

Abstract
Purpose
EZH2 and EZH1, the catalytic components of polycomb repressive complex 2 (PRC2), trigger trimethylation of H3K27 (H3K27me3) to repress the transcription of target genes and are implicated in the pathogenesis of various cancers including multiple myeloma and prostate cancer. Here, we investigated the preclinical effects of UNC1999, a dual inhibitor of EZH2 and EZH1, in combination with proteasome inhibitors on multiple myeloma and prostate cancer.
Experimental Design
In vitro and in vivo efficacy of UNC1999 and the combination with proteasome inhibitors was evaluated in multiple myeloma cell lines, primary patient cells, and in a xenograft model. RNA-seq and ChIP-seq were performed to uncover the targets of UNC1999 in multiple myeloma. The efficacy of the combination therapy was validated in prostate cancer cell lines.
Results
Proteasome inhibitors repressed EZH2 transcription via abrogation of the RB-E2F pathway, thereby sensitizing EZH2-dependent multiple myeloma cells to EZH1 inhibition by UNC1999. Correspondingly, combination of proteasome inhibitors with UNC1999, but not with an EZH2-specific inhibitor, induced synergistic antimyeloma activity in vitro. Bortezomib combined with UNC1999 remarkably inhibited the growth of myeloma cells in vivo. Comprehensive analyses revealed several direct targets of UNC1999 including the tumor suppressor gene NR4A1. Derepression of NR4A1 by UNC1999 resulted in suppression of MYC, which was enhanced by the combination with bortezomib, suggesting the cooperative blockade of PRC2 function. Notably, this combination also exhibited strong synergy in prostate cancer cells.
Conclusions
Our results identify dual inhibition of EZH2 and EZH1 together with proteasome inhibition as a promising epigenetics-based therapy for PRC2-dependent cancers.
Introduction
Polycomb repressive complex 2 (PRC2) represses the transcription of target genes through its catalytic components: enhancer of zeste homolog 2 (EZH2) and its homolog EZH1, catalyzing trimethylation of H3K27 (H3K27me3) (1). This repressive mark is removed by the histone demethylases ubiquitously transcribed tetratricopeptide repeat X chromosome (UTX; also known as KDM6A) and jumonji domain-containing protein 3 (JMJD3; also known as KDM6B) (2). PRC2 is closely linked to the control of stem cells and tumorigenesis (3). Specifically, EZH2 is frequently deregulated in cancer, and its overexpression was reported in solid malignancies such as prostate cancer (4) as well as in hematologic malignancies such as lymphoma in which EZH2-activating mutations were identified (5), suggesting that EZH2 is a new potential therapeutic target. Indeed, clinical trials of EZH2 inhibitors in a variety of cancers are ongoing (6). Recently, dual inhibitors of EZH2 and EZH1 have been developed to conquer PRC2-dependent cancers (7, 8). Noteworthy, it was shown that UNC1999 (9), a novel small-molecule inhibitor that is active against EZH2 (both wild-type and mutant) and EZH1, has a promising preclinical activity against MLL-rearranged leukemia in vitro and in vivo (7).
Multiple myeloma, which accounts for more than 1% of all cancer-related deaths (10), remains an incurable disease, thereby emphasizing the need for novel therapeutic approaches to improve patient outcome (11). In multiple myeloma, EZH2 overexpression correlates with the progression from monoclonal gammopathy of undetermined significance (MGUS) to multiple myeloma (12). Significantly, the identification of inactivating mutations in UTX in 10% of myeloma samples underscores the important role of H3K27me3 in myelomagenesis (13). Moreover, EZH2 knockdown using siRNA leads to the inhibition of multiple myeloma cell growth (14). These data propose a possible use of EZH2 inhibitors in the treatment of multiple myeloma. Indeed, a few reports of the use of EZH2 inhibitors in multiple myeloma have recently emerged with varying degrees of success (15–17). However, neither detailed molecular mechanisms of action of the novel agents nor the combination with currently available agents in vitro and in vivo has been thoroughly investigated. In the last decade, proteasome inhibitors such as bortezomib and carfilzomib together with other innovative therapeutics have dramatically improved the life expectancy of multiple myeloma patients (11). Despite this breakthrough, patients eventually develop resistance to treatment, therefore combining proteasome inhibitors with novel agents is one option to improve patient outcome (18). Whether the combination of proteasome inhibitors and novel PRC2 inhibitors constitutes a new therapeutic strategy has not yet been explored.
Prostate cancer, a leading cause of death in men (10), is another malignancy in which EZH2 plays a crucial role through its part as the catalytic unit of PRC2 (4) and also through PRC2-independent coactivation of transcription factors such as androgen receptor (19). The success of bortezomib in multiple myeloma increased the interest in using it in nonhematologic malignancies (20). Thus, several phase I/II clinical trials were conducted using bortezomib alone and in combination with other agents for the treatment of prostate cancer; however, these studies reported only moderate to no improvement in patient outcome (21).
In this study, we investigated the potential of the dual inhibition of EZH2 and EZH1 together with proteasome inhibitors as a novel mechanistic approach for the treatment of PRC2-dependent tumors such as multiple myeloma and prostate cancer.
Materials and Methods
Human samples from patients and healthy volunteers
Multiple myeloma cells and bone marrow stromal cells (BMSC) were collected from the bone marrow of newly diagnosed multiple myeloma patients at Chiba University Hospital. All patients provided written informed consent in accordance with the declaration of Helsinki, and patient anonymity was ensured. This study was approved by the Institutional Review Committee at Chiba University (Approval #532). Plasma cells were purified, and BMSCs were generated as previously described (22, 23). Peripheral blood samples collected from healthy volunteers were processed by Ficoll-Paque (GE Healthcare) gradient to obtain peripheral blood mononuclear cells.
Murine xenograft models of human multiple myeloma
Male NOD/Shi-scid, IL-2RgKOJic (NOG) mice were purchased from CLEA Japan Inc. Animal studies using MM.1S xenograft model were conducted according to Chiba University guidelines for the use of laboratory animals and approved by the Review Board for Animal Experiments of Chiba University (approval ID: 27-213). For single-agent UNC1999 model, mice were inoculated subcutaneously in the right flank with 5 × 106 MM.1S cells in 100 μL RPMI1640. After detection of tumors, mice were treated for 3 weeks with 25 mg/kg intraperitoneal UNC1999 twice a week (n = 7). A vehicle control group (n = 10) received intraperitoneal vehicle (5% DMSO in corn oil).
For the combination xenograft model, mice were inoculated subcutaneously in the right flank with 4 × 106 MM.1S cells in 100 μL RPMI1640. After detection of tumors, mice were treated for 5 weeks with 15 mg/kg intraperitoneal UNC1999 3 days a week (n = 14); 0.5 mg/kg subcutaneous bortezomib (Velcade) in the left flank twice a week (n = 13); or 15 mg/kg intraperitoneal UNC1999 3 days a week and 0.5 mg/kg subcutaneous bortezomib twice a week (n = 13). A vehicle control group received intraperitoneal vehicle (5% DMSO in corn oil) and subcutaneous saline (n = 14).
