

Abstract
The reovirus fusion-associated small transmembrane (FAST) proteins are the smallest known viral fusogens (∼100–150 amino acids) and efficiently induce cell-cell fusion and syncytium formation in multiple cell types. Syncytium formation enhances cell-cell virus transmission and may also induce immunogenic cell death, a form of apoptosis that stimulates immune recognition of tumor cells. These properties suggest that FAST proteins might serve to enhance oncolytic virotherapy. The oncolytic activity of recombinant VSVΔM51 (an interferon-sensitive vesicular stomatitis virus [VSV] mutant) encoding the p14 FAST protein (VSV-p14) was compared with a similar construct encoding GFP (VSV-GFP) in cell culture and syngeneic BALB/c tumor models. Compared with VSV-GFP, VSV-p14 exhibited increased oncolytic activity against MCF-7 and 4T1 breast cancer spheroids in culture and reduced primary 4T1 breast tumor growth in vivo. VSV-p14 prolonged survival in both primary and metastatic 4T1 breast cancer models, and in a CT26 metastatic colon cancer model. As with VSV-GFP, VSV-p14 preferentially replicated in vivo in tumors and was cleared rapidly from other sites. Furthermore, VSV-p14 increased the numbers of activated splenic CD4, CD8, natural killer (NK), and natural killer T (NKT) cells, and increased the number of activated CD4 and CD8 cells in tumors. FAST proteins may therefore provide a multi-pronged approach to improving oncolytic virotherapy via syncytium formation and enhanced immune stimulation.
Keywords: oncolytic virotherapy, vesicular stomatitis virus, FAST protein, syncytium formation, 4T1 mammary adenocarcinoma, CT26 colon carcinoma
Graphical Abstract
Introduction
Oncolytic viruses (OVs) constitute a form of cancer immunotherapy exhibiting promising results in a broad range of cancers.1 OVs are naturally occurring or genetically engineered viruses that preferentially replicate within and kill cancer cells because of signaling defects in cellular metabolism and innate immunity.2 OVs have moved from theory to practice with the recent licensing of a second-generation oncolytic herpes simplex virus 1 (HSV-1) expressing granulocyte macrophage colony-stimulating factor (GM-CSF; Imlygic) to treat inoperable melanoma.3, 4 Other OVs based on several different virus platforms, including vaccinia virus, rhabdoviruses, adenovirus, and reovirus, are undergoing advanced clinical trials.5, 6 Selective destruction of cancer cells in the tumor microenvironment results in perturbation of tumor vasculature, release of inflammatory cytokines, and activation of innate and adaptive immune responses against the tumor.7, 8, 9 These direct and indirect effects of virus-mediated cancer cell killing are responsible for tumor destruction, and new approaches are being pursued to enhance both cytocidal and immune-mediated tumor destruction by OVs.
In this study, we describe the use of an unusual viral membrane fusion protein to enhance the oncolytic activity of an interferon (IFN)-sensitive vesicular stomatitis virus mutant (VSVΔM51). The reovirus fusion-associated small transmembrane (FAST) proteins are the only examples of membrane fusion proteins encoded by nonenveloped viruses, and they evolved specifically to induce cell-cell rather than virus-cell membrane fusion.10 Unlike enveloped virus fusion proteins, FAST proteins are not components of the virus particle and hence play no role in virus entry into cells. Instead, FAST proteins are expressed inside virus-infected cells and trafficked through the endoplasmic reticulum (ER)-Golgi pathway to the plasma membrane, where they mediate cell-cell fusion and syncytium formation in multiple cell types and at physiological pH.11, 12, 13, 14, 15, 16 There are six distinct members of the FAST protein family encoded by numerous species of orthoreoviruses and aquareoviruses.17 The different FAST proteins share little to no sequence similarity, but all are small (95–198 residues), single-pass transmembrane proteins with exceedingly small N-terminal ectodomains (∼20–40 residues) external to the plasma membrane and equal-sized or longer C-terminal cytoplasmic tails. All three domains function as fusion modules and contain motifs essential for membrane fusion activity.18, 19, 20, 21, 22, 23
FAST proteins are the smallest known membrane fusion proteins. They are not reliant on specific cell receptors, and thus fuse numerous cell types. Humans have no pre-existing immunity against FAST proteins (they derive from non-human viruses), and they promote localized and disseminated virus transmission.24, 25, 26 We exploited these desirable features of the FAST proteins by engineering a recombinant, IFN-sensitive oncolytic vesicular stomatitis virus (VSVΔM51) to express the p14 FAST protein of reptilian reovirus.27 We show that addition of p14 to VSV increases cancer cell death and virus transmission within tumors, reduces tumor growth and metastases, and stimulates more robust innate and adaptive immune responses, all while maintaining a favorable safety profile. The FAST proteins therefore provide a novel approach to enhance oncolytic virotherapy by increasing cytocidal and immune-mediated tumor cell killing.
Results
FAST Protein p14 Increases VSV Dissemination, Replication, and Cytolytic Activity in Cell Culture
To determine whether a FAST protein could increase the efficacy of oncolytic virotherapy by promoting virus spread and/or anti-tumor immune responses, we used a VSV OV containing a methionine deletion in its matrix protein (VSVΔM51) that renders the virus highly susceptible to host IFN responses.28 The VSVΔM51 backbone was used to create recombinant viruses encoding either GFP (VSV-GFP) or the 125-residue p14 FAST protein from reptilian reovirus (VSV-p14) (Figure 1A). At a low MOI infection, VSV-p14 induced extensive cell-cell fusion and syncytium formation in Vero cells (Figure 1B), consistent with the ability of FAST proteins to fuse multiple cell types at neutral pH. No syncytium formation occurred in cells infected with VSV-GFP because the VSV G protein induces membrane fusion efficiently at only acidic pH. Syncytium formation induced by VSV-p14 resulted in increased dissemination of the infection and a 20-fold increase in virus yield (Figure 1C). Similar results were obtained following infection of MCF-7 or 4T1 breast cancer spheroid cultures (Figure 2). Whereas syncytium formation was difficult to detect in these three-dimensional cultures, spheroids infected with VSV-p14 contained extensive, soap-bubble-like membrane blebs not observed in spheroids infected with VSV-GFP (Figure 2A). The recombinant VSV-p14 also replicated more efficiently than VSV-GFP in spheroid cultures, resulting in a 10- to 20-fold increase in virus titers (Figure 2B, left panels), and dramatically decreased spheroid viability compared with VSV-GFP (Figure 2B, right panels).
Figure 1.

