

Abstract
Purpose
Azacitidine is standard, first-line therapy in higher-risk myelodysplastic syndromes (MDS). Whether azacitidine-based combinations with lenalidomide or vorinostat produce superior overall response rates (ORRs) to azacitidine is not known.
Patients and Methods
North American Intergroup Study S1117 is a phase II/III trial that randomly assigned patients with higher-risk MDS and chronic myelomonocytic leukemia (CMML) 1:1:1 to azacitidine (75 mg/m2/day on days 1 to 7 of a 28-day cycle); azacitidine plus lenalidomide (10 mg/day on days 1 to 21); or azacitidine plus vorinostat (300 mg twice daily on days 3 to 9). The primary phase II end point was improved ORR.
Results
Of 277 patients from 90 centers, 92 received azacitidine, 93 received azacitidine plus lenalidomide, and 92 received azacitidine plus vorinostat. Median age was 70 years (range, 28 to 93 years), 85 patients (31%) were female, and 53 patients (19%) had CMML. Serious adverse events were similar across arms, although combination-arm patients were more likely to undergo nonprotocol-defined dose modifications (P < .001).With a median follow-up of 23 months (range, 1 to 43 months), the ORR was 38% for patients receiving azacitidine, 49% for azacitidine plus lenalidomide (P = .14 v azacitidine), and 27% for azacitidine plus vorinostat (P = .16 v azacitidine). For patients with CMML, ORR was higher for azacitidine plus lenalidomide versus azacitidine (68% v 28%, P = .02) but similar for all arms across cytogenetic subgroups, as was remission duration and overall survival. ORR was higher with mutations in DNMT3A and lower for SRSF2, whereas ORR duration improved with fewer mutations. Lenalidomide dose reduction was associated with worse overall survival (hazard ratio, 1.30; P = .05).
Conclusion
Patients with higher-risk MDS treated with azacitidine-based combinations had similar ORR to azacitidine monotherapy, although patients with CMML benefitted from azacitidine plus lenalidomide. The efficacy of combination regimens may have been affected by dose modifications.
INTRODUCTION
The myelodysplastic syndromes (MDS) and chronic myelomonocytic leukemias (CMML; an MDS/myeloproliferative neoplasm [MPN] overlap) comprise a spectrum of distinct bone marrow disorders associated with cytopenias, a consequent increased risk of bleeding and infection, and, in higher-risk subtypes, a high likelihood of transformation to acute myeloid leukemia.1-3 They are the most common myeloid malignancies, with approximately 15,000 to 20,000 new diagnoses in the United States yearly, 25% to 30% of which constitute higher-risk disease.4
Three drugs, azacitidine, decitabine, and lenalidomide, were approved by the US Food and Drug Administration for the treatment of MDS or one of its subtypes.5-7 Lenalidomide purportedly works through inhibition of phosphatase activity in the common deleted region, which plays a key role in cell cycle regulation; through a defect in ribosomal protein function via ubiquitination and degradation of CK1 alpha in patients with the deletion 5q cytogenetic abnormality; and through bone marrow microenvironment effects in patients without the deletion 5q cytogenetic abnormality.8-10 Lenalidomide has demonstrated transfusion independence response rates of 67% in patients with lower-risk MDS with the deletion 5q abnormality and of 26% in lower-risk patients without the abnormality.6,11 It has also shown activity in patients with MPN.12,13 Azacitidine and decitabine exert their effects via DNA methyltransferase inhibition and direct cytotoxicity.14 Each drug can affect peripheral cytopenias, transfusion needs, and quality of life.5,7,15 In addition, azacitidine improves overall survival (OS) in patients with higher-risk MDS or CMML.15
Histone acetylation facilitates active gene transcription and is highly regulated by histone deacetylases (HDACs).16 HDAC inhibition can restore normal acetylation of histone proteins and transcription factors. Vorinostat, a small molecule inhibitor of class I and II HDAC enzymes, has been combined with azacitidine in phase I and II studies in higher-risk MDS,17 with an overall response rate (ORR) among 33 patients of 70%.18 Lenalidomide has also been combined with azacitidine in the phase I and II setting,19 with an ORR among 36 patients of 72%.20
We conducted a multicenter, randomized, three-arm, phase II/III study of azacitidine combined with lenalidomide or with vorinostat versus azacitidine monotherapy in patients with higher-risk MDS and CMML. The phase II analysis was to determine whether combination therapy could improve the response rate compared with azacitidine to justify phase III evaluation for OS. Secondary objectives included assessing outcomes in predefined subgroups, such as CMML and common MDS cytogenetic and molecular abnormalities.
PATIENTS AND METHODS
Study Design and Treatment
In the North American Intergroup Study S1117 (ClinicalTrials.gov identifier NCT01522976), patients were randomly assigned 1:1:1 using a dynamic allocation scheme stratified on MDS versus CMML to one of three study arms: azacitidine monotherapy (75 mg/m2/day intravenously or subcutaneously on days 1 to 7 of a 28-day cycle for both monotherapy and combination arms), with allowance for azacitidine to be administered on either a 7-day continuous or a 7-day interrupted schedule (eg, 5-2-2: azacitidine administered on days 1 to 5, followed by 2 days of no treatment, followed by 2 days of azacitidine)21,22; azacitidine plus lenalidomide (10 mg/day orally on days 1 to 21); or azacitidine plus vorinostat (300 mg twice daily orally on days 3 to 9; Fig 1). Dose modification or interruption guidelines are listed in Appendix Table A1 (online only). Antibiotic prophylaxis could be used per individual institutional practices.
Fig 1.

CONSORT diagram. ALL, acute lymphatic leukemia; AML, acute myelogenous leukemia; CAD, coronary artery disease; DVT, deep vein thrombosis.
Patients
Eligibility criteria included higher-risk MDS (International Prognostic Scoring System [IPSS] Intermediate-2 or High and/or bone marrow blasts ≥ 5%) or CMML with < 20% blasts, reviewed centrally; and age ≥ 18 years (Appendix, online only).