For all mice groups, tumor volume was calculated from caliper measurements every 3 to 4 days until day of first death in each group; mice were sacrificed when tumors reached 2,000 cm3 or were ulcerated. Survival was evaluated from the first day of treatment until death.
Reagents
UNC1999 was produced at Icahn School of Medicine at Mount Sinai (9) and was diluted in DMSO to a stock of 10 mmol/L for cell culture experiments. For in vivo experiments, UNC1999 was slowly dissolved in 5% DMSO in corn oil with vigorous vortex followed by rotation to achieve a homogenous suspension of 60 or 100 mg/mL. GSK126 was purchased from CHEMIETEK and was diluted in DMSO to a stock of 20 mmol/L. Bortezomib and carfilzomib for cell culture experiments were obtained from Selleck Chemicals and were diluted in DMSO to stocks of 100 μmol/L. Bortezomib was purchased from Janssen Pharmaceutical KK for in vivo experiments and was diluted in normal saline to a 1 mg/mL stock. MG132 was obtained from Cayman Chemical and was diluted in DMSO to a stock of 10 mmol/L.
RNA-seq library construction and sequencing analysis
Total RNA was purified from 2 × 106 MM.1S cells using the RNeasy plus Micro Kit (Qiagen). RNA concentration and integrity were verified using Agilent 2100 Bioanalyzer. Amplification, construction of the libraries, and sequencing were performed as previously described (24). TopHat (version 1.3.2; with default parameters) was used to align to the human reference genome (hg19 from University of California, Santa Cruz Genome Browser; http://genome.ucsc.edu/). Then, gene expression values were calculated as reads per kilobase of exon unit per million mapped reads (RPKM) using cufflinks (version 2.0.2).
Chromatin immunoprecipitation sequencing
Chromatin immunoprecipitation (ChIP) was performed using a previously described protocol (24), with the following modifications: MM.1S cells were digested with micrococcal nuclease (MNase). DNA libraries were prepared from 3 ng immunoprecipitated DNA and input samples using a ThruPLEX DNA-seq Kit (Rubicon Genomics) according to the manufacturer’s instructions.
Accession numbers
RNA-sequencing (RNA-seq) and ChIP-sequencing (ChIP-seq) data obtained in this study were deposited in DNA Data Bank of Japan (DDBJ; DDBJ, accession number DRA004880).
Statistical analysis
Statistical significance of difference was measured by unpaired two-tailed Student t test or Welch test when the variance was judged as significantly different. P values less than 0.05 were considered significant, using StatMate III version 3.18. Survival was assessed using Kaplan–Meier curves (Graph Pad Prism, version 4) and log-rank analysis using StatMate III version 3.18. The Kruskal–Wallis test was used when more than two groups needed to be compared. Pearson’s product–moment correlation was utilized to determine the presence of correlation. The combined effect of UNC1999 with bortezomib or carfilzomib was analyzed by isobologram analysis using the Compu-Syn software program (ComboSyn, Inc.; ref. 25).
Methods for human cell lines, supplementary reagents, immunoblot analysis, assay of apoptosis, assays of cytotoxicity, quantitative reverse transcription PCR (RT-PCR), ChIP assays, gene set enrichment analysis (GSEA), vectors, and luciferase assay are detailed in the Supplementary Methods on the Clinical Cancer Research website.
Results
UNC1999, a dual inhibitor of EZH2 and EZH1, inhibits the growth of multiple myeloma cells
Confirming a previous report by an siRNA method (14), knockdown of EZH2 by shRNA using lentiviral vectors leads to growth of inhibition in H929 myeloma cells following reduction in H3K27me3 levels with no significant change in H3K36me2 and H3K4me3 histone modifications (Fig. 1A–C; Supplementary Fig. S1A), suggesting EZH2 dependency in multiple myeloma. Interestingly, we found that EZH1 knockdown by shRNA also induced growth inhibition in H929 cells, albeit less severe than that observed with EZH2 knockdown (Supplementary Fig. S1B). These data indicate that both EZH2 and EZH1 are essential for the growth of multiple myeloma cells. To examine the impact of pharmacologic inhibition of PRC2 activity on multiple myeloma cells, we used UNC1999 (7, 9) which exerts dual inhibition of the enzymatic activities of EZH2 and EZH1 (IC50, EZH2 <10 nmol/L; EZH1 45 nmol/L). UNC1999 potently inhibited the growth of several multiple myeloma cell lines including drug-resistant cells (DOX40) in a dose- and time-dependent manner (Fig. 1D; Supplementary Fig. S1C and S1D). Moreover, UNC1999 treatment resulted in a significant concentration- and time-dependent reduction in H3K27me3 level with little to no effect on EZH2 protein level nor on H3K36me2 and H3K4me3 histone modifications (Fig. 1E; Supplementary Fig. S1E). Importantly, UNC1999 induced significant cytotoxicity in CD138+ bone marrow plasma cells (BMPC) of multiple myeloma patients (Fig. 1F). When tested on mononuclear cells from healthy donors, UNC1999 caused minimal cytotoxicity at concentrations up to 5 μmol/L. (Supplementary Fig. S1F).
Figure 1.