VSV-p14 Induces Syncytium Formation and Enhances Virus Dissemination and Yield
(A) Schematic of recombinant VSV containing gene insertions encoding either GFP or the p14 FAST protein. (B) Vero cells were mock-infected or infected with the indicated recombinant viruses at an MOI of 0.1 and Giemsa-stained at 24 hpi to detect syncytium formation. (C) As in (B), using the supernatant from infected cultures to determine the virus yield by plaque assay at the indicated times post-infection. Results are mean ± SEM from duplicate experiments. *p < 0.05 compared with VSV-GFP.
Figure 2.

VSV-p14 Has Enhanced Oncolytic Activity in Breast Cancer Spheroids
(A) 4T1 and MCF-7 breast cancer cells growing as spheroids were mock-infected or infected with 1 × 105 PFU/mL recombinant VSV-GFP or VSV-p14, and phase-contrast images of the infected spheroids were captured at 24 hpi. Arrows indicate large membrane blebs induced by VSV-p14. Scale bar, 200 μm. (B) As in (A), quantifying virus yields in the supernatants (mean ± SEM from n = 3 experiments) at the indicated times post-infection by TCID50 in permissive Vero cells (left panels), or quantifying percent cell viability at 24 hpi (mean ± SD of a representative experiment in quadruplicate; n = 2–4 experiments at 24–40 hpi) using a phosphatase assay (right panels). *p < 0.05 compared with VSV-GFP.
VSV-p14 Slows Primary Tumor Growth and Prolongs Survival in a Syngeneic Breast Cancer Model
A syngeneic 4T1 primary mammary adenocarcinoma BALB/c mouse model was used to assess the in vivo efficacy of VSV-p14 as an oncolytic virotherapy. The triple-negative 4T1 model has been shown in numerous studies to respond relatively poorly to oncolytic virotherapy using the IFN-sensitive VSVΔM51 virus,29, 30 providing a useful and challenging immunocompetent model to look for improved outcomes. Tumor cells were implanted in the mammary fat pad, and when tumors became palpable (10 days post-implantation) animals were mock-injected or injected with one intravenous injection of VSV-GFP or VSV-p14 (1 × 108 plaque-forming units [PFUs]) followed by three intratumoral injections at the same virus dose. As shown, VSV-GFP had only modest effects on slowing the rate of tumor growth, whereas VSV-p14 was significantly more effective at slowing tumor development, particularly over the first 35 days post-treatment (Figure 3B). VSV-p14 was also significantly better than VSV-GFP at prolonging survival (Figure 3C). To assess the safety profile of VSV-p14, we injected 4T1 tumor-bearing immunocompetent mice with a single dose of VSV-p14 or VSV-GFP via the tail vein, and virus titers in the tumor and various organs were quantified at 24 and 48 hr post-infection (hpi). Biodistribution of both viruses was similar at 24 hpi in tumors, lungs, liver, and spleen, with no virus detected in the heart and low levels of VSV-GFP, but not VSV-14, detectable in the brain (Figure 4A). Virus titers in tumors were approximately 10- to 100-fold higher for both viruses than titers in the other organs at 24 hpi, consistent with previous reports showing the predilection of VSVΔM51 to replicate in cancer cells.28 By 48 hpi, virus titers were undiminished in tumors but were 3–4 logs lower in the lungs and liver, and were undetectable in the spleen, heart, and brain (Figure 4B). Thus, p14 enhances the efficacy of oncolytic VSV against primary tumors without altering VSV biodistribution.
Figure 3.

VSV-p14 Slows Growth of Primary Mammary Tumors and Improves Survival
(A) Syngeneic 4T1 subcutaneous mammary tumors were established in BALB/c mice, and 10 days later animals were mock-treated or treated by one intravenous injection of VSV-GFP or VSV-p14 (1 × 108 PFU) followed by three intratumoral injections at the same virus dose (n = 5 per treatment group). (B) Tumor size was monitored over time, and average tumor volumes ± SEM were calculated for each treatment group. Statistical analysis used ANOVA to compare the treatment groups. *p < 0.05 compared with control; †p < 0.05 compared with VSV-GFP. (C) Survival data were assessed by a log rank test. *p < 0.05 compared with control.
Figure 4.
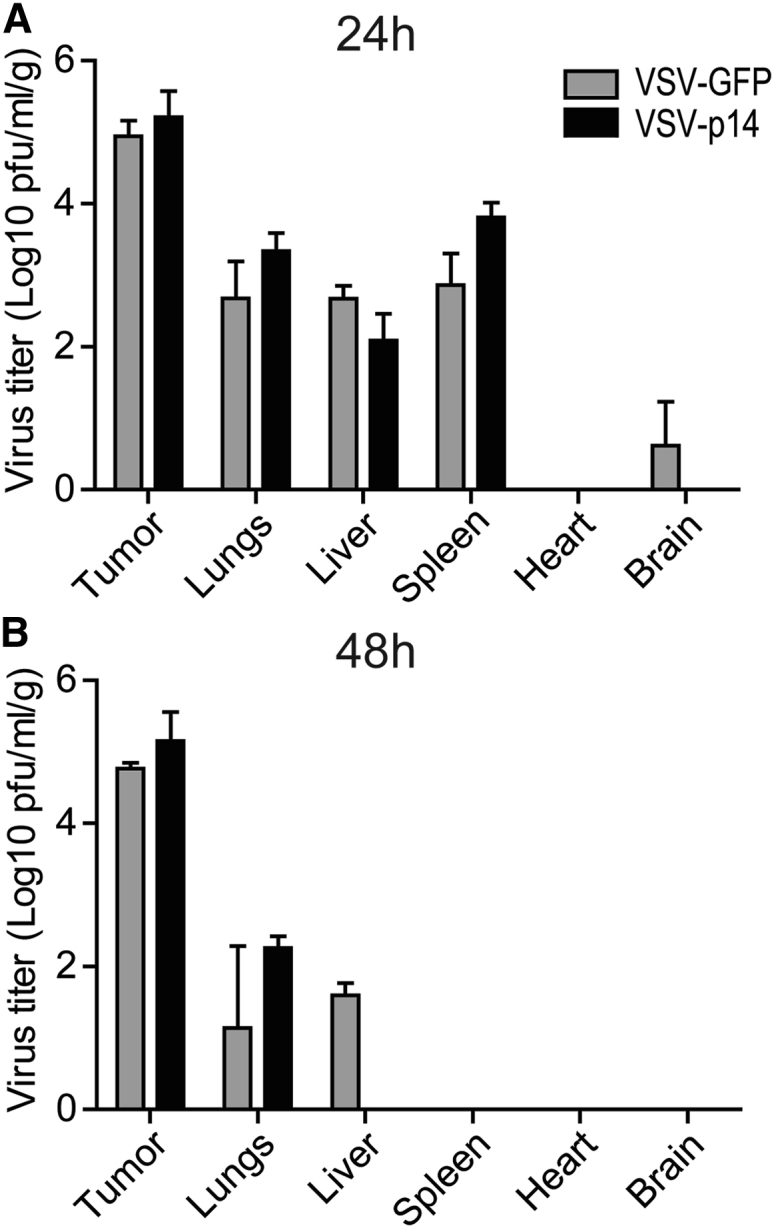
VSV-p14 and VSV-GFP Show Similar Biodistribution and Safety Profiles
Subcutaneous 4T1 mammary tumors were established in BALB/c mice; then animals were treated with one intravenous injection (1 × 108 PFU) of VSV-p14 or VSV-GFP. (A and B) Mice were sacrificed at (A) 24 hpi (n = 3 per treatment group) or (B) 48 hpi (n = 2 per treatment group), tumors and the indicated organs were harvested, and virus titers were quantified by plaque assay. Results are the average titer per gram of tissue ± SEM.