Outcomes
The primary phase II end point was ORR reviewed centrally on a patient-by-patient basis per 2006 International Working Group MDS response criteria (complete response [CR] plus partial response [PR] plus hematologic improvement [HI]).23 OS was measured from study entry, response duration from the time response was first documented to loss of response, progression, or death. MDS Centers of Excellence were defined per the MDS Foundation24; center volume was defined as low (one to four patients enrolled) or high (five to 17 patients enrolled) on the basis of the median volume in this study (n = 4; Appendix).
Mutation Analysis
Genomic DNA was isolated from cryopreserved peripheral blood or bone marrow mononuclear cells and subjected to targeted, amplicon-based, next-generation sequencing of coding regions of MDS-associated genes, using Ion Torrent (National Cancer Institute of Canada/Canadian Cancer Trials Group Alliance and Nationwide samples; n = 97) or Illumina (San Diego, CA) platforms (Cleveland Clinic and Albert Einstein University; n = 16). Somatic mutation calls were made (present or absent) with a minimum average depth of coverage of 500× magnification. The lower limit of detection was estimated between variant allele fractions of 0.02 to 0.05 (Supplemental Methods in the Appendix).
Statistics
The phase II objective of this phase II/III study was to select, on the basis of ORR, one combination arm for phase III evaluation of OS compared with azacitidine. A total of 240 eligible patients (80 per arm) were required for the phase II analysis. If the true ORR of azacitidine was 35% (null hypothesis) and the true ORR of a combination arm was 55% (alternative hypothesis), each comparison of a combination arm versus azacitidine had a power of 81% with a one-sided alpha of 5%. Additional statistics are described in the Supplemental Methods section in the Appendix.
RESULTS
Baseline Characteristics
From June 2012 through June 2014, 282 patients were enrolled from 90 centers in the United States and Canada. Five patients were ineligible because of a diagnosis other than MDS or CMML or pre-existing toxicity, leaving 277 eligible patients: 92 received azacitidine, 93 received azacitidine plus lenalidomide, and 92 received azacitidine plus vorinostat. Baseline characteristics were similar across arms (Table 1). The median age was 70 years (range, 28 to 93 years), 85 patients (31%) were female, 53 patients (19%) had CMML, and 18 patients (6%) had treatment-related MDS. IPSS risk group distribution was similar across arms, with all patients with Low/Intermediate-1 IPSS classifications having excess blasts or nonproliferative CMML-1. The revised IPSS-defined cytogenetic risk group distribution and distinct cytogenetic abnormalities of interest occurred at similar rates across treatment arms, and baseline characteristics of patients were similar whether they were treated at an MDS Center of Excellence or a high volume center (Appendix Table A2). A majority of patients (58%) were dependent on packed RBC transfusions at baseline.
Table 1.
Patient Characteristics

Adverse Events
Serious adverse events attributed to therapy were similar across arms (Table 2), with two exceptions: patients in the azacitidine plus vorinostat arm had more grade 3 or higher gastrointestinal toxicities (14 patients [15%] v four patients [4%] in the azacitidine arm; P = .02), whereas patients in the azacitidine plus lenalidomide arm had more grade 3 or higher rash (14 patients [16%] v three patients [3%] in the azacitidine arm; P = .005). Rates of grade 3 or higher febrile neutropenia were similar across the three arms, as were rates of infection and infestations for all three cohorts, across grades: 89% for azacitidine monotherapy, 91% for azacitidine plus lenalidomide, and 91% for azacitidine plus vorinostat. Patients in combination arms were significantly more likely to have therapy stopped because of toxicities or complications than were patients receiving azacitidine monotherapy (8% for azacitidine, 20% for azacitidine plus lenalidomide [P = .05 v azacitidine], and 21% for azacitidine plus vorinostat [P = .03 v azacitidine, with P = .02 for both combination arms v azacitidine]) and to undergo nonprotocol-defined dose modifications (24% for azacitidine, 43% for azacitidine plus lenalidomide [P = .002], and 42% for azacitidine plus vorinostat [P = .01, with P < .001 for combinations v azacitidine]).
Table 2.
Toxicity

ORR and Duration
Patients received a median of 22 weeks of therapy: 23 weeks for patients receiving azacitidine, 25 weeks for those receiving azacitidine plus lenalidomide (P = .61 v azacitidine), and 20 weeks for azacitidine plus vorinostat (P = .33 v azacitidine, P = .9 for combinations v azacitidine). With a median follow-up among patients still alive of 23 months (range, 1 to 43 months), the ORR for the entire cohort was 38%: 38% for patients receiving azacitidine; 49% for patients receiving azacitidine plus lenalidomide (P = .14 v azacitidine); and 27% for patients receiving azacitidine plus vorinostat (P = .16 v azacitidine; Table 3). Rates of CR/PR/HI and marrow CR were also not significantly different across groups, although within HI, patients receiving azacitidine plus lenalidomide had higher rates of HI-neutrophil than did azacitidine monotherapy patients (19% v 5%; P = .007). Among previously untreated patients, there was a trend toward improved ORR for those treated with azacitidine plus lenalidomide (n = 81) versus azacitidine (n = 79; 49% v 35%; P = .08). Time to best response did not differ across treatment arms. On the basis of the phase II analysis, neither combination arm was selected for phase III testing of OS. The median response duration for the cohort was 15 months: 10 months for azacitidine, 14 months for azacitidine plus lenalidomide (P = .85 v azacitidine), and 18 months for azacitidine plus vorinostat (P = .37). For patients remaining on therapy for ≥ 6 months (n = 119), patients receiving azacitidine plus lenalidomide had a higher ORR (87%) versus patients receiving azacitidine (62%; P = .01), although no difference in response duration (P = .98).
Table 3.