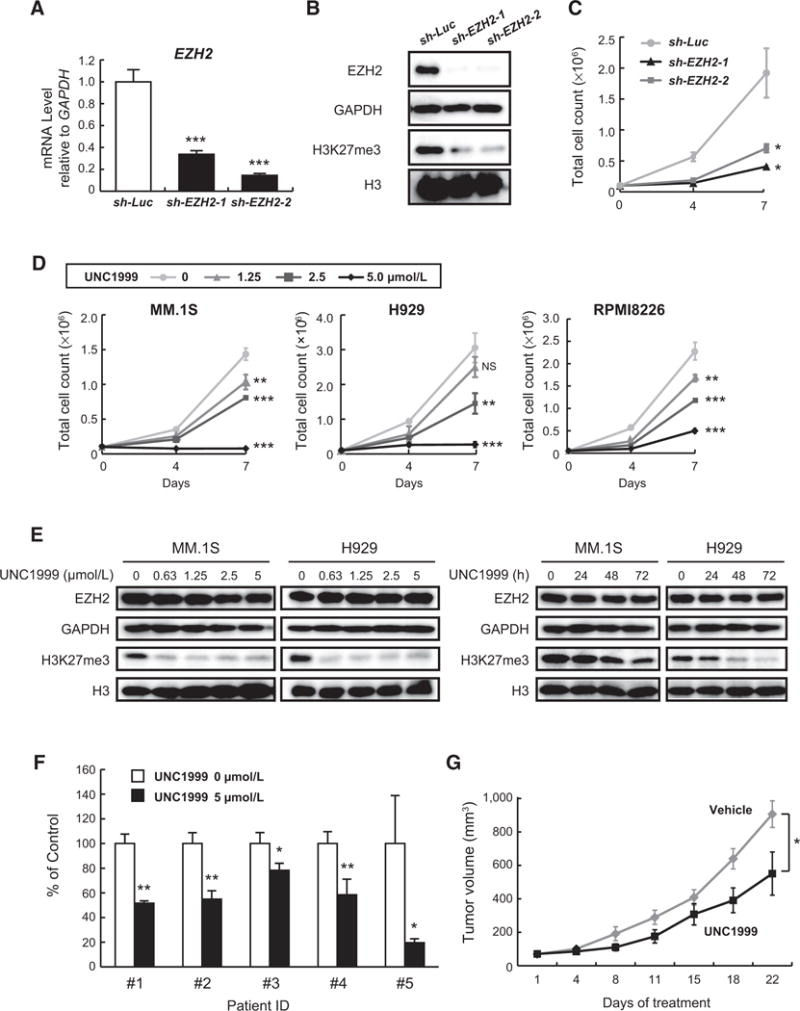
UNC1999 blocks the trimethylation of H3K27 with subsequent inhibition of the growth of multiple myeloma cells. H929 cells transduced with the indicated lentiviruses were selected by cell sorting for GFP expression and subjected to (A) quantitative RT-PCR analysis of EZH2 mRNA expression. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. B, Immunoblot analysis for the indicated proteins. GAPDH and H3 served as loading controls. C, Cell proliferation assay of H929 cells transduced with the indicated lentiviruses. Cell counting was performed using Trypan blue on the indicated days of cultures. Data represent mean ± SD of triplicate cultures. D, Cell proliferation assays of MM.1S, H929, and RPMI8226 human myeloma cell lines treated with a range of concentrations of UNC1999 for 4 or 7 days. Cell counting was performed using Trypan blue staining. The y-axis is presented as the mean cell count ± SD of triplicate cultures. E, Immunoblot analyses for EZH2 and H3K27me3 after treatment of multiple myeloma cells with a range of concentrations of UNC1999 for 72 hours (left) or 5 μmol/L of UNC1999 for the indicated times (right). H3 and GAPDH served as loading controls. F, MTS assay showing viability of primary multiple myeloma cells isolated from patients treated with 5 μmol/L of UNC1999 for 48 hours relative to untreated control. Data represent mean ± SD (n = 2~4). G, In vivo analysis using murine xenograft model of human myeloma MM.1S cells inoculated into the flanks of NOG mice. When the tumor volume reached approximately 50 mm3, UNC1999 was administered intraperitoneally (25mg/kg), twice per week (n = 7) for 21 days. Tumor volume was calculated from caliper measurements and compared with vehicle-treated tumors (n = 10) at the indicated time points. Data represent mean ± SE. *, P < 0.05; **, P < 0.01;***, P < 0.001; NS, not significant.
To determine the impact of dual inhibition of EZH2 and EZH1 on multiple myeloma cells in vivo, we treated MM.1S xenograft–bearing NOG mice with 25 mg/kg of UNC1999 intraperitoneally (IP) twice a week for 3 weeks. The growth of xenografts treated with UNC1999 was significantly reduced compared with control (P < 0.05 on day 22; Fig. 1G). Taken together, these data show that PRC2 components EZH2 and EZH1 are valid therapeutic targets in multiple myeloma both in vitro and in vivo.
Bortezomib transcriptionally downregulates EZH2 via E2F inactivation
A recent study showed that bortezomib reduces EZH2 protein (26), which prompted us to thoroughly investigate the impact of proteasome inhibitors on EZH2. Interestingly, bortezomib downregulated not only EZH2 protein but also its mRNA in multiple myeloma cells in dose- and time-dependent manners (Fig. 2A–C). This was also observed using two other proteasome inhibitors, namely carfilzomib and MG132 (Fig. 2D and E; Supplementary Fig. S2A and S2B). However, EZH2 protein in bortezomib-resistant cell line KMS11/BTZ was not down regulated by bortezomib compared with the parental cell line KMS11 (Supplementary Fig. S2C and S2D), suggesting a possible role of EZH2 downregulation in bortezomib-induced cytotoxicity.
Figure 2.
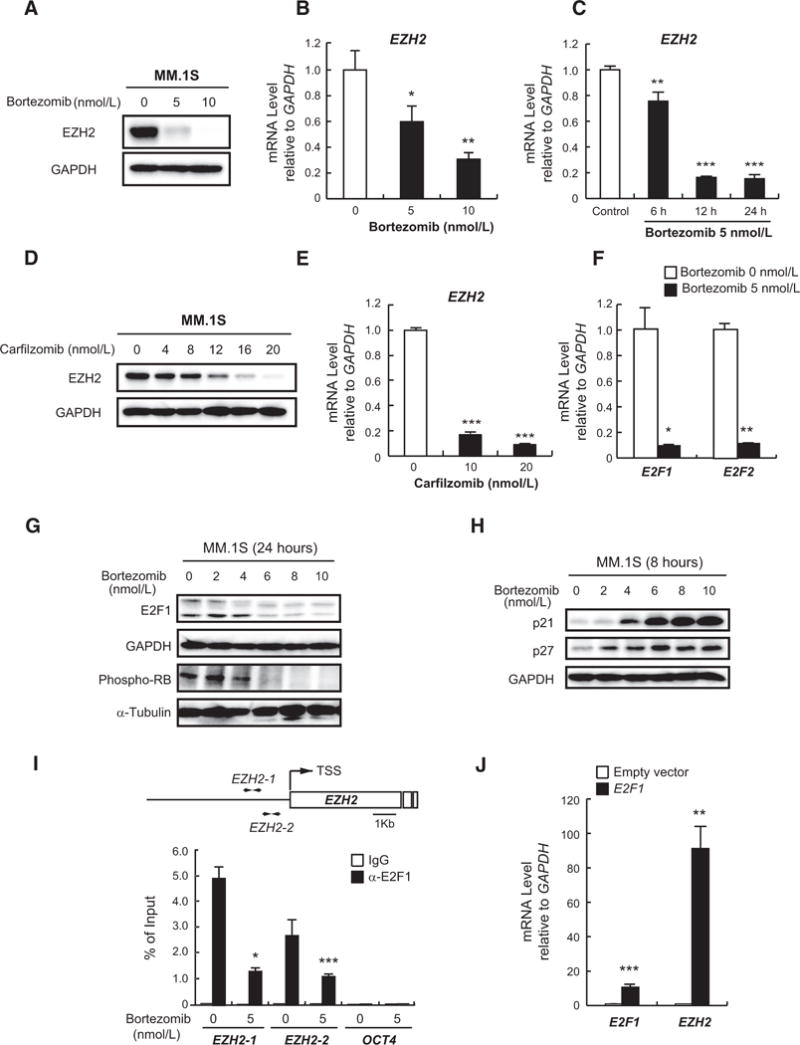
Bortezomib down regulates EZH2 via E2F inactivation. A, Immunoblot analysis for EZH2 after treatment of MM.1S cells with the indicated concentrations of bortezomib for 24 hours. GAPDH served as a loading control. B and C, Quantitative RT-PCR analysis of EZH2 mRNA expression in MM.1S cells treated with the indicated doses of bortezomib for 8 hours (B) or 5 nmol/L of bortezomib for the indicated times (C). The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. D, Immunoblot analysis of EZH2 in MM.1S cells treated with the indicated doses of carfilzomib for 24 hours. GAPDH served as a loading control. E, Quantitative RT-PCR analysis of EZH2 mRNA expression in MM.1S cells treated with the indicated doses of carfilzomib for 12 hours. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. F, Quantitative RT-PCR analysis of E2F1 and E2F2 mRNA expression in MM.1S cells treated with 5 nmol/L of bortezomib for 12 hours. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. G and H, Immunoblot analyses for the indicated proteins in MM.1S cells upon (G) 24-hour or (H) 8-hour treatment with a range of concentrations of bortezomib. GAPDH and α-tubulin served as loading controls. I, ChIP analysis for E2F1 occupancy on EZH2 promoter in MM.1S cells treated with 5 nmol/L of bortezomib or DMSO for 24 hours. OCT4 was used as a negative control. Values correspond to mean percentage of input enrichment ± SD of triplicate qPCR reactions of a single replicate. Schematic representation of the location of the primer sets is depicted (top). J, Quantitative RT-PCR analysis of mRNA expression of E2F1 and EZH2 in H929 cells transduced with E2F1-overexpressing or empty vectors. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates.*, P < 0.05;**, P < 0.01; and***, P < 0.001.