VSV-p14 Shows Greater Efficacy against Metastatic Disease
To examine whether the delayed tumor growth observed in 4T1 primary tumors would translate to similar protection in models of metastatic disease, we first assessed the effect of systemic administration of VSV-GFP or VSV-p14 in an established post-surgical 4T1 metastasis model.31 Primary 4T1 mammary tumors were established in BALB/c mice and resected 12 days later, a time point when micrometastatic nodes have already been seeded at distant sites. On days 13, 15, and 17, animals were intravenously administered PBS, VSV-GFP, or VSV-p14, and survival was monitored over time. As shown (Figure 5A), VSV-GFP and VSV-p14 both significantly prolonged survival compared with PBS control, and VSV-p14 also mediated significant survival enhancement compared with VSV-GFP (median survival time of 33.5 days for PBS group, 41.5 days for VSV-GFP group, and 61 days for VSV-p14 group). Given the aggressive nature of the 4T1 metastasis model, the enhanced protection observed with VSV-p14 is quite striking.
Figure 5.

VSV-p14 Demonstrates Better Protection against Metastatic Disease Compared with VSV-GFP
(A) Subcutaneous 4T1 mammary tumors were established in BALB/c mice, and primary tumors were resected on day 12. Mice were treated on days 13, 15, and 17 with PBS, VSV-GFP, or VSV-p14 (n = 5 per group). Survival data were assessed by a log rank test. *p < 0.016 compared with PBS; †p < 0.016 compared with VSV-GFP. (B) CT26LacZ colon carcinoma cells (2 × 105 cells) were injected intravenously into BALB/c mice to establish lung metastases. Animals were injected intravenously with PBS (control, n = 8) or with 1 × 107 PFU of VSV-p14 (n = 8) or VSV-GFP (n = 7) on days 3, 5, and 7; lungs were removed 7 days following the last virus injection; and the mean numbers ± SEM of surface lung metastases were visually quantified following staining of the excised lungs for β-galactosidase. *p < 0.05 compared with control.
To extend the results beyond breast cancer, we employed the CT26 syngeneic metastatic colon carcinoma model in BALB/c mice. Following tail-vein injection of the cancer cells, metastases rapidly develop in the lung.32 Using CT26 cells stably transduced to express β-galactosidase (CT26LacZ), we injected mice intravenously with 2 × 105 cells and subsequently injected them with 1 × 107 PFU of VSV-p14 or VSV-GFP on days 3, 5, and 7 after tumor cell injection. Quantifying the number of surface lung metastases 7 days after the last virus treatment revealed that VSV-p14 significantly reduced the number of lung metastases by approximately 3-fold compared with controls (Figure 5B). VSV-GFP also showed a trend toward decreased metastases compared with control, but this effect did not reach statistical significance (Figure 5B). VSV-p14 was therefore significantly more effective than VSV-GFP at reducing metastases in two different metastatic models.
VSV-p14 Enhances Immune Cell Activation
Although tumor tropism and lytic activity are important for effective OV therapy,1, 28 increasing evidence indicates OVs work in part by activating and directing the immune system to target tumors.33 To determine the effect of VSV-p14 on the immune system, we examined the frequency and activation status of lymphocytes in the primary tumors, draining lymph node, and spleen. VSV-p14 treatment induced a statistically significant increase in the number of splenic CD8 T cells and a trend toward increased numbers of splenic CD4 T cells, natural killer T (NKT) cells, and natural killer (NK) cells (Figure 6). When these same cell populations were examined for expression of the early activation marker CD69, VSV-p14 treatment was shown to induce significant upregulation of CD69 expression on all four splenic immune cell populations (Figure 6). In contrast, VSV-GFP did not induce accumulation of T cell or NK cell populations in the spleen, but an upward trend in the level of CD69 expression was observed on CD4 T cells, CD8 T cells, and NKT cells, but not on NK cells (Figure 6). Similar results were observed when examining CD4 and CD8 lymphocyte populations in the tumor and draining lymph node. An upward trend in the numbers of CD4 and CD8 T cells in the draining lymph node and an upward trend in tumor-infiltrating CD8 cells were observed in animals treated with VSV-p14, but not in animals treated with VSV-GFP (Figure 7). VSV-p14 also significantly increased the frequency of activated CD69+ CD4 and CD8 T cells in both the draining lymph node and tumor compared with control animals, and significantly increased the frequency of activated CD8 cells in the lymph node compared with VSV-GFP (Figure 7).
Figure 6.

VSV-p14 Induces Activation of Splenic T Cells and NK Cells
4T1 tumor-bearing mice (n = 9–10 per treatment group) received one intravenous injection of PBS (control) or VSV-p14 or VSV-GFP (1 × 108 PFU) on day 12, followed by similar intratumoral inoculations on days 13, 14, and 15. Spleen cells were isolated 24 hr following the final injection. The mean ± SEM number of splenic CD4 T cells (CD4+ TCRβ+), CD8 T cells (CD8+ TCRβ+), NKT cells (CD1d-tetramer+ TCRβ+), and NK cells (CD49b+ TCRβ−) (top row) and the expression of CD69 by CD4 T cells, CD8 T cells, NKT cells, and NK cells (bottom row) were assessed by flow cytometry. *p < 0.05 compared with control.
Figure 7.

VSV-p14 Increases the Frequency of Activated T Cells in the Tumors and Draining Lymph Node
4T1 tumor-bearing mice (n = 9–10 per treatment group) received one intravenous injection of PBS (control) or VSV-p14 or VSV-GFP (1 × 108 PFU) on day 12, followed by similar intratumoral inoculations on days 13, 14, and 15. The draining lymph node and tumors were isolated 24 hr after the final injection. The mean ± SEM number of CD4 T cells (CD4+ TCRβ+) and CD8 T cells (CD8+ TCRβ+) in the draining lymph node and tumors (top row), or the frequency of the same cells expressing the early activation marker CD69+ was assessed using flow cytometry. *p < 0.05 compared with control; †p < 0.016 compared with VSV-GFP.
