. QJM. 2008;101(11):881–887. doi: 10.1093/qjmed/hcn112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunzerath L, Hewitt BG, Li TK, Warren KR. Alcohol research: past, present and future. Ann N Y Acad Sci. 2011;1216(6648):1–23. doi: 10.1111/j.1749-6632.2010.05832.x. 32. [DOI] [PubMed] [Google Scholar]
- Halsted CH, Villanueva JA, Devlin AM. Folate deficiency, methionine metabolism and alcoholic liverdisease. Alcohol. 2002;27:169–172. doi: 10.1016/s0741-8329(02)00225-2. [DOI] [PubMed] [Google Scholar]
- Herrmann W, Quast S, Ullrich M, Schultze H, Bodis M, Geisel J. Hyperhomocysteinemia in high-aged subjects: relation of B-vitamins, folic acid, renal function and the methylenetetrahydrofolate reductase mutation. Atherosclerosis. 1999;144:91–101. doi: 10.1016/s0021-9150(99)00036-2. [DOI] [PubMed] [Google Scholar]
- Ho PI, Collins SC, Dhitavat S, Ortiz D, Ashline D, Rogers E, Shea TB. Homocysteine potentiates beta-amyloid neurotoxicity: role of oxidative stress. J Neurochem. 2001;78:249–253. doi: 10.1046/j.1471-4159.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- Ho PI, Ashline D, Dhitavat S, Ortiz D, Collins SC, Shea TB, Rogers E. Folate deprivation induces neurodegeneration: roles of oxidative stress and increased homocysteine. Neurobiol Dis. 2003;14:32–42. doi: 10.1016/s0969-9961(03)00070-6. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- Jara-Prado A, Ortega-Vazquez A, Martinez-Ruano L, Rios C, Santamaria A. Homocysteine-induced brain lipid peroxidation: effects of NMDA blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox Res. 2003;5:237–243. doi: 10.1007/BF03033381. [DOI] [PubMed] [Google Scholar]
- Kamat PK, Kalani A, Givvimani S, Sathnur PB, Tyagi SC, Tyagi N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience. 2013;252:302–319. doi: 10.1016/j.neuroscience.2013.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Dargusch R, Schubert D, Kimura H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxid Redox Signal J. 2006;8:661–670. doi: 10.1089/ars.2006.8.661. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal J. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- Kohn C, Dubrovska G, Huang Y, Gollasch M. Hydrogen sulfide: potent regulator of vascular tone and stimulator of angiogenesis. Int J Biomed Sci. 2012;8:81–86. [PMC free article] [PubMed] [Google Scholar]
- Koike S, Ogasawara Y. Sulfur atom in its bound state is a unique element involved in physiological functions in mammals. Molecules. 2016;21(12) doi: 10.3390/molecules21121753. http://dx.doi.org/10.3390/molecules21121753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1 B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4(203):ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Mouton PR, Emokpae R, Jr, Cutler RG, Mattson MP. Folate deficiency inhibits proliferation of adult hippocampal progenitors. Neuroreport. 2005;16:1055–1059. doi: 10.1097/00001756-200507130-00005. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal J. 2007;9:2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- Meigs JB, Jacques PF, Selhub J, Singer DE, Nathan DM, Rifai N, D’Agostino RB, Sr, Wilson PWF. Fasting plasma homocysteine levels in the insulin resistance syndrome - the Framingham offspring study. Diabetes Care. 2001;24:1403–1410. doi: 10.2337/diacare.24.8.1403. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Omura S, Takahashi Y. Generation of superoxide anions by a glycation reaction in conventional laboratory media. J Biosci Bioeng. 2012;114:275–280. doi: 10.1016/j.jbiosc.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Nurk E, Refsum H, Tell GS, Engedal K, Vollset SE, Ueland PM, Nygaard HA, Smith AD. Plasma total homocysteine and memory in the elderly: the Hordaland homocysteine study. Ann Neurol. 2005;58:847–857. doi: 10.1002/ana.20645. [DOI] [PubMed] [Google Scholar]
- Placido AI, Pereira CM, Duarte AI, Candeias E, Correia SC, Carvalho C, Cardoso S, Oliveira CR, Moreira PI. Modulation of endoplasmic reticulum stress: an opportunity to prevent neurodegeneration? CNS Neurol Disord Drug Targets. 2015;14:518–533. doi: 10.2174/1871527314666150429112353. [DOI] [PubMed] [Google Scholar]