To dissect the mechanism underlying the repression of EZH2 by bortezomib, we focused on the RB-E2F pathway as EZH2 is a downstream target of E2F transcription factors (27). We found that bortezomib down regulated E2F1 and E2F2 mRNA as well as E2F1 and phosphorylated RB proteins, suggesting that the RB-E2F pathway was abrogated by bortezomib (Fig. 2F and G). It is known that bortezomib blocks the degradation of cyclin-dependent kinase inhibitors (CDKI), resulting in their accumulation and subsequent induction of cell-cycle arrest (28). Our data confirmed that CDKIs p21 and p27 were stabilized by bortezomib (Fig. 2H). We next confirmed the previous finding that E2F1 transactivates EZH2 promoter (27) using luciferase reporter assay (Supplementary Fig. S3A). Moreover, ChIP assays in MM.1S cells revealed that bortezomib significantly inhibited the binding of E2F1 to EZH2 promoter (Fig. 2I). Notably, overexpression of E2F1 leads to remarkable upregulation of EZH2 in H929 cells (Fig. 2J). In addition, E2F inhibitor, HLM006474, significantly down regulated EZH2 mRNA confirming that EZH2 repression by bortezomib is mediated through E2F1 (Supplementary Fig. S3B). These results indicate that bortezomib treatment leads to accumulation of CDKIs p21 and p27 with subsequent reduction of RB phosphorylation, resulting in inactivation of E2F family, which in turn down regulates EZH2 in multiple myeloma cells.
UNC1999 enhances cytotoxicity induced by proteasome inhibitors
To determine if EZH2 plays a role in the response and/or resistance to bortezomib, we next studied the gene expression of pretreatment samples from multiple myeloma patients enrolled on the APEX 039 clinical study who received bortezomib treatment (29). Our analysis using this database revealed that patients with higher levels of EZH2 expression have poorer response to bortezomib (Supplementary Fig. S4A and S4B). Furthermore, EZH2 overexpression using a lentiviral vector in RPMI8226 and H929 cells conferred resistance to bortezomib compared with cells transduced with an empty vector (Fig. 3A; Supplementary Fig. S5A). Interestingly, RPMI8226 cells transduced with EZH2 lentivirus vector gained growth advantage over those transduced with the empty vector (Supplementary Fig. S5B). To unveil the molecular mechanism underlying EZH2-dependent bortezomib resistance, we performed RNA-seq of RPMI8226 cells transduced with EZH2-overexpressing or empty vectors. Analysis of RNA-seq data showed that MYC targets, ribosome, oxidative phosphorylation, and G1–S transition gene sets were strongly enriched in RPMI8226 cells overexpressing EZH2 (Supplementary Fig. S5C; Supplementary Table S5). These aggressive features of multiple myeloma cells overexpressing EZH2 might be associated with acquired resistance to bortezomib. Importantly, UNC1999 treatment overcame this resistance to bortezomib conferred by EZH2 (Fig. 3A; Supplementary Fig. S5A), clearly supporting the rationale of the combination of UNC1999 and proteasome inhibitors such as bortezomib in multiple myeloma cells.
Figure 3.
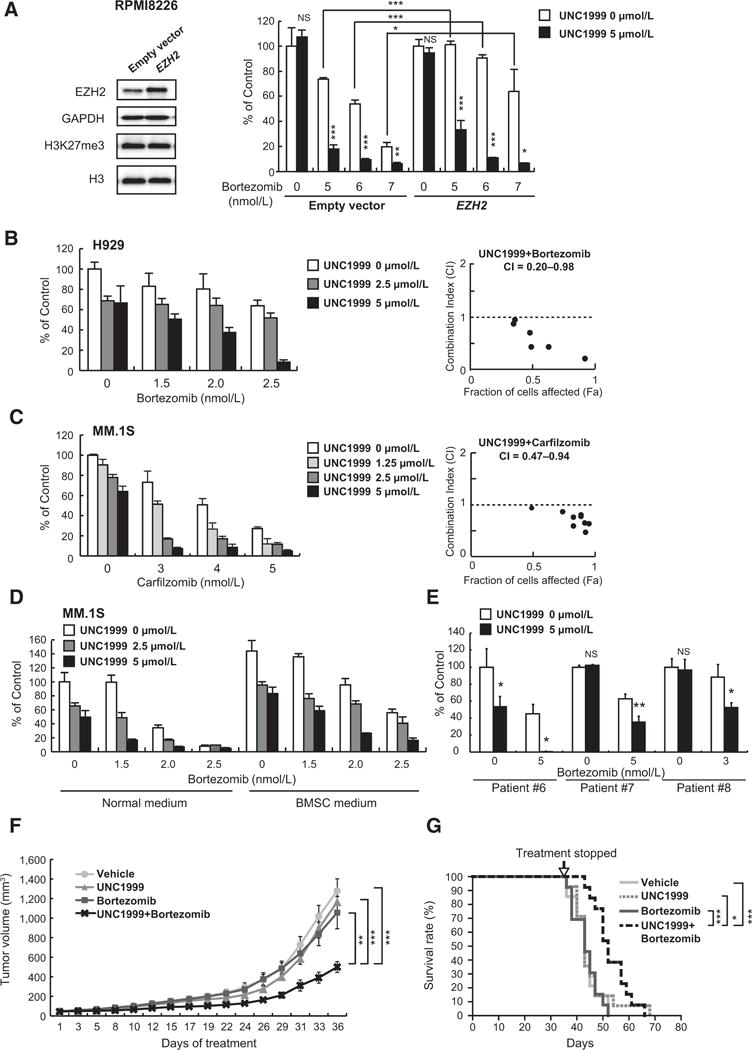
UNC1999 augments bortezomib-induced cytotoxicity both in vitro and in vivo. A, MTS assay showing the viability of RPMI8226 cells transduced with EZH2-overexpressing or empty vectors upon treatment with 5 μmol/L of UNC1999 (72 hours) and the indicated doses of bortezomib (last 48 hours) relative to untreated cells. Data represent mean ± SD of triplicates. Immunoblot analyses of the indicated proteins in EZH2-overexpressing or empty vector–transduced RPMI8226 cells are shown to the left of the graph. GAPDH and H3 served as loading controls. B and C, MTS assays showing viability of (B) H929 and (C) MM.1S cells upon treatment with the indicated doses of UNC1999 (72 hours) in combination with the indicated doses of bortezomib (B) or carfilzomib (C) (last 48 hours), respectively, relative to untreated control. Data represent mean ± SD of triplicates. Calculation of CI is shown to the right of each graph. D, MTS assay showing the viability of MM.1S cells upon treatment with the indicated doses of UNC1999 (72 hours) in combination with the indicated doses of bortezomib (last 48 hours) in the presence of normal medium or BMSC medium, relative to untreated control in normal medium. Data represent mean ± SD of triplicates. E, MTS assay showing the viability of primary multiple myeloma cells isolated from patients treated simultaneously with 5 μmol/L of UNC1999 and the indicated doses of bortezomib for 48 hoursrelative to untreated control. Data represent mean ± SD (n = 3~4). F, MM.1S cells were inoculated into the flanks of NOG mice. When the tumor volume reached approximately 40 mm3, mice were segregated into groups that received either vehicle (n = 14), 15 mg/kg of UNC1999 intraperitoneally 3 times per week (n = 14), 0.5 mg/kg of bortezomib subcutaneously twice per week (n = 13), or the combination of UNC1999 and bortezomib (n = 13) for 5 weeks. Tumor volume was calculated from caliper measurements at the indicated time points. Data represent mean ± SE. G, Kaplan–Meier survival analysis of mice described in F. Statistical significance of survival difference was determined by the log-rank test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and NS, not significant.