We also examined myeloid cell populations in the spleen, draining lymph node, and tumors. Although VSV-p14 did not significantly alter the frequency of splenic myeloid-derived suppressor cells (MDSCs), macrophages, or dendritic cells (DCs), there was a slight but significant increase in the expression of CD80 on DCs in the spleen (Figure S1).
Discussion
Since first discovered as a promising OV,34 VSV has been engineered in various ways to enhance its efficacy in oncolytic virotherapy and advance clinical trials.35 Development of an IFN-sensitive VSVΔM51 platform decreased neurotoxicity and increased specificity for cancer cells that are commonly defective in IFN response pathways.28 Subsequent recombinant VSVΔM51 constructs expressing transgenes that modulate immune responses,36, 37, 38, 39, 40 or substitute the VSV G protein with other receptor-binding proteins that reduce neurotoxicity or target specific cancer cell types,41, 42, 43, 44 have all been shown to enhance oncolytic virotherapy to some extent. The present results indicate that VSV expressing a reovirus FAST protein induced syncytium formation, enhanced tumor cell killing, increased immune activation, and prolonged survival in primary and metastatic immunocompetent mouse cancer models. With the concept that a single oncolytic virotherapy is unlikely to be the “be all and end all” approach to translating oncolytic virotherapy into improved clinical outcomes, this alternate approach to enhance VSV-mediated oncolysis offers several potential advantages for developing clinically applicable rhabdovirus oncolytic immunotherapies.
Expression of the p14 FAST protein by a replication-defective adenovirus vector was recently shown not to provide a therapeutic benefit in a murine cancer model.45 However, in cella and ex vivo coinfection experiments with VSV-p14 and an oncolytic vaccinia virus showed a synergistic interaction that increased cell killing and spread of both viruses.29 This synergistic effect was attributed to p14-induced syncytium formation enhancing cell-cell spread of the mostly cell-associated vaccinia virus, with expression of the vaccinia virus B18R gene that locally inhibits type I IFN responses promoting replication of VSV-p14. Considering this prior study, we examined whether VSV-p14 by itself could function as an enhanced OV platform. VSV-p14 induced robust cell-cell fusion and cell death, and resulted in increased virus yields in breast cancer spheroids (Figure 2). These in cella results were extended in vivo using a primary breast cancer model, where enhanced destruction of tumor cells correlated with a statistically significant increase in survival relative to the control VSV-GFP virus (Figure 3). The absence of any discernible change in the relative biodistribution of VSV-p14 in vivo (Figure 4) implied enhanced oncolytic activity did not occur at the expense of specific cancer cell killing or biosafety considerations. Furthermore, VSV-p14 was significantly more effective than VSV-GFP at reducing metastases and prolonging survival in two different metastatic cancer models (Figure 5), indicating FAST proteins can directly enhance the therapeutic activity of an OV.
OVs also stimulate anti-tumor immunity, which can at least partially be attributed to virus-mediated immunogenic cell death (ICD).46, 47, 48 ICD releases damage-associated molecular patterns that increase recruitment and activation of antigen-presenting cells, thereby enhancing tumor antigen uptake and presentation.49, 50, 51 FAST proteins enhance cytolysis and disrupt calcium homeostasis,26, 52, 53 suggesting they are likely to trigger ICD and consistent with our present findings showing FAST proteins increased immune cell activation following oncolytic virotherapy. VSV-p14 treatment significantly increased the proportion of activated T cells and NK cells in the spleen (Figure 6), and the proportion of activated CD4 and CD8 cells in the draining lymph node and tumor (Figure 7). In the case of the draining lymph nodes, VSV-p14 treatment significantly increased the frequency of CD69-positive CD4 and CD8 cells compared with VSV-GFP. Although VSV-GFP showed a trend toward increased proportions of activated T cells and NK cells, these increases were less than those observed with VSV-p14 and were not statistically significant compared to those with no treatment (Figure 7). Although some OVs enhance recruitment of immunosuppressive MDSC populations leading to tumor-associated immunosuppression,54 we did not observe an increased frequency of myeloid cells in the spleen, lymph nodes, or 4T1 tumors following VSV treatments (Figure S1). Interestingly, there was a significant decrease in the frequency of DCs in the draining lymph nodes of animals treated with VSV-p14, but not VSV-GFP, with a trend toward increased infiltration into tumors (Figure S1), possibly an indication of increased DC migration from the draining lymph nodes into the tumor. Furthermore, we observed a small increase in the level of CD80 expression on DCs in the spleen, consistent with increased activation of these antigen-presenting cells. Immune activation is further consistent with the increased CD8 T cell expansion and upregulation of the activation marker CD69 on multiple immune cell populations. Thus, enhanced oncolytic virotherapy using VSV-p14 correlates with both increased virus dissemination and cell killing via syncytium formation and with robust systemic immune responses, in keeping with a dual mechanism of action of OVs.
The concept of using syncytiogenic proteins to enhance oncolytic virotherapy has previously been employed using enveloped virus membrane fusion proteins that function at physiological pH to induce syncytium formation.55 Several different enveloped virus fusion proteins have been engineered into OVs and shown to enhance tumor cell killing via direct cytotoxic effects and by stimulating more potent anti-tumor immune responses via ICD and enhanced delivery of tumor antigens to antigen-presenting cells.56, 57, 58, 59, 60 However, the large size of enveloped virus fusion proteins restricts their application to OVs whose genome size is not constrained by the dimensions of the virus particle. Other potential limitations with use of some enveloped virus proteins in oncolytic virotherapy are potent neutralizing immune responses induced by these proteins, a reliance on species- or tissue-specific cell receptors, or extensive cytotoxicity that can reduce the extent of OV replication.57, 58, 61, 62
The reovirus FAST proteins are the only examples of nonenveloped virus proteins that induce cell-cell fusion and syncytium formation.10 FAST proteins are the smallest known membrane fusion proteins (p14 is only 125 residues), rendering them weakly immunogenic, and their small size facilitates incorporation of FAST protein genes into almost any OV platform, alone or in combination with other immunostimulatory genes. FAST proteins are also not reliant on specific cell receptors, and thus fuse numerous cell types; humans have no pre-existing immunity against FAST proteins (they derive from non-human viruses); and FAST proteins promote localized and disseminated virus transmission. As we now show, these desirable properties translate into improved outcomes using an oncolytic rhabdovirus platform to express the p14 FAST protein, suggesting this as an alternate potential OV platform for cancer treatment.