- Qureshi M, Khsandwala H, Haq IU, Prasad K. Elevated levels of plasma homocysteine in hypertensive patients with diabetes mellitus. J Cardiovasc Pharmacol Ther. 2003;8:261–266. doi: 10.1177/107424840300800403. [DOI] [PubMed] [Google Scholar]
- Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223–2233. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- Rosselli M, Keller PJ, Dubey RK. Role of nitric oxide in the biology, physiology and pathophysiology of reproduction. Hum Reprod Update. 1998;4(1):3–24. doi: 10.1093/humupd/4.1.3. [DOI] [PubMed] [Google Scholar]
- Sachdev PS. Homocysteine and brain atrophy. Prog Neuro-Psychopharmacol Biol Psychiatry. 2005;29:1152–1161. doi: 10.1016/j.pnpbp.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Santos CX, Tanaka LY, Wosniak J, Laurindo FR. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal J. 2009;11:2409–2427. doi: 10.1089/ars.2009.2625. [DOI] [PubMed] [Google Scholar]
- Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N, Tyagi SC. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am J Physiol Cell Physiol. 2012;303:C41–C51. doi: 10.1152/ajpcell.00398.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N. Functional and molecular insights of hydrogen sulfide signaling and protein sulfhydration. J Mol Biol. 2016 doi: 10.1016/j.jmb.2016.12.015. http://dx.doi.org/10.1016/j.jmb.2016.12.015. [DOI] [PMC free article] [PubMed]
- Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. New Engl J Med. 2002;346(7):476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- Tchantchou F, Graves M, Ortiz D, Rogers E, Shea TB. Dietary supplementation with 3-deaza adenosine, N-acetyl cysteine, and S-adenosyl methionine provide neuroprotection against multiple consequences of vitamin deficiency and oxidative challenge: relevance to age-related neurodegeneration. Neuromol Med. 2004;6:93–103. doi: 10.1385/NMM:6:2-3:093. [DOI] [PubMed] [Google Scholar]
- Tyagi N, Sedoris KC, Steed M, Ovechkin AV, Moshal KS, Tyagi SC. Mechanisms of homocysteine-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2005;289:2649–2656. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7(1):65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197(7):857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Wright CB, Paik MC, Brown TR, Stabler SP, Allen RH, Sacco RL, DeCarli C. Total homocysteine is associated with white matter hyperintensity volume: the Northern Manhattan Study. Stroke. 2005;36:1207–1211. doi: 10.1161/01.STR.0000165923.02318.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57(4):585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- Xie L, et al. SIRT3 mediates the antioxidant effect of hydrogen sulfide in endothelial cell. Antioxid Redox Signal J. 2016;24(6):329–343. doi: 10.1089/ars.2015.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Luo J. Endoplasmic reticulum stress and ethanol neurotoxicity. Biomolecules. 2015;14:2538–2553. doi: 10.3390/biom5042538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Yang W, Wu L, Wang R. H2S, endoplasmic reticulum stress and apoptosis of insulin-secreting beta cells. J Biol Chem. 2007;282(22):16567–16576. doi: 10.1074/jbc.M700605200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Xu C, Yang G, Wu L, Wang R. Interaction of H2S with calcium permeable channels and transporters. Oxid Med Cell Longev. 2015 doi: 10.1155/2015/323269. http://dx.doi.org/10.1155/2015/323269. [DOI] [PMC free article] [PubMed]
- Zhao H, Chen MH, Shen ZM, Kahn PC, Lipke PN. Environmentally induced reversible conformational switching in the yeast cell adhesion protein alpha-agglutinin A Publication of the Protein Society. Protein Sci. 2001;10:1113–1123. doi: 10.1110/ps.41701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou CG, Banerjee R. Homocysteine and redox signaling. Antioxid Redox Signal J. 2005;7:547–559. doi: 10.1089/ars.2005.7.547. [DOI] [PubMed] [Google Scholar]
- den Heijer T, Vermeer SE, Clarke R, Oudkerk M, Koudstaal PJ, Hofman A, Breteler MM. Homocysteine and brain atrophy on MRI of non-demented elderly. Brain. 2003;126:170–175. doi: 10.1093/brain/awg006. [DOI] [PubMed] [Google Scholar]