We found that UNC1999 enhanced the cytotoxicity induced by bortezomib or carfilzomib in multiple myeloma cell lines with significant suppression of EZH2 and H3K27me3 (Fig. 3B and C; Supplementary Fig. S6A–S6D), even in the presence of conditioned media derived from BMSCs to mimic the microenvironment of the bone marrow (Fig. 3D). Moreover, the enhanced cytotoxicity was similarly observed in DOX40 and KMS11/BTZ cell lines which are resistant to doxorubicin and bortezomib, respectively (Supplementary Fig. S6B and S6D). These results were validated using the combination index (CI; ref.25), which confirmed the synergism between UNC1999 and bortezomib or carfilzomib (CI < 1.0; Fig. 3B and C; Supplementary Fig. S6A and S6B; and Supplementary Table S1A–S1E). In addition, UNC1999 enhanced the cytotoxicity induced by bortezomib in CD138+ BMPCs derived from multiple myeloma patients (Fig. 3E). In contrast, minimal cytotoxicity was observed in peripheral blood mononuclear cells isolated from healthy donors treated with the combination of UNC1999 and bortezomib (Supplementary Fig. S6E).
To elucidate the mechanisms of the cytotoxicity induced by this combination, we conducted flow cytometric analysis using Annexin V staining and found that the percentage of apoptotic cells had dramatically increased with the combination (Supplementary Fig. S7A). We also confirmed the induction of apoptosis as evidenced by the cleavage of caspases-3, -8, -9, and PARP in multiple myeloma cell lines treated with the combination (Supplementary Fig. S7B). These results indicate that increased apoptosis is one mechanism through which UNC1999 enhances the cytotoxicity induced by bortezomib in multiple myeloma cells.
We further investigated the efficacy of combined treatment of UNC1999 (15 mg/kg IP 3 times a week) and low-dose subcutaneous bortezomib (0.5 mg/kg twice a week) in MM.1S xenograft model in NOG mice for 5 weeks. Notably, the combination significantly reduced the size of the tumors as compared with either single agent (Fig. 3F). Importantly, the combination treatment significantly prolonged the survival of mice without overt weight loss (Fig. 3G; Supplementary Fig. S7C). These results illustrate that UNC1999 enhances bortezomib-induced cytotoxicity of myeloma cells not only in vitro but also in vivo.
Genome-wide analyses unveil the UNC1999-target genes in multiple myeloma cells
To explore the genome-wide effects and target genes of UNC1999 and the combination with bortezomib, we performed RNA-seq of MM.1S cells treated with 5 μmol/L of UNC1999 for 72 hours, 5 nmol/L of bortezomib for 48 hours, or the combination of both agents (UNC1999 for 72 hours with bortezomib in the last 48 hours) versus DMSO-treated cells, and ChIP-seq for H3K27me3 of UNC1999 versus DMSO-treated control cells of the same experiment. We defined “PRC2 targets” as those with H3K27me3 enrichment greater than 2.0-fold over the input signal at the promoter region (transcriptional start site ± 2.0 kb) in MM.1S cells (Fig. 4A). ChIP-seq revealed that H3K27me3 levels at the promoters significantly decreased following UNC1999 treatment. Within PRC2 target genes, we defined genes that showed more than 2-fold reduction in H3K27me3 levels compared with DMSO-treated cells as “UNC1999 target genes” (Fig. 4A). GSEA using our RNA-seq data confirmed that PRC2 target gene sets were significantly enriched in both UNC1999- and combination-treated cells (Fig. 4B; Supplementary Tables S2 and S3). Correspondingly, the expression of PRC2 target genes was significantly elevated in UNC1999- and combination-treated MM.1S cells as compared with DMSO-treated control cells (Fig. 4C). Among the PRC2 target genes, we selected 74 genes with significantly enhanced expression (>1.5-fold UNC1999/Control) and remarkable reduction of H3K27me3 (≥2-fold) upon UNC1999 treatment (Fig. 4D and Supplementary Table S4) as major UNC1999 target genes in MM.1S cells. These genes included nuclear receptor transcription factor, NR4A1 (also known as NUR77), cell growth–, differentiation-, and apoptosis-related, EGR1, the cell-cycle regulator, CDKN1C (also known as p57, KIP2), and component of NF-κB, LTB. Although LTB and EGR1 are novel tumor-suppressor candidates in multiple myeloma (30, 31), CDKN1C is known as a direct target of EZH2 (32) and a tumor suppressor with prognostic value in several cancers (33).
Figure 4.