Materials and Methods
Cells
African green monkey (Vero) cells were maintained in Medium 199 supplemented with 5% fetal bovine serum (FBS). QM5 (Quail muscle fibrosarcoma) cells were cultured in DMEM supplemented with 10% FBS. Mouse mammary epithelial (4T1) tumor cells were maintained in complete RPMI 1640 media supplemented with 10% FBS. MCF-7 cells were cultured in DMEM/Nutrient Mixture F-12 (DMEM/F-12) supplemented with 10% FBS. Mouse colon carcinoma (CT26LacZ) cells were cultured in DMEM with 10% FBS. All culture reagents were obtained from GIBCO, and all cells were cultured as monolayers at 37°C with 5% CO2.
Mice
Female BALB/c mice were purchased form Charles River Laboratories (Senneville, Canada) and used at 8–12 weeks of age. All animal protocols followed the guidelines of the Canadian Council on Animal Care and were approved by the University Committee on Laboratory Animals.
Generation of Recombinant VSV
The p14 FAST protein gene in pcDNA327 and the EGFP gene in pEGFP-N1 were amplified by PCR and subcloned into the XhoI and NheI sites located between the G and L genes in pVSVΔM51-XN28 to generate pVSVΔM51-XN-p14 and pVSVΔM51-XN-GFP (referred to as VSV-p14 and VSV-GFP). QM5 cells were infected with the modified vaccinia virus Ankara strain expressing T7 RNA polymerase (MVA-T7),63 and 4 hr later co-transfected with four plasmids at a ratio of 2:2:1.25:0.25 μg: pVSVΔ51-XN-p14 or pVSVΔ51-XN-GFP, and pBS-N, pBS-P, and pBS-L, encoding the VSV N, P, and L proteins, respectively, under the control of a CMV promoter. Two days later, cell culture supernatants were harvested, filtered through a 0.2-μm filter to remove vaccinia virus, and then used to infect Vero cells. Vero cell supernatants were harvested 3 days post-infection, and the recombinant VSV particles were isolated by plaque purification on Vero cells. The identities of the recombinant viruses were confirmed by sequencing cDNA amplicons obtained by PCR using primers complementary to VSV sequences flanking the insertion site. Virus stocks were amplified and titered by plaque assay using Vero cells.
Oncolytic Activity in Cell Culture
Recombinant viruses were tested for their cytolytic activity in cell culture using Vero cells and breast cancer spheroids. Vero cells cultured in 12-well plates were infected with recombinant viruses at an MOI of 0.1 for 1 hr at 37°C; then cells were washed with PBS to remove unbound virus. Cells were then cultured in fresh medium for 24 hr. Culture supernatants were harvested to quantify virus yield by plaque assay, and monolayers were stained with Wright-Giemsa to view cell death and syncytium formation under bright-field microscopy. To obtain spheroid cultures, we seeded MCF-7 and 4T1 mammary cancer cells into ultra-low attachment Costar six-well plates using 3 × 104 cells/well and cultured them for 6 days in a mammosphere medium (DMEM/F-12 supplemented with 20 ng/mL basic fibroblast growth factor (bFGF), 20 ng/mL epidermal growth factor (EGF), 100 U/mL penicillin, 100 μg/mL streptomycin, and 1× B27 serum-free supplement), replacing the medium with fresh medium every 72 hr. Spheroid cultures were infected using 1 × 105 PFU/well of VSV-GFP or VSV-p14; culture supernatants were harvested at 16–24 hpi, and virus yields were determined by tissue culture infective dose 50% (TCID50) in permissive Vero cells. Cell viability was assessed using a phosphatase assay. Spheroids were disassociated in trypsin for 5 min, washed in PBS, resuspended in a 1:1 ratio (final volume 1 mL) of PBS and phosphatase solution (0.1 M sodium acetate [pH 5.5], 0.1% Triton X-100, 4 mg/mL phosphatase substrate), and incubated for 90 min at 37°C in the dark. 50 μL of 1 N NaOH was then added to each sample to stop the reaction, samples were cleared by centrifugation at 1,000 × g for 5 min, supernatants were transferred to 96-well plates, and absorbance was measured at 405 nm using an Asys Expert 96 Microplate Reader.
Immunocompetent Animal Tumor Models
Primary Breast Cancer Model
4T1 breast cancer cells were harvested in the logarithmic growth phase, resuspended in saline, and injected subcutaneously (2 × 105 cells in 50 μL) into the mammary fat pad of female BALB/c mice (n = 5/treatment group). Palpable tumors formed within 10 days after seeding. Mice were injected intravenously with VSV-GFP or VSV-p14 (1 × 108 PFU/mouse in 50 μL), followed by four similar intratumoral injections 1 day apart. For the efficacy studies, 4T1 tumors were measured every 2–4 d using an electronic caliper, and tumor volume was calculated as (W × W × L)/2. For biodistribution studies, BALB/c mice with established 4T1 subcutaneous breast tumors were injected intravenously with VSV-GFP or VSV-p14 (1 × 108 PFU/mouse in 50 μL), mice were sacrificed 24–48 hpi, and normal organs (lungs, liver, spleen, heart, brain) and tumor tissues were harvested for virus titration by plaque assay, as previously described.64
Post-surgical Breast Cancer Metastasis Model
4T1 tumors were established in mice, as described above, and primary tumors were resected 12 days following tumor inoculation, as previously described.31 On days 13, 15, and 17, mice received 100 μL of intravenous injections of PBS or 1 × 108 PFUs of VSV-GFP or VSV-p14. Survival was monitored over time.
Lung Metastasis Model
CT26-LacZ colon carcinoma cells (2 × 105 in 50 μL) were injected intravenously into BALB/c mice, and at days 3, 5, and 7, mice were injected intravenously with VSV-GFP or VSV-p14 (1 × 108 PFU/mouse) or with PBS. Mice were sacrificed 7 days following the last virus injection, lungs were harvested, and lung metastases were quantified visually following staining of the tumors using 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal; Sigma-Aldrich).
Immune Phenotyping
BALB/c mice were inoculated with 2 × 105 4T1 cells in the fourth mammary fat pad. Twelve days after inoculation, mice received intravenous injections of PBS, VSV-GFP, or VSV-p14. On days 13, 14, and 15, mice received intratumoral injections of PBS, VSV-GFP, or VSV-p14. Spleens, draining lymph nodes, and primary tumors were isolated on day 16. Following mechanical dispersion, tumor-infiltrating lymphocytes were enriched by centrifugation through a 33% Percoll gradient (GE Healthcare, Baie d’Urfe, Canada). Red blood cells were lysed with ammonium chloride buffer, and cells were washed by centrifugation. The immune profile of lymphoid and myeloid populations was examined by flow cytometry.