Responses

Response Rates and Duration Within Predefined Subgroups
For patients with CMML, the ORR (38% for the entire cohort) was significantly higher for those receiving azacitidine plus lenalidomide than for those receiving azacitidine monotherapy (68% v 28%; P = .02). Median response duration for patients with CMML was 19 months and similar across arms. No differences in ORR were seen for therapy-related MDS, IPSS subgroups, transfusion-dependent patients, or allogeneic transplantation rates.
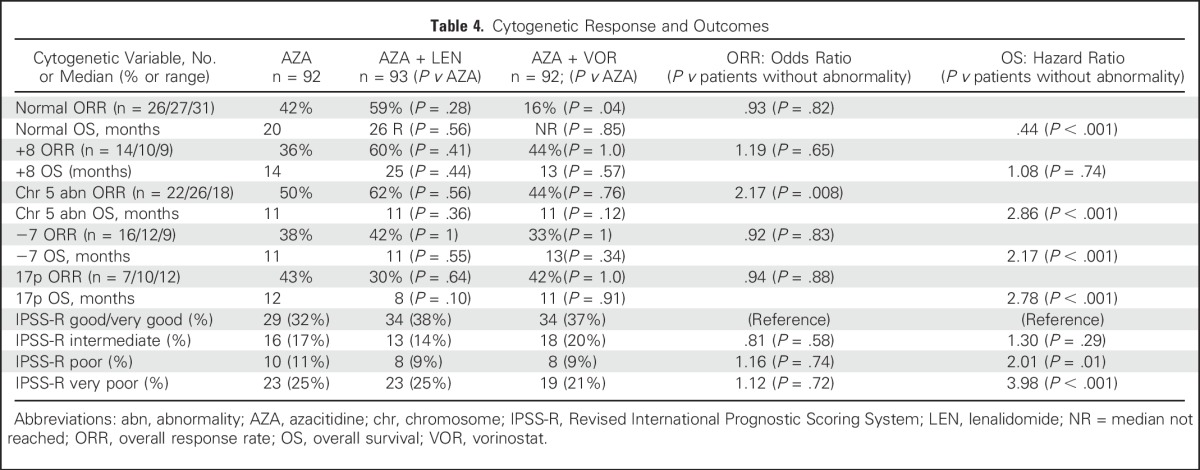
Within the Revised IPSS–defined cytogenetic risk groups and for distinct cytogenetic abnormalities of interest, ORR across treatment arms were, in general, similar (Table 4). ORR across arms was better for patients with chromosome 5 abnormality versus those without (odds ratio, 2.17; P = .008). Patients with chromosome 5 abnormalities and +8 receiving azacitidine plus lenalidomide had a nonsignificant higher ORR than did patients receiving azacitidine (62% v 50%, P = .56, and 60% v 36%, P = .41, respectively), with no significant difference in response duration.
Table 4.
Cytogenetic Response and Outcomes
Early predictors of ORR, which did not differ across arms, included doubling of platelet count and increase in platelet count > 30,000 after the first cycle (n = 45 and n = 74, P < .001 and P < .001, respectively). Early increase in neutrophil count or hemoglobin were not associated with ORR.
Of 113 patients with available mutational data, 103 (91%) had at least one mutation, the most common being ASXL1 (n = 31), TET2 (n = 26), SRSF2 (n = 23), TP53 (n = 22), RUNX1 (n = 21), and U2AF1 (n = 19; Table 5). The median number of mutations was two (range, 0 to 7). This mutation profile was consistent with other studies enriched for higher-risk MDS and CMML.25,26
Table 5.
Mutation Incidence and Response Among 113 Tested Patients

Compared with patients without mutations, ORR was significantly higher for those with mutations in DNMT3A (67% v 34%; P = .025) and numerically higher for those with BCOR (57% v 34%; P = .23) and NRAS (60% v 36%; P = .28), but lower for SRSF2 (17% v 41%; P = .037) and ASXL1 (23% v 43%; P = .049; Appendix Table A3). Response duration was worse for those with mutations in TET2 (P = .046) and TP53 (P = .003), with a trend for ASXL1 (P = .069). Response duration improved significantly with fewer mutations (hazard ratio [HR], 6.86 for two or more mutations v 0; P = .01; Appendix Fig A1, online only).
Overall Survival
The median OS for the entire cohort was 17 months: 15 months for azacitidine patients; 19 months for azacitidine plus lenalidomide patients (P = .68 v azacitidine); and 17 months for those receiving azacitidine plus vorinostat (P = .22 v azacitidine; Fig 2).The median OS after treatment failure for the entire cohort was 9 months: 7 months for azacitidine patients; 9 months for azacitidine plus lenalidomide patients (P = .74 v azacitidine); and 10 months for azacitidine plus vorinostat patients (P = .07 v azacitidine; P = .21 for combination arms after failure v azacitidine; Fig 3). For patients receiving therapy for > 6 months, the median OS for the cohort was 25 months: 20 months for those receiving azacitidine; 26 months for those receiving azacitidine plus lenalidomide (P = .74 v azacitidine); and 27 months for those receiving azacitidine plus vorinostat (P = .40 v azacitidine). There was a significant association between mean dose reduction of lenalidomide during the first four cycles of therapy and worse OS in multivariable analyses adjusting for IPSS and age (HR, 1.30; P = .05), but not for vorinostat (HR, 1.21; P = .13).
Fig 2.

Overall survival. Comparisons are between combination arms and azacitidine (AZA) monotherapy. LEN, lenalidomide; VOR, vorinostat.
Fig 3.

Overall survival after failure. AZA, azacitidine; LEN, lenalidomide; VOR, vorinostat.