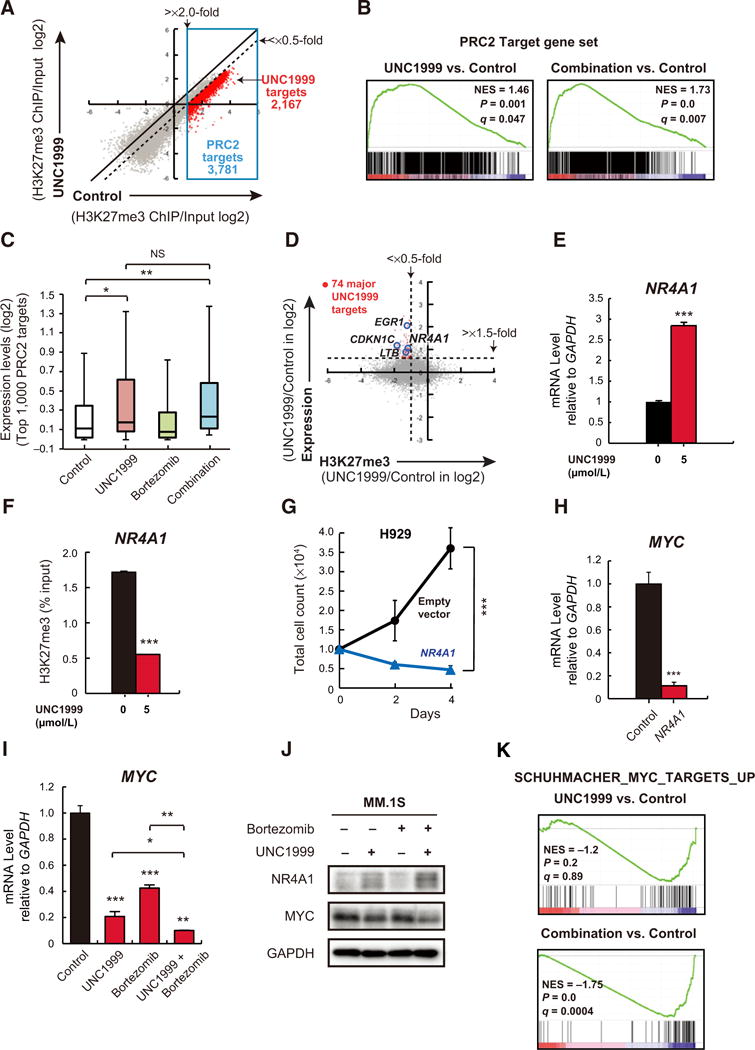
Identification of UNC1999-target genes in MM.1S cells. A, A scatter plot showing the correlation of the fold enrichment values (ChIP/input; TSS ± 2.0 kb) of H3K27me3 against the input signals of RefSeq genes between DMSO (control)- and UNC1999-treated MM.1S cells. Dotted diagonal line indicates 2-fold change. Blue box indicates PRC2 target genes (3,781 genes) with greater than 2-fold enrichment in H3K27me3 mark in the control. Red dots represent UNC1999 target genes with more than 2-fold reduction in H3K27me3 levels (2,167 genes). B, Gene set enrichment plots for PRC2 target genes defined in (A) in MM.1S cells treated with UNC1999 alone (left) and combination (right) versus DMSO-treated control cells. NES, normalized enrichment score; q, FDR q value; P, P value. C, Box-and-whisker plots showing the expression changes of the top 1,000 PRC2 target genes defined in (A) in DMSO-, UNC1999-, bortezomib-, and combination-treated MM.1S cells. Boxes represent 25 to 75 percentile ranges. Vertical lines represent 10 to 90 percentile ranges. Horizontal bars represent median. D, A scatter plot showing the correlation between the expression of genes and H3K27me3 levels in UNC1999- vs. DMSO-treated control MM.1S cells. Red dots indicate major UNC1999 target genes with significantly enhanced expression (>1.5-fold UNC1999/Control). Representative genes are highlighted. E, Quantitative RT-PCR analysis of NR4A1 mRNA expression in MM.1S cells following treatment with 5 μmol/L of UNC1999 for 72 hours. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. F, ChIP analysis for H3K27me3 occupancy loss in promoter regions (TSS ± 2 kb) of NR4A1 in MM.1S cells treated with 5 μmol/L of UNC1999 for 72 hours versus DMSO-treated cells. Values correspond to mean percentage of input enrichment ± SD of triplicate qPCR reactions of a single replicate. G, Cell proliferation assay of H929 cells (10,000 cells per well in 96-well plate) transduced with the indicated retroviruses for 48 hours prior to cell sorting for GFP expression. Cell counting was performed using Trypanblue atthe indicated times. The y-axis is presented as the mean cell number ± SD of quadruplicates. H, Quantitative RT-PCR analysis of MYC mRNA expression in H929 cells transduced with NR4A1-overexpressing or empty vectors. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. I, Quantitative RT-PCR analysis of MYC mRNA expression in MM.1S cells upon 72 hours of 5 μmol/L of UNC1999 and/or 5 nmol/L of bortezomib treatment for the last 12 hours. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. J, Immunoblot analysis for the indicated proteins in MM.1S cells upon 72 hours of 5 μmol/L of UNC1999 and/or 5 nmol/L of bortezomib treatment for the last 12 hours. GAPDH was used as a loading control. K, A representative MYC target gene set significantly enriched in MM.1S cells treated with UNC1999 alone (top) and combination (bottom) versus DMSO-treated cells. NES, normalized enrichment score; q, FDR q value; P, P value. *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
UNC1999-induced upregulation of NR4A1 suppresses MYC with resultant multiple myeloma growth suppression
Among the major UNC1999 target genes, we focused on NR4A1. The upregulation of NR4A1 by UNC1999 was confirmed by RT-PCR (Fig. 4E), and the other NR4A family genes NR4A2 (also known as NURR1) and NR4A3 (also known as NOR1) were also up regulated by UNC1999 (Supplementary Fig. S8A and S8B). Reduction of H3K27me3 levels at the NR4A1 promoter was confirmed by a ChIP assay (Fig. 4F). Remarkably, overexpression of NR4A1 leads to growth arrest in multiple myeloma cells (Fig. 4G), indicating a tumor-suppressive function of NR4A1 in multiple myeloma as previously implied (34). MYC (also known as c-Myc), one of the key genes in multiple myeloma pathogenesis, is reportedly a direct target of NR4As which repress its expression, and NR4A1 specifically occupies MYC promoter region upon NR4A expression (35). Importantly, we found that overexpression of NR4A1 resulted in remarkable downregulation of MYC (Fig. 4H). Moreover, MYC mRNA and its protein were suppressed by UNC1999 (Fig. 4I and J), leading to repression of MYC target gene sets (Fig. 4K). This suppression of MYC was further enhanced by the combination treatment (Fig. 4I–K). To determine the clinical relevance of these findings, we examined the gene expression of NR4A1 and MYC in multiple myeloma samples of patients treated with bortezomib in the APEX 039 clinical study (29). Responsive (R) patients to bortezomib tended to have higher levels of NR4A1 than nonresponsive (NR) ones. In addition, MYC expression was significantly higher in nonresponsive than responsive patients, suggesting an association between bortezomib-resistance and MYC. (Supplementary Fig. S8C). Consistent with our in vitro data, EZH2 levels were negatively correlated with NR4A1, albeit not statistically significant, and MYC and NR4A1 showed a significant inverse correlation (Supplementary Fig. S8D). Taken together, these data indicate that NR4A1 is one of the target genes of UNC1999 in multiple myeloma and triggers MYC downregulation which is enhanced by the combination with bortezomib.