Flow Cytometry
All antibodies were purchased from eBioscience or BioLegend (San Diego, CA): purified CD16/32 (clone 97); fluorescein isothiocyanate (FITC)-conjugated CD3 (145-2C11), CD49b (DX5), CD11b (M1/70); phycoerythrin (PE)-labeled CD69 (H1.2F3), CD86 (GL1), Gr-1 (RB6-8C5); peridinin chlorophyll (PERCP)-labeled CD4 (RM4-5), CD11c (H13), TCR-β (H57-597), F4/80 (BM8); allophycocyanin (APC)-labeled CD8α (53-6.7), CD80 (16-10A1). To examine NKT cells by flow cytometry, we stained cells with allophycocyanin-labeled CD1d tetramers loaded with the glycolipid PBS57 (NIH Tetramer Core Facility, Emory Vaccine Center at Yerkes, Atlanta, GA). All cell samples were pre-incubated with anti-CD16/32 antibody to block non-specific binding. Following Fc-receptor blocking, cells were incubated at 4°C for 20 min with surface-staining antibody panels, washed, and fixed in 2% paraformaldehyde. Data acquisition was performed using a two-laser FACSCalibur flow cytometer (BD Biosciences, San Jose, CA), and data analysis was performed using FlowJo (V10.2; FlowJo, Ashland, OR).
Statistical Analyses
Data are expressed as mean ± SEM for replicate experiments or mean ± SD for a single representative experiment. A non-parametric two-tailed Mann-Whitney U test was used to compare between two groups. Comparisons between more than two groups were made using a Kruskal-Wallis non-parametric ANOVA with Dunn’s post-test. Statistical significance was set at p < 0.05. Survival data were analyzed by log rank (Mantel-Cox) significance test, and the statistical significance level was set using the Bonferroni corrected threshold [p < (0.05/K), where K is the number of comparisons performed]. Statistical computations were carried out using GraphPad Instat 3.02 and GraphPad Prism 7.02.
Author Contributions
F.L.B., S.G., J.C.B., B.J., C.P., and R.D. conceived the study and evaluated the data. F.L.B., S.G., A.L.G., and C.P. designed and conducted the experiments. N.M. and H.H. provided technical support. F.L.B., S.G., B.J., C.P., and R.D. wrote the manuscript. All authors critically reviewed the manuscript.
Conflicts of Interest
R.D. is a consultant with Innovascreen, which holds the patents relating to the p14 FAST protein.
Acknowledgments
R.D. was supported by grants from the Canadian Institutes of Health Research (CIHR) (MOP-13723 and MOP-57881). J.C.B. was supported by a grant from the Terry Fox Foundation (TFF 122868). B.J. was supported by a CIHR grant (MOP-110988). C.P. was supported by a Beatrice Hunter Cancer Research Institute (BHCRI) Cancer Research Training Program (CRTP) postdoctoral fellowship. S.G. was supported by doctoral studentships from CIHR, BHCRI, and the Killam Trust. A.L.G. was supported by postdoctoral fellowships from the Canadian Breast Cancer Foundation and Queen Elizabeth II Hospital Foundation.
Footnotes
Supplemental Information includes one figure and can be found with this article online at http://dx.doi.org/10.1016/j.omto.2017.08.001.
Contributor Information
Chungen Pan, Email: chungenp@gmail.com.
Roy Duncan, Email: roy.duncan@dal.ca.
Supplemental Information
References
- 1.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell J., McFadden G. Viruses for tumor therapy. Cell Host Microbe. 2014;15:260–265. doi: 10.1016/j.chom.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 4.Harrington K.J., Puzanov I., Hecht J.R., Hodi F.S., Szabo Z., Murugappan S., Kaufman H.L. Clinical development of talimogene laherparepvec (T-VEC): a modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev. Anticancer Ther. 2015;15:1389–1403. doi: 10.1586/14737140.2015.1115725. [DOI] [PubMed] [Google Scholar]
- 5.Fukuhara H., Ino Y., Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–1379. doi: 10.1111/cas.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pol J., Buqué A., Aranda F., Bloy N., Cremer I., Eggermont A., Erbs P., Fucikova J., Galon J., Limacher J.M. Trial Watch—oncolytic viruses and cancer therapy. OncoImmunology. 2015;5:e1117740. doi: 10.1080/2162402X.2015.1117740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breitbach C.J., Arulanandam R., De Silva N., Thorne S.H., Patt R., Daneshmand M., Moon A., Ilkow C., Burke J., Hwang T.H. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013;73:1265–1275. doi: 10.1158/0008-5472.CAN-12-2687. [DOI] [PubMed] [Google Scholar]
- 8.Sobol P.T., Boudreau J.E., Stephenson K., Wan Y., Lichty B.D., Mossman K.L. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol. Ther. 2011;19:335–344. doi: 10.1038/mt.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wongthida P., Diaz R.M., Galivo F., Kottke T., Thompson J., Pulido J., Pavelko K., Pease L., Melcher A., Vile R. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 2010;70:4539–4549. doi: 10.1158/0008-5472.CAN-09-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciechonska M., Duncan R. Reovirus FAST proteins: virus-encoded cellular fusogens. Trends Microbiol. 2014;22:715–724. doi: 10.1016/j.tim.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Corcoran J.A., Clancy E.K., Duncan R. Homomultimerization of the reovirus p14 fusion-associated small transmembrane protein during transit through the ER-Golgi complex secretory pathway. J. Gen. Virol. 2011;92:162–166. doi: 10.1099/vir.0.026013-0. [DOI] [PubMed] [Google Scholar]
- 12.Corcoran J.A., Salsman J., de Antueno R., Touhami A., Jericho M.H., Clancy E.K., Duncan R. The p14 fusion-associated small transmembrane (FAST) protein effects membrane fusion from a subset of membrane microdomains. J. Biol. Chem. 2006;281:31778–31789. doi: 10.1074/jbc.M602566200. [DOI] [PubMed] [Google Scholar]