The OS for patients with CMML was similar across treatment arms: median not reached for those receiving azacitidine, those receiving azacitidine plus lenalidomide (P = .87 v azacitidine), and those receiving azacitidine plus vorinostat (P = .78 v azacitidine). Within cytogenetic risk categories, the OS (compared with Very Good/Good) was worse for Poor (HR, 2.01; P = .01) and Very Poor (HR, 3.98; P < .001), without significant modification by treatment arm (Table 4). Compared with patients without a given cytogenetic abnormality, OS was better for normal (HR, 0.45; P < .001) and worse for chromosome 5 abnormalities (HR, 2.86; P < .001), −7 (HR, 2.18; P < .001), and 17p (HR, 2.81; P < .001). Although small numbers prevented definitive conclusions, combinations hinted at better OS in patients with chromosome 5 (P = .15) and for patients without 17p (P = .22) abnormalities. Patients with fewer mutations had better OS (HR, 4.55; P = .04); those with SETBP1 (P = .03) and TP53 (P < .001) had worse OS, with trends for worse OS in those with mutations in CUX1 (log-rank P = .08), and TET2 (P = .07).
Treatment Center Effect
Controlling for treatment arm, the baseline characteristics and outcome of all patients and patients on discrete study arms treated at MDS Centers of Excellence (n = 75) or high-volume (n = 137) sites were similar to other centers (Appendix Table A2).
DISCUSSION
Patients with higher-risk MDS and CMML have limited treatment options. Although hematopoietic cell transplantation is potentially curative, it is implemented in < 5% of patients.27 Because these diseases are biologically and prognostically similar to acute myeloid leukemia in older adults, it is appealing to consider more aggressive therapeutic approaches, such as combinations of drugs that work in theoretically complementary or synergistic ways.
We randomly assigned patients with higher-risk MDS or CMML to receive azacitidine, azacitidine plus lenalidomide, or azacitidine plus vorinostat on the basis of single-arm phase II trials in which the ORR for each of the combinations was approximately double what had been seen previously for azacitidine monotherapy.15,18,20 Unfortunately, those outcomes were not realized in the current study, with one exception: patients with CMML treated with azacitidine plus lenalidomide had twice the ORR as with azacitidine. This makes some sense in the context of previous trials, which have demonstrated activity of azacitidine in MDS and of lenalidomide in MPNs for this overlap disorder.
The single-arm studies on which this trial was based were small phase II trials with larger variances, which may explain higher ORR. It is also possible that results were affected by variations in patient selection and treatment practices. Patients on combination arms may have been undertreated. They were significantly more likely to undergo nonprotocol-defined treatment modifications and to be withdrawn from therapy because of toxicities, despite the overall similarity in adverse events across arms. This is not entirely surprising, because one study found that the physicians were twice as likely as patients to attribute treatment toxicities to poor drug tolerability, leading to treatment discontinuation.28 There was a significant association between lenalidomide dose reductions and worse OS similar to what was seen in patients with deletion 5q lower-risk MDS treated with lenalidomide.29 It is thus unresolved whether combination therapies can be realistically implemented on a broad scale. Finally, the response criteria have limitations in higher-risk patients treated with a hypomethylating agent, including the response assessment being confounded by temporary treatment-related cytopenias, and best response does not always reflect the most common response.
This study was not powered to assess OS differences among treatment groups, the ultimate measure of a clinically meaningful end point and one for which it is not entirely clear that ORR is an adequate interim marker. It is intriguing that median OS was 3 to 4 months longer for combination arms compared with azacitidine and that a trend for OS improvement emerged for patients treated with azacitidine plus vorinostat compared with azacitidine after treatment failure, possibly indicating a deeper response for combination therapies. Still, any effects on OS can only be validated in an adequately powered prospective study.
For cytogenetic subgroups, ORR was better for patients with chromosome 5 abnormalities compared with those without these abnormalities, but essentially did not differ across treatment arms. For the entire cohort, OS was worse for patients with abnormalities of chromosome 5, −7, and 17p and may be improved by combinations in patients with chromosome 5 or absence of 17p abnormalities. The distribution of molecular mutations was similar to previous reports, with DNMT3A lesions and fewer mutations associated with significantly higher ORR and TET2, and TP53 (P = .001) associated with compromised response durations.30-32 Patients with fewer mutations had better OS, whereas those with SETBP1 and TP53 had worse OS. Although a direct comparison with the recent study by Welch et al33 is challenging given different patient populations (22% MDS), treatment (decitabine for 10 days), and definitions of response, the ORR for patients with TP53 mutations in this study (45% v 35% for wild-type) seemed to be similar, as did the negative impact on OS.
In conclusion, patients with higher-risk MDS treated with azacitidine plus lenalidomide or azacitidine plus vorinostat had a similar ORR to patients treated with azacitidine monotherapy. Although specific patient subgroups, (CMML, normal cytogenetics, or chromosome 5 abnormalities), may derive benefit from azacitidine-based combinations, this should be confirmed in studies focused on these subgroups. Because underdosing may have been associated with compromised response and survival in combination arms, in most circumstances, patients with higher-risk MDS should be treated without dose adjustment for an induction phase of the first 4 months of therapy. Future studies in higher-risk MDS and CMML should be adequately powered to demonstrate an improvement in OS as a primary end point, or at minimum, response duration for molecular subtypes between study arms, in which innovative designs, such as Bayesian randomization, could optimize therapies for uncommon molecular lesions.
ACKNOWLEDGMENT
Sequencing was performed by Brooke Snetsinger, A.J.M. McNaughton, MD, and X. Liu, MD, at the Queen’s Genomics Laboratory at Ongwanada (Q-GLO).
Appendix
Supplemental Methods
Patients.
Eastern Cooperative Oncology Group performance status of 0 to 2; no previous treatment with study drugs or allogeneic transplantation, although prior autologous transplantation was allowed; no radiation therapy or chemotherapy within the previous 12 months, although therapy-related myelodysplastic syndrome (MDS) was allowed; and no specific level of organ dysfunction or abnormality of blood counts was excluded. Cytogenetic risk groups were defined per the Revised International Prognostic Scoring System and reviewed centrally.
Statistics.