UNC1999 and bortezomib cooperatively suppress PRC2 function
Although bortezomib down regulated EZH2, it did not significantly enrich the polycomb gene set in GSEA of our RNA-seq data (NES 0.635, FDR q value 1.0, P value 1.0). Immunoblotting and RT-PCR confirmed the downregulation of EZH2 following bortezomib treatment; in contrast, EZH1, the homolog of EZH2, was maintained and the global H3K27me3 levels were not reduced at all (Fig. 5A and B). Correspondingly, the sensitivity of H929 cells to bortezomib was only slightly enhanced following shRNA knockdown of EZH2 (Supplementary Fig. S9A). Therefore, we examined the effect of simultaneous knockdown of EZH2 and EZH1 on myeloma cells. Although EZH1 knockdown alone impaired the growth of multiple myeloma cells, double knockdown of EZH2 and EZH1 induced rapid cell death of H929 cells (Supplementary Fig. S1B). These results indicate that dual inhibition of EZH2 and EZH1 is required to obtain maximal anti-myeloma effect. Next, we performed side-by-side experiments to compare the dual inhibition of EZH2 and EZH1 with the specific inhibition of EZH2 as a partner of bortezomib, using UNC1999 and GSK126 (IC50, EZH2 9.9 nmol/L; EZH1 680 nmol/L; ref. 36), respectively. We confirmed that GSK126, as a single agent, induces modest cytotoxicity in multiple myeloma cell lines (Supplementary Fig. S9B). UNC1999 exhibited much better combination effects than GSK126 in several cell lines as evidenced by combination index (Fig. 5C; Supplementary Fig. S9C; Supplementary Table S1F and S1G). In agreement with these findings, the combination of bortezomib with UNC1999, but not GSK126, further reduced the levels of EZH2 and H3K27me3 (Fig. 5D). These results strongly suggest that coinhibition of EZH2 and its homolog EZH1 is necessary to fully block the activity of PRC2 and effectively enhance the sensitivity of myeloma cells to bortezomib.
Figure 5.
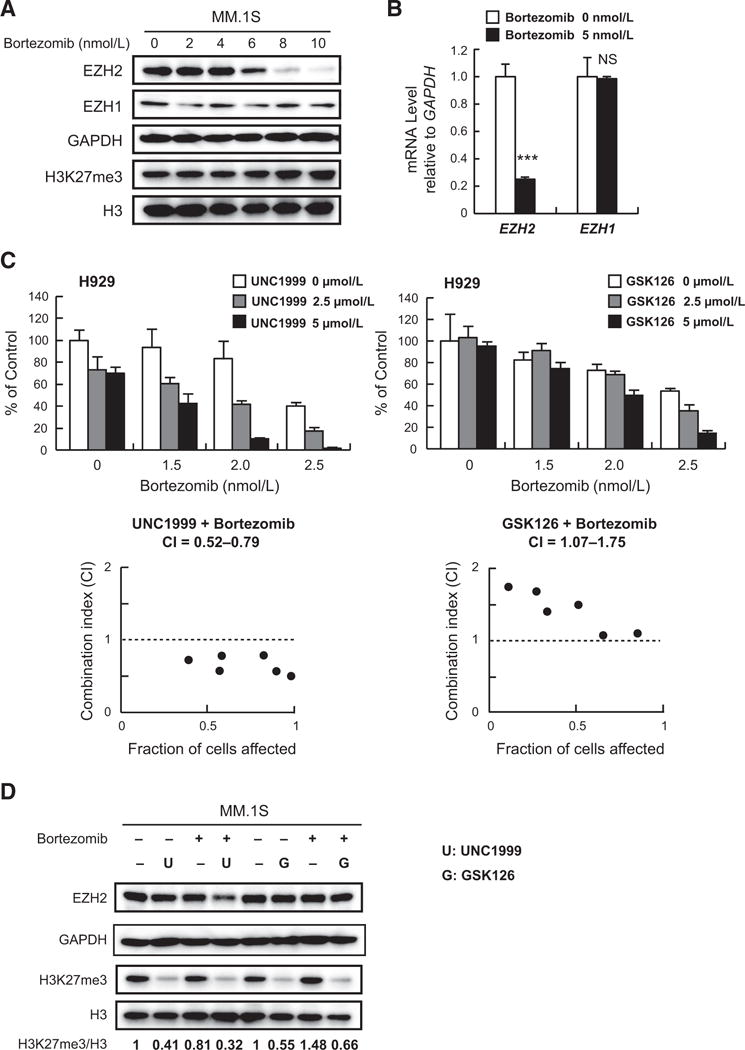
UNC1999 and bortezomib cooperatively suppress PRC2 function. A, Immunoblot analysis for the indicated proteins in MM.1S cells after treatment with the indicated concentrations of bortezomib for 24 hours. GAPDH and H3 served as loading controls. B, Quantitative RT-PCR analysis of mRNA expression of EZH2 and EZH1 in MM.1S cells treated with 5 nmol/L of bortezomib for 12 hours. The y-axis represents fold-change after normalization to GAPDH, and error bars represent SD of triplicates. C, MTS assays performed side by side showing the viability of H929 cells upon treatment for 72 hours with the indicated doses of UNC1999 (left) or GSK126 (right) with the indicated doses of bortezomib in the last 48 hours relative to untreated control. Data represent mean ± SD of triplicate cultures. CI calculation is shown below each graph. D, Immunoblot analysis for the indicated proteins in MM.1S cells upon 72 hours of 5 μmol/L of UNC1999 or GSK126 and/or 5 nmol/L of bortezomib treatment for the last 12 hours. GAPDH and H3 were used as loading controls. H3K27me3 amounts relative to total H3 are shown.
UNC1999 demonstrates synergistic effects with bortezomib in prostate cancer
EZH2 overexpression was found to be associated with disease progression in advanced prostate cancer (4). We therefore studied the efficacy of the combination treatment of UNC1999 and bortezomib in prostate cancer cells. UNC1999 exhibited strong synergistic effects with bortezomib in LNCaP and DU145 cell lines (Fig. 6A; Supplementary Fig. S10; Supplementary Table S1H and S1I). Noticeably, side-by-side experiments clearly showed that combining bortezomib with UNC1999 demonstrated superior synergism than combining it with GSK126, the specific inhibitor of EZH2, emphasizing the importance of EZH1 inhibition in prostate cancer. (Fig. 6A; Supplementary Table S1H). Moreover, we observed EZH2 downregulation by bortezomib also in LNCaP cells (Fig. 6B). These results indicate that the strategy of dual inhibition of EZH2 and EZH1 in combination with proteasome inhibitors can be broadly applied to the treatment of PRC2-dependent tumors such as prostate cancer.