- 13.Dawe S., Corcoran J.A., Clancy E.K., Salsman J., Duncan R. Unusual topological arrangement of structural motifs in the baboon reovirus fusion-associated small transmembrane protein. J. Virol. 2005;79:6216–6226. doi: 10.1128/JVI.79.10.6216-6226.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parmar H.B., Barry C., Kai F., Duncan R. Golgi complex-plasma membrane trafficking directed by an autonomous, tribasic Golgi export signal. Mol. Biol. Cell. 2014;25:866–878. doi: 10.1091/mbc.E13-07-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Racine T., Hurst T., Barry C., Shou J., Kibenge F., Duncan R. Aquareovirus effects syncytiogenesis by using a novel member of the FAST protein family translated from a noncanonical translation start site. J. Virol. 2009;83:5951–5955. doi: 10.1128/JVI.00171-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shmulevitz M., Duncan R. A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. EMBO J. 2000;19:902–912. doi: 10.1093/emboj/19.5.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nibert M.L., Duncan R. Bioinformatics of recent aqua- and orthoreovirus isolates from fish: evolutionary gain or loss of FAST and fiber proteins and taxonomic implications. PLoS ONE. 2013;8:e68607. doi: 10.1371/journal.pone.0068607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barry C., Duncan R. Multifaceted sequence-dependent and -independent roles for reovirus FAST protein cytoplasmic tails in fusion pore formation and syncytiogenesis. J. Virol. 2009;83:12185–12195. doi: 10.1128/JVI.01667-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clancy E.K., Duncan R. Reovirus FAST protein transmembrane domains function in a modular, primary sequence-independent manner to mediate cell-cell membrane fusion. J. Virol. 2009;83:2941–2950. doi: 10.1128/JVI.01869-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corcoran J.A., Syvitski R., Top D., Epand R.M., Epand R.F., Jakeman D., Duncan R. Myristoylation, a protruding loop, and structural plasticity are essential features of a nonenveloped virus fusion peptide motif. J. Biol. Chem. 2004;279:51386–51394. doi: 10.1074/jbc.M406990200. [DOI] [PubMed] [Google Scholar]
- 21.Key T., Duncan R. A compact, multifunctional fusion module directs cholesterol-dependent homomultimerization and syncytiogenic efficiency of reovirus p10 FAST proteins. PLoS Pathog. 2014;10:e1004023. doi: 10.1371/journal.ppat.1004023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shmulevitz M., Epand R.F., Epand R.M., Duncan R. Structural and functional properties of an unusual internal fusion peptide in a nonenveloped virus membrane fusion protein. J. Virol. 2004;78:2808–2818. doi: 10.1128/JVI.78.6.2808-2818.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Top D., Read J.A., Dawe S.J., Syvitski R.T., Duncan R. Cell-cell membrane fusion induced by p15 fusion-associated small transmembrane (FAST) protein requires a novel fusion peptide motif containing a myristoylated polyproline type II helix. J. Biol. Chem. 2012;287:3403–3414. doi: 10.1074/jbc.M111.305268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clancy E.K., Barry C., Ciechonska M., Duncan R. Different activities of the reovirus FAST proteins and influenza hemagglutinin in cell-cell fusion assays and in response to membrane curvature agents. Virology. 2010;397:119–129. doi: 10.1016/j.virol.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 25.Salsman J., Top D., Barry C., Duncan R. A virus-encoded cell-cell fusion machine dependent on surrogate adhesins. PLoS Pathog. 2008;4:e1000016. doi: 10.1371/journal.ppat.1000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salsman J., Top D., Boutilier J., Duncan R. Extensive syncytium formation mediated by the reovirus FAST proteins triggers apoptosis-induced membrane instability. J. Virol. 2005;79:8090–8100. doi: 10.1128/JVI.79.13.8090-8100.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corcoran J.A., Duncan R. Reptilian reovirus utilizes a small type III protein with an external myristylated amino terminus to mediate cell-cell fusion. J. Virol. 2004;78:4342–4351. doi: 10.1128/JVI.78.8.4342-4351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stojdl D.F., Lichty B.D., tenOever B.R., Paterson J.M., Power A.T., Knowles S., Marius R., Reynard J., Poliquin L., Atkins H. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 29.Le Boeuf F., Diallo J.S., McCart J.A., Thorne S., Falls T., Stanford M., Kanji F., Auer R., Brown C.W., Lichty B.D. Synergistic interaction between oncolytic viruses augments tumor killing. Mol. Ther. 2010;18:888–895. doi: 10.1038/mt.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bourgeois-Daigneault M.C., Roy D.G., Falls T., Twumasi-Boateng K., St-Germain L.E., Marguerie M., Garcia V., Selman M., Jennings V.A., Pettigrew J. Oncolytic vesicular stomatitis virus expressing interferon-γ has enhanced therapeutic activity. Mol Ther Oncolytics. 2016;3:16001. doi: 10.1038/mto.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gebremeskel S., Clattenburg D.R., Slauenwhite D., Lobert L., Johnston B. Natural killer T cell activation overcomes immunosuppression to enhance clearance of postsurgical breast cancer metastasis in mice. OncoImmunology. 2015;4:e995562. doi: 10.1080/2162402X.2014.995562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khanna C., Hunter K. Modeling metastasis in vivo. Carcinogenesis. 2005;26:513–523. doi: 10.1093/carcin/bgh261. [DOI] [PubMed] [Google Scholar]
- 33.Lichty B.D., Breitbach C.J., Stojdl D.F., Bell J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 34.Stojdl D.F., Lichty B., Knowles S., Marius R., Atkins H., Sonenberg N., Bell J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 35.Melzer M.K., Lopez-Martinez A., Altomonte J. Oncolytic vesicular stomatitis virus as a viro-immunotherapy: defeating cancer with a “hammer” and “anvil.”. Biomedicines. 2017;5:8. doi: 10.3390/biomedicines5010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller J.M., Bidula S.M., Jensen T.M., Reiss C.S. Vesicular stomatitis virus modified with single chain IL-23 exhibits oncolytic activity against tumor cells in vitro and in vivo. Int. J. Interferon Cytokine Mediat. Res. 2010;2010:63–72. doi: 10.2147/ijicmr.s9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obuchi M., Fernandez M., Barber G.N. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J. Virol. 2003;77:8843–8856. doi: 10.1128/JVI.77.16.8843-8856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsburg E., Publicover J., Buonocore L., Poholek A., Robek M., Palin A., Rose J.K. A vesicular stomatitis virus recombinant expressing granulocyte-macrophage colony-stimulating factor induces enhanced T-cell responses and is highly attenuated for replication in animals. J. Virol. 2005;79:15043–15053. doi: 10.1128/JVI.79.24.15043-15053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin E.J., Wanna G.B., Choi B., Aguila D., 3rd, Ebert O., Genden E.M., Woo S.L. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope. 2007;117:210–214. doi: 10.1097/01.mlg.0000246194.66295.d8. [DOI] [PubMed] [Google Scholar]
- 40.Stephenson K.B., Barra N.G., Davies E., Ashkar A.A., Lichty B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012;19:238–246. doi: 10.1038/cgt.2011.81. [DOI] [PubMed] [Google Scholar]