Fisher’s exact tests and Wilcoxon rank-sum tests were used to compare categorical and quantitative factors between combination arms and azacitidine. Survival end points were estimated using the Kaplan-Meier method and analyzed with the log-rank test and Cox proportional hazard regression models. A two-sided alpha value of .05 denoted significance, P values were not adjusted for multiple comparisons. The following results were based on data available as of February 12, 2016, with survival updated as of April 13, 2015.
Outcomes.
Baseline transfusion requirements were recorded during the 8 weeks preceding study registration to determine whether transfusion reduction or independence occurred. In instances in which therapy-related cytopenias were suspected, peripheral blood counts before and after bone marrow assessments or 8-week duration time points were also reviewed. Blood counts were assessed at least every 4 weeks, with bone marrow biopsies after cycles 4 and 7 and at suspicion of progression. Among 66 patients without a bone marrow biopsy or with an inadequate biopsy after cycle 4, there were no significant differences across study arms. Disease progression was defined as > 50% increase in myeloblasts from baseline; non–treatment-related ≥ 50% decrement in neutrophil or platelet count; reduction of hemoglobin of ≥ 2g/dL from baseline; or becoming transfusion dependent. Treatment failure with azacitidine failure was defined as a lack or loss of response or disease progression.
Response Definitions
-
Accurate counts of RBC units and platelet transfusions received before and after starting protocol treatment are necessary to determine patients’ responses to therapy. The numbers of RBC units and platelet transfusions during the 8 weeks before registration in the study will be recorded for use as baselines. Only RBC transfusions given for hemoglobin < 9 g/dL or platelet transfusions for platelets < 50,000/mm3 before registration will be considered in the RBC transfusion response evaluation.
Transfusion dependence: Patients who receive one or more RBC or platelet transfusions will be considered RBC or platelet transfusion dependent, respectively, in the absence of another explanation, such as gastrointestinal bleeding, hemolysis, etc.
Relevant reduction in RBC transfusion requirement: This is defined for patients who were RBC transfusion dependent and received ≥ 4 units during the 8 weeks before registration on study. If, during an 8-week period after entering the study, the total number of RBC units transfused has decreased by at least 4 units compared with the number transfused during the 8 weeks before registration, then the patient will have a relevant reduction in RBC transfusion requirement.
Transfusion independence: For assessment of response, RBC or platelet transfusion independence requires that the patient receive no RBC or platelet transfusions, respectively, for a period of at least 8 weeks.
Accurate measurements of hemoglobin and platelet counts before and after starting protocol treatment are also necessary to determine patients’ responses to therapy. Whenever patients are transfusion dependent, hemoglobin and platelet counts should be measured immediately before transfusions to ensure that they reflect the patient’s true hematologic status.
Complete Remission.
The patient must satisfy all of the following bone marrow and peripheral blood criteria, and the patient must not receive RBC or platelet transfusions, erythropoietin, myeloid growth factor, or thrombopoietic agent within 28 days before this disease assessment:
Bone marrow evaluation must meet the following criteria:
Myeloblasts must be ≤ 50%. (NOTE: Persistence of dysplasia will be noted but does not preclude achievement of complete response [CR].)
b. Peripheral blood evaluation: The patient must satisfy all of the following for blood examinations performed during at least a 4-week period:
Hemoglobin ≥ 11.0 g/dl.
Neutrophils ≥ 1,000/mm3.
Platelets ≥ 100,000/mm3.
Blasts = 0%.
No evidence of dysplasia. (NOTE: The presence of mild megaloblastoid changes may be permitted if they are thought to be consistent with treatment effect. However, persistence of pretreatment abnormalities, for example, pseudo–Pelger-Hüet cells, ringed sideroblasts, or dysplastic megakaryocytes, is not consistent with CR.) Note: Transient cytopenias during repeated chemotherapy courses should not be considered as interrupting durability of response, as long as they recover to the improved counts of the previous course.
Marrow CR.
The patient must satisfy the definition of CR for the bone marrow examination, and the marrow myeloblasts must have decreased by > 50% from pretreatment. Marrow CR may be achieved with or without improved blood counts, and any hematologic improvement will be noted.
Partial Remission.
The patient must satisfy all of the following criteria, and the patient must not receive RBC or platelet transfusions, erythropoietin, myeloid growth factor, or thrombopoietic agent within 28 days before this disease assessment:
Bone marrow evaluation: blasts > 5% but decreased by > 50% from pretreatment or a WHO subtype of MDS that is less advanced than pretreatment; cellularity and morphology are not relevant.
Peripheral blood evaluation: all of the peripheral blood results required for CR during at least a 4-week period.
Stable Disease.
Failure to achieve at least a partial remission (PR), but with no evidence of progression for at least 8 weeks.
Failure.
Death during treatment or disease progression or progression to acute myeloid leukemia or a WHO subtype of MDS or chronic myelomonocytic leukemia that is more advanced than pretreatment.
Relapse (after a CR, marrow CR, or PR).
One or more of the following criteria (a. to e.) in the absence of another explanation, such as acute infection, gastrointestinal bleeding, hemolysis, etc. Note that the detection of circulating blasts is not by itself a sufficient criterion for relapse, but should trigger a bone marrow examination to determine whether relapse has occurred.
Return to pretreatment bone marrow blast percentage.
50% or greater decrement from the maximum absolute granulocyte count during CR or PR. Granulocyte counts during periods of active infection will not be considered in determining the maximum.
50% or greater decrement from the maximum platelet count during CR or PR.
A reduction in hemoglobin concentration by at least 1.5 g/dL from the maximum level during CR or PR.
Becoming transfusion dependent.
DNA Sequencing
DNA sequencing methods for each participating institution are summarized below.
Canadian Sites, Alliance, and Nationwide.