Figure 6.
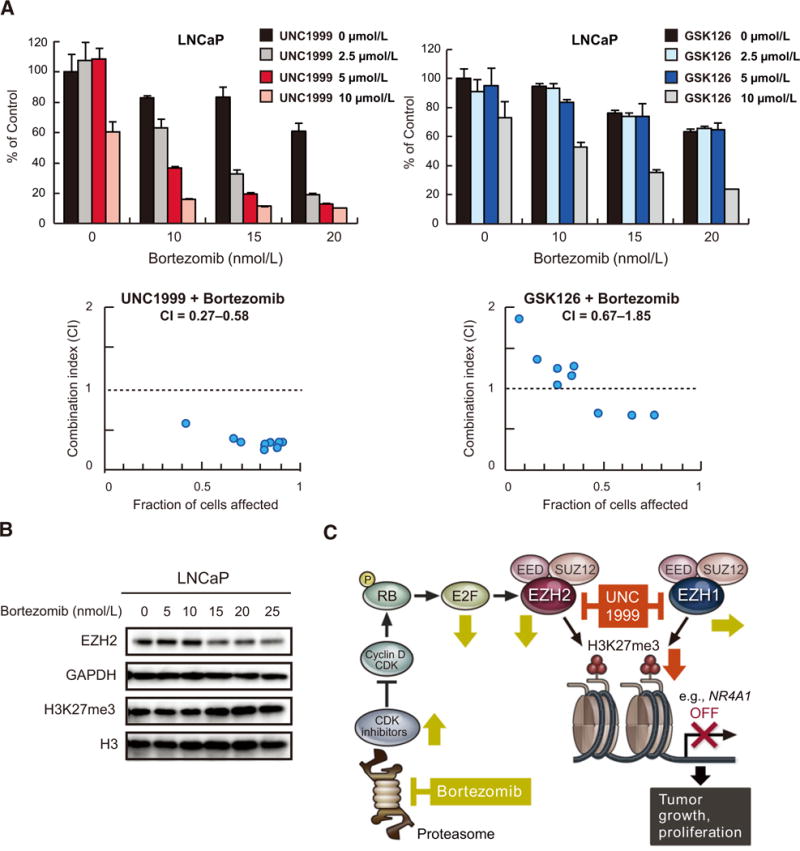
UNC1999 enhances bortezomib-induced cytotoxicity in prostate cancer cells. A, MTS assays performed side-by-side showing viability of LNCaP cells upon simultaneous treatment for 72 hours with the indicated doses of UNC1999 (left) or GSK126 (right) in combination with the indicated doses of bortezomib relative to untreated control. Data represent mean ± SD of triplicate cultures. CI calculation is shown below each graph. B, Immunoblot analysis for the indicated proteins in LNCaP cells after treatment with the indicated concentrations of bortezomib for 72 hours. GAPDH and H3 served as loading controls. C, A proposed model for the synergistic activity of UNC1999 and bortezomib. Bortezomib down regulates EZH2 through stabilization of CDK inhibitors and inhibition of E2F1, whereas UNC1999 enhances the effect of bortezomib by suppressing both EZH2 and EZH1. This leads to derepression of PRC2 target genes such as NR4A1 resulting in inhibition of growth and proliferation of tumor cells.
Discussion
In this study, we investigated the use of UNC1999, a dual inhibitor of EZH2 and EZH1, alone and in combination with proteasome inhibitors in multiple myeloma both in vitro and in vivo, as well as in prostate cancer. Our analysis illustrated that multiple myeloma patients with higher levels of EZH2 expression tended to respond poorly to bortezomib. In line with these observations, EZH2 overexpression by lentiviral vectors triggered bortezomib resistance in multiple myeloma cells which could be overcome by UNC1999. Most importantly, we have demonstrated potent synergistic cytotoxic effects between UNC1999 and proteasome inhibitors, and dissected the underlying mechanism of the synergy and its molecular signature in multiple myeloma cells. Furthermore, we showed that bortezomib markedly down regulated EZH2 transcription and its protein expression. Undeniably, the mechanisms of action of proteasome inhibitors are multi-faceted and include cell-cycle arrest through the accumulation of CDK inhibitors (37). Bortezomib stabilizes CDK inhibitors p21 and p27 leading to hypophosphorylation of RB, which in turn prevents E2F1 from binding to its target genes including the E2F1 promoter, thereby repressing E2F1 transcription (38). Importantly, E2F1 is known to transactivate EZH2 (27). As expected, bortezomib treatment down regulated E2F1 and decreased the binding of E2F1 to EZH2 promoter.
We have previously shown that substantial amounts of H3K27me3 persist after deletion of Ezh2 in a mouse model of myelodysplastic syndrome (MDS) (39). This fact points out the role that EZH1 plays in maintaining reduced but notable levels of H3K27me3 in the absence of EZH2. In agreement, bortezomib-induced downregulation of EZH2 did not significantly impair PRC2 function. Importantly, we have demonstrated that the combination of UNC1999 and bortezomib resulted in superior cytotoxic effects than the combination of GSK126 and bortezomib in multiple myeloma and prostate cancer cells. This could be explained by the fact that UNC1999 effectively blocks PRC2 activity by inhibiting both EZH2 and EZH1, whereas GSK126 only inhibits EZH2, allowing residual PRC2 activity through EZH1. PRC2 targets tumor-suppressor genes and developmental regulator genes, thereby playing an oncogenic role in EZH2-dependent cancers (3). As expected and previously reported in leukemic cells (7), transcriptional profiling showed derepression of PRC2 target genes following UNC1999 treatment. Of note, we observed more significant derepression of PRC2 target genes in the combination of UNC1999 and bortezomib, suggesting cooperative inhibition of PRC2 function by the combination. Taken together, these observations suggest that the dual inhibition of EZH2 and EZH1 sensitizes multiple myeloma and prostate cancer cells to proteasome inhibition, and that epigenetic therapies targeting both EZH2 and EZH1 may be required to achieve a sustainable effect on PRC2-dependent cancers (Fig. 6C).
Among the most upregulated genes in UNC1999-treated cells was NR4A1, and its upregulation was enhanced in cells treated with the combination of UNC1999 and bortezomib. Other members of the orphan nuclear receptor NR4A subgroup, NR4A2 and NR4A3, were also up regulated by UNC1999. NR4As are immediate early or stress response genes that can be induced by a vast number of stimuli such as growth factors, inflammatory cytokines, and mitogenic, and apoptotic signals (40). Of note, reduction of Nr4a1/3 gene expression leads to development