- 41.Ayala-Breton C., Barber G.N., Russell S.J., Peng K.W. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum. Gene Ther. 2012;23:484–491. doi: 10.1089/hum.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergman I., Griffin J.A., Gao Y., Whitaker-Dowling P. Treatment of implanted mammary tumors with recombinant vesicular stomatitis virus targeted to Her2/neu. Int. J. Cancer. 2007;121:425–430. doi: 10.1002/ijc.22680. [DOI] [PubMed] [Google Scholar]
- 43.Muik A., Kneiske I., Werbizki M., Wilflingseder D., Giroglou T., Ebert O., Kraft A., Dietrich U., Zimmer G., Momma S., von Laer D. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J. Virol. 2011;85:5679–5684. doi: 10.1128/JVI.02511-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wollmann G., Drokhlyansky E., Davis J.N., Cepko C., van den Pol A.N. Lassa-vesicular stomatitis chimeric virus safely destroys brain tumors. J. Virol. 2015;89:6711–6724. doi: 10.1128/JVI.00709-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong C.M., Nash L.A., Del Papa J., Poulin K.L., Falls T., Bell J.C., Parks R.J. Expression of the fusogenic p14 FAST protein from a replication-defective adenovirus vector does not provide a therapeutic benefit in an immunocompetent mouse model of cancer. Cancer Gene Ther. 2016;23:355–364. doi: 10.1038/cgt.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diaz R.M., Galivo F., Kottke T., Wongthida P., Qiao J., Thompson J., Valdes M., Barber G., Vile R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 47.Prestwich R.J., Ilett E.J., Errington F., Diaz R.M., Steele L.P., Kottke T., Thompson J., Galivo F., Harrington K.J., Pandha H.S. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. 2009;15:4374–4381. doi: 10.1158/1078-0432.CCR-09-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J., Tai L.H., Ilkow C.S., Alkayyal A.A., Ananth A.A., de Souza C.T., Wang J., Sahi H., Ly L., Lefebvre C. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol. Ther. 2014;22:1320–1332. doi: 10.1038/mt.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gebremeskel S., Johnston B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies. Oncotarget. 2015;6:41600–41619. doi: 10.18632/oncotarget.6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Workenhe S.T., Mossman K.L. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014;22:251–256. doi: 10.1038/mt.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Workenhe S.T., Simmons G., Pol J.G., Lichty B.D., Halford W.P., Mossman K.L. Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy. Mol. Ther. 2014;22:123–131. doi: 10.1038/mt.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown C.W., Stephenson K.B., Hanson S., Kucharczyk M., Duncan R., Bell J.C., Lichty B.D. The p14 FAST protein of reptilian reovirus increases vesicular stomatitis virus neuropathogenesis. J. Virol. 2009;83:552–561. doi: 10.1128/JVI.01921-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciechonska M., Key T., Duncan R. Efficient reovirus- and measles virus-mediated pore expansion during syncytium formation is dependent on annexin A1 and intracellular calcium. J. Virol. 2014;88:6137–6147. doi: 10.1128/JVI.00121-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clements D.R., Sterea A.M., Kim Y., Helson E., Dean C.A., Nunokawa A., Coyle K.M., Sharif T., Marcato P., Gujar S.A., Lee P.W. Newly recruited CD11b+, GR-1+, Ly6C(high) myeloid cells augment tumor-associated immunosuppression immediately following the therapeutic administration of oncolytic reovirus. J. Immunol. 2015;194:4397–4412. doi: 10.4049/jimmunol.1402132. [DOI] [PubMed] [Google Scholar]
- 55.Hastie E., Grdzelishvili V.Z. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 2012;93:2529–2545. doi: 10.1099/vir.0.046672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin E.J., Chang J.I., Choi B., Wanna G., Ebert O., Genden E.M., Woo S.L. Fusogenic vesicular stomatitis virus for the treatment of head and neck squamous carcinomas. Otolaryngol. Head Neck Surg. 2007;136:811–817. doi: 10.1016/j.otohns.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 57.Bateman A.R., Harrington K.J., Kottke T., Ahmed A., Melcher A.A., Gough M.J., Linardakis E., Riddle D., Dietz A., Lohse C.M. Viral fusogenic membrane glycoproteins kill solid tumor cells by nonapoptotic mechanisms that promote cross presentation of tumor antigens by dendritic cells. Cancer Res. 2002;62:6566–6578. [PubMed] [Google Scholar]
- 58.Ebert O., Shinozaki K., Kournioti C., Park M.S., García-Sastre A., Woo S.L. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res. 2004;64:3265–3270. doi: 10.1158/0008-5472.can-03-3753. [DOI] [PubMed] [Google Scholar]
- 59.Lin E.H., Salon C., Brambilla E., Lavillette D., Szecsi J., Cosset F.L., Coll J.L. Fusogenic membrane glycoproteins induce syncytia formation and death in vitro and in vivo: a potential therapy agent for lung cancer. Cancer Gene Ther. 2010;17:256–265. doi: 10.1038/cgt.2009.74. [DOI] [PubMed] [Google Scholar]
- 60.Nakamori M., Fu X., Rousseau R., Chen S.Y., Zhang X. Destruction of nonimmunogenic mammary tumor cells by a fusogenic oncolytic herpes simplex virus induces potent antitumor immunity. Mol. Ther. 2004;9:658–665. doi: 10.1016/j.ymthe.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 61.Guedan S., Grases D., Rojas J.J., Gros A., Vilardell F., Vile R., Mercade E., Cascallo M., Alemany R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012;19:1048–1057. doi: 10.1038/gt.2011.184. [DOI] [PubMed] [Google Scholar]
- 62.Errington F., Jones J., Merrick A., Bateman A., Harrington K., Gough M., O’Donnell D., Selby P., Vile R., Melcher A. Fusogenic membrane glycoprotein-mediated tumour cell fusion activates human dendritic cells for enhanced IL-12 production and T-cell priming. Gene Ther. 2006;13:138–149. doi: 10.1038/sj.gt.3302609. [DOI] [PubMed] [Google Scholar]
- 63.Sutter G., Ohlmann M., Erfle V. Non-replicating vaccinia vector efficiently expresses bacteriophage T7 RNA polymerase. FEBS Lett. 1995;371:9–12. doi: 10.1016/0014-5793(95)00843-x. [DOI] [PubMed] [Google Scholar]
- 64.Le Bœuf F., Batenchuk C., Vähä-Koskela M., Breton S., Roy D., Lemay C., Cox J., Abdelbary H., Falls T., Waghray G. Model-based rational design of an oncolytic virus with improved therapeutic potential. Nat. Commun. 2013;4:1974. doi: 10.1038/ncomms2974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.