Genomic DNA (gDNA) was extracted after thawing viably frozen bone marrow mononuclear cells, using the DNeasy Blood and Tissue Kit (69504; Qiagen, Hilden, Germany) per the manufacturer’s protocol, but with the substitution of proteinase K from Sigma (P5568, Sigma-Aldrich, St. Louis, MO) and with the substitution of 50 μL low Tris-EDTA buffer (602-1297-01; Life Technologies, Carlsbad, CA) as the final suspension solution. gDNA was quantified using the TaqManRNase P qPCR Detection Kit (4316831; Life Technologies). Barcoded libraries were prepared from 15 ng of extracted gDNA, profiling 589 coding regions in 48 recurrently mutated genes, using a custom, pan-myeloid, two-tube, 1,552-amplicon, Ion Torrent AmpliSeq polymerase chain reaction (PCR) panel (ThermoFisher Scientific, Carlsbad, CA) and Ion AmpliSeq Library Kit 2.0 (4475435; ThermoFisher Scientific). Targets included all coding exons or hotspots for ASXL1, BCOR, BCORL1, BOD1L, BRAF, BRCC3, CALR, CBL, CEBPA, CSF3R, CUX1, DNMT3A, ETV6, EZH2, FLT3, GATA1, GATA2, GNAS, GNB1, IDH1, IDH2, JAK2, KDM6A, KIT, KRAS, MPL, NF1, NF-E2, NPM1, NRAS, PHF6, PTPN11, RAD21, RIT1, RUNX1, SETBP1, SF3B1, SH2B3, SMC1A, SMC3, SRSF2, STAG2, TET2, TLR2, TP53, U2AF1, WT1, and ZRSR2 (information provided on request). Libraries were then quantified using the Ion Library TaqMan Quantitation Kit (4468802; ThermoFisher Scientific), diluted to 100 pM, pooled and sent to the Queen’s Genomics Laboratory at Ongwanada in Kingston, Canada. Libraries were templated using the Ion OneTouch 2 system and Ion PI Template OT2 200 Kit v3 (4488318; Life Technologies), then sequenced using the Ion Proton System and Ion PI Sequencing 200 Kit v3 (4488315; Life Technologies). Barcoded libraries, in a batch of 12 to 30 libraries, were run together on a single Ion PI v3 chip (4488315; Life Technologies). Sequences were aligned to the human genome hg19, and variants were called in Ion Torrent Suite (Version 3.2.0). Files were uploaded into Ion Reporter (Version 5.2), and each sample was independently filtered through a workflow of optimized variant calling geared to our custom AmpliSeq panel. Variants were further filtered through the Strict filter to exclude UCSC Common SNPs (University of California, Santa Cruz, common single nucleotide polymorphisms) and nonexonic and synonymous variants, and to initially include only variants with an allele ratio of variant allele fractions 0.2 to 1.0 and a depth coverage of > 25. Next, we searched for lower-frequency variants (variant allele fractions 0.02 to 0.19) reported in the Catalogue of Somatic Mutations in Cancer (COSMIC; http://cancer.sanger.ac.uk/cosmic) database and/or with Ion Reporter P ≤ .001. Next, candidate variants were visually inspected using the Integrative Genomics Viewer (Broad Institute, Cambridge, MA). Variants were excluded if they appeared only in the ends of short sequence reads or consistently exhibited forward or reverse strand bias. Finally, all known false discoveries (as confirmed through independent PCR and Sanger sequencing, not shown), and suspected mis-priming events were removed from the final variant calls.
Cleveland Clinic.
Genomic DNA was extracted from peripheral blood or bone marrow mononuclear cells in blood samples that were stored at the Stem Cell Tissue Bank at the Cleveland Clinic. Direct sequencing was performed on coding exons of 62 genes: APC, ASXL1, BCOR, BCORL1, BTRC, C7orf55, LUC7L2, CBL, CCDC42B, CDH23, CEBPA, CFTR, CSF1R, CUX1, DDX41, DDX54, DHX29, DNMT3A, EED, ERBB4, ETV6, EZH2, FLT3, GATA2, GLI1, GLI2, GNB1, GPR98, IDH1, IDH2, IRF4, JAK2, JAK3, KDM6A, KIT, KRAS, MECOM, MED12, MLL, NF1, NPM1, NRAS, OGT, PHF6, PRPF8, PTCH1, PTPN11, RAD21, RNF25, RUNX1, SETBP1, SF3B1, SMC3, SRSF2, STAG2, STAT3, SUZ12, TET2, TP53, U2AF1, WT1, ZRSR2, and SRSF2 using IlluminaTrueSeq Custom Amplicon kit per manufacturer protocol. For germline confirmation, mutations were analyzed in nonclonal CD3þ cells whenever DNA was available. Bidirectional sequencing was performed by standard techniques using an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA). All mutations were scored as pathogenic on the basis of the observation that they were not detected in normal samples, in germline source, or in published SNP databases (dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP), and/or they were not reported as SNPs in previous publications.
Albert Einstein University.
Genomic DNA was isolated from bone marrow aspiration or peripheral blood and then coding regions of 21 genes were assessed as follows: ASXL1, EZH2, ETV6, RUNX1, TP53, CBL, DNMT3A, IDH1, IDH2, JAK2, KIT, MPL, NPM1, NRAS, PHF6, SETBP1, SF3B1, SRSF2, TET2, U2AF1, and ZRSR2. These 21 genes amplified by PCR. The DNA sequences of these regions were then determined using next-generation sequencing technology. Somatic mutations consistent with MDS or other myeloid neoplasms within these samples were identified after cross-referencing all identified sequence variants with selected databases, including but not limited to COSMIC, dbSNP, and the listing of MDS mutations as identified in the numerous publications. The limit of detection of this assay is 5% (ie, if 5% of the gene copies in a specimen contain the mutant allele, then the mutated base should be consistently detected). Other assay limitations may include potential mis-calls due to limited depth of coverage (target of 500× minimum for this assay), amplicon design limitations (read quality can decrease toward the middle of amplicons), and platform basis. Poor-quality DNA resulting from poor specimen quality may cause assay failures.
Fig A1.

Response duration by number of mutations.
Table A1.
Dose Modifications for Adverse Events

Table A2.
Patient Characteristics for Those Treated at MDS CEs or Not and Treated and at Higher and Lower Volume Centers

Table A3.
Association Between Mutation and Outcomes

Footnotes
Supported in part by the following Public Health Service/US Department of Health and Human Services grants awarded by the National Cancer Institute (NCI), National Clinical Trials Network Grants No. CA180888, CA180861, CA180819,CA180834, CA21115, CA180820, CA180821, CA180863, CA180834,CA180818, CA180855, CA180830, CA180798, CA140158; by the NCI Community Oncology Research Program Grants No. CA189830, CA189853,CA189971, CA189972, CA189860,CA189821, CA189804, CA189856, CA189954, CA189872, CA189858; by the Coleman Leukemia Research Foundation; and in part by the Edward P. Evans Foundation and the Canadian Cancer Society Grant No. 021039. Funding was provided by the Southeastern Ontario Academic Medical Organization Innovation Fund, the University Hospitals Kingston Fund/Women’s Giving Circle, and the Ontario Institute for Cancer Research.
Presented in part as a late-breaking abstract and an oral presentation at the 56th and 57th annual meetings of the American Society of Hematology, San Francisco and Orlando, December 9, 2014, and December 7, 2015, respectively.
Clinical trial information: NCT01522976.
See accompanying Editorial on page 2729
AUTHOR CONTRIBUTIONS
Conception and design: Mikkael A. Sekeres, Megan Othus, Alan F. List, Olatoyosi Odenike, Richard M. Stone, Steven D. Gore, Mark R. Litzow, Rena Buckstein, Min Fang, Clara D. Bloomfield, Mario R. Velasco, Frederick R. Appelbaum, Harry P. Erba
Collection and assembly of data: Mikkael A. Sekeres, Megan Othus, Diane Roulston, Aziz Nazha, Mario R. Velasco, Rakesh Gaur, Eyal C. Attar, Elina K. Cook, Michael J. Rauh, Harry P. Erba
Data analysis and interpretation: Mikkael A. Sekeres, Megan Othus, Diane Roulston, Anna Moseley, Aziz Nazha, Yanming Zhang, Ehab Atallah, Elina K. Cook, Alyssa H. Cull, Michael J. Rauh, Harry P. Erba
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Randomized Phase II Study of Azacitidine Alone or in Combination With Lenalidomide or With Vorinostat in Higher-Risk Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia: North American Intergroup Study SWOG S1117
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc
Mikkael A. Sekeres
Consulting or Advisory Role: Celgene, Millennium
Megan Othus
No relationship to disclose
Alan F. List
Leadership: Cell Biomedicine Group, Cell Therapeutics
Consulting or Advisory Role: Celgene, Fuji Pharma
Research Funding: Celgene
Patents, Royalties, Other Intellectual Property: Patent for myelodysplastic syndrome diagnostic testing
Olatoyosi Odenike
Consulting or Advisory Role: Incyte, CTI/Baxalta, Celgene, Abbvie
Research Funding: Celgene (Inst), Incyte (Inst), Astex Pharmaceuticals (Inst), NS Pharma (Inst), Agios (Inst), Abbvie (Inst), Janssen Pharmaceuticals (Inst), Millennium (Inst), OTS (Inst)
Richard M. Stone
Consulting or Advisory Role: Amgen, Celgene, Astellas Pharma, Agios, Karyopharm Therapeutics, Pfizer, Novartis, Orsenix, Jazz Pharmaceuticals, Abbvie/Genentech
Research Funding: Novartis (Inst)
Steven D. Gore
Consulting or Advisory Role: Celgene
Research Funding: Celgene (Inst)
Mark R. Litzow
Honoraria: Amgen
Consulting or Advisory Role: Amgen
Research Funding: Amgen
Travel, Accommodations, Expenses: Amgen
Rena Buckstein
Honoraria: Celgene, Novartis
Consulting or Advisory Role: Celgene, Novartis
Research Funding: Celgene
Min Fang
Research Funding: Affymetrix (Inst), Nucleix (Inst)
Diane Roulston
No relationship to disclose
Clara D. Bloomfield
No relationship to disclose
Anna Moseley
Employment: Cook Medical
Travel, Accommodations, Expenses: Cook Medical
Aziz Nazha
No relationship to disclose
Yanming Zhang
No relationship to disclose
Mario R. Velasco
Employment: Cancer Care Specialists of Central IL
Rakesh Gaur
Honoraria: Medtrial
Research Funding: Amgen (Inst), Celgene (Inst), Bristol-Myers Squibb (Inst), Cyclacel (Inst)
Ehab Atallah
Consulting or Advisory Role: Bristol-Myers Squibb, Pfizer, Amgen, Novartis
Research Funding: Takeda (Inst)
Eyal C. Attar
Employment: Agios
Stock or Other Ownership: Agios, Ariad
Elina K. Cook
No relationship to disclose
Alyssa H. Cull
No relationship to disclose
Michael J. Rauh
No relationship to disclose
Frederick R. Appelbaum
Stock or Other Ownership: Adaptive Biotechnologies, Igenica
Consulting or Advisory Role: National Marrow Donor Program, Igenica
Harry P. Erba
Honoraria: Novartis, Celgene, Incyte, Amgen, Jazz Pharmaceuticals, Daiichi Sankyo, Immunogen, Millennium/Takeda, Ono Pharmaceutical, Pfizer, Seattle Genetics, Sunesis Pharmaceuticals
Consulting or Advisory Role: Daiichi Sankyo, Sunesis Pharmaceuticals, Pfizer, Novartis, Incyte, Celgene, Seattle Genetics, Amgen, Ono Pharmaceutical, Jazz Pharmaceuticals, Immunogen, Millennium/Takeda
Speakers' Bureau: Novartis, Incyte, Celgene
Research Funding: Seattle Genetics (Inst), Amgen (Inst), Millennium/Takeda (Inst), Celator (Inst), Astellas Pharma (Inst), Agios (Inst), Daiichi Sankyo (Inst), Immunogen (Inst), Janssen Oncology (Inst), Juno Therapeutics
REFERENCES
- 1.Greenberg P, Cox C, LeBeau MM, et al. : International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 89:2079-2088, 1997 [PubMed] [Google Scholar]
- 2.Greenberg PL, Tuechler H, Schanz J, et al. : Revised international prognostic scoring system for myelodysplastic syndromes. Blood 120:2454-2465, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sekeres MA, Cutler C: How we treat higher-risk myelodysplastic syndromes. Blood 123:829-836, 2014 [DOI] [PubMed] [Google Scholar]
- 4.Sekeres MA: Epidemiology, natural history, and practice patterns of patients with myelodysplastic syndromes in 2010. J Natl Compr Canc Netw 9:57-63, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, Issa JP, Rosenfeld CS, et al. : Decitabine improves patient outcomes in myelodysplastic syndromes: Results of a phase III randomized study. Cancer 106:1794-1803, 2006 [DOI] [PubMed] [Google Scholar]
- 6.List A, Dewald G, Bennett J, et al. : Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med 355:1456-1465, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Silverman LR, Demakos EP, Peterson BL, et al. : Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J Clin Oncol 20:2429-2440, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Ebert BL, Pretz J, Bosco J, et al. : Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451:335-339, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krönke J, Fink EC, Hollenbach PW, et al. : Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523:183-188, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wei S, Chen X, Rocha K, et al: A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc Natl Acad Sci USA 106:12974-12979, 2009 [Erratum: Proc Natl Acad Sci USA 110:14504, 2013] [DOI] [PMC free article] [PubMed]
- 11. Santini V, Almeida A, Giagounidis A, et al: Randomized phase III study of lenalidomide versus placebo in RBC transfusion-dependent patients with lower-risk non-del(5q) myelodysplastic syndromes and ineligible for or refractory to erythropoiesis-stimulating agents. J Clin Oncol 34:2988-2996, 2016. [DOI] [PubMed]
- 12.Mesa RA, Yao X, Cripe LD, et al. : Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) phase 2 trial E4903. Blood 116:4436-4438, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quintás-Cardama A, Kantarjian HM, Manshouri T, et al. : Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol 27:4760-4766, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saunthararajah Y, Triozzi P, Rini B, et al. : p53-Independent, normal stem cell sparing epigenetic differentiation therapy for myeloid and other malignancies. Semin Oncol 39:97-108, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. : Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol 10:223-232, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks P, Rifkind RA, Richon VM, et al. : Histone deacetylases and cancer: Causes and therapies. Nat Rev Cancer 1:194-202, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Ruefli AA, Ausserlechner MJ, Bernhard D, et al. : The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci USA 98:10833-10838, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silverman LR, Verma A, Odchimar-Reissig R, et al. : A phase II trial of epigenetic modulators vorinostat in combination with azacitidine (azaC) in patients with the myelodysplastic syndrome (MDS): Initial results of study 6898 of the New York Cancer Consortium. Blood 122:386-393, 2013. 23719299 [Google Scholar]
- 19.Sekeres MA, List AF, Cuthbertson D, et al. : Phase I combination trial of lenalidomide and azacitidine in patients with higher-risk myelodysplastic syndromes. J Clin Oncol 28:2253-2258, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sekeres MA, Tiu RV, Komrokji R, et al. : Phase 2 study of the lenalidomide and azacitidine combination in patients with higher-risk myelodysplastic syndromes. Blood 120:4945-4951, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grinblatt DL, Sekeres MA, Komrokji RS, et al. : Patients with myelodysplastic syndromes treated with azacitidine in clinical practice: The AVIDA registry. Leuk Lymphoma 56:887-895, 2015 [DOI] [PubMed] [Google Scholar]
- 22.Lyons RM, Cosgriff TM, Modi SS, et al. : Hematologic response to three alternative dosing schedules of azacitidine in patients with myelodysplastic syndromes. J Clin Oncol 27:1850-1856, 2009 [DOI] [PubMed] [Google Scholar]
- 23.Cheson BD, Greenberg PL, Bennett JM, et al. : Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 108:419-425, 2006 [DOI] [PubMed] [Google Scholar]
- 24. Myelodysplastic Syndromes Foundation: MDS Centers of Excellence. https://www.mds-foundation.org/mds-centeres-of-excellence/
- 25.Bejar R, Stevenson KE, Caughey B, et al. : Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol 32:2691-2698, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walter MJ, Shen D, Shao J, et al. : Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 27:1275-1282, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekeres MA, Schoonen WM, Kantarjian H, et al. : Characteristics of US patients with myelodysplastic syndromes: Results of six cross-sectional physician surveys. J Natl Cancer Inst 100:1542-1551, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steensma DP, Komrokji RS, Stone RM, et al. : Disparity in perceptions of disease characteristics, treatment effectiveness, and factors influencing treatment adherence between physicians and patients with myelodysplastic syndromes. Cancer 120:1670-1676, 2014 [DOI] [PubMed] [Google Scholar]
- 29.Sekeres M, Swern A, List A, et al. : Effect of lenalidomide (LEN) exposure on AML-free survival and overall survival in LEN-treated patients (pts) with IPSS Low- or Intermediate-1-risk myelodysplastic syndromes (MDS) with del(5q). Blood 126:2870a, 2015 [Google Scholar]
- 30.Bejar R, Lord A, Stevenson K, et al. : TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 124:2705-2712, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haferlach T, Nagata Y, Grossmann V, et al. : Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28:241-247, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traina F, Visconte V, Elson P, et al. : Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 28:78-87, 2014 [DOI] [PubMed] [Google Scholar]
- 33.Welch JS, Petti AA, Miller CA, et al. : TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med 375:2023-2036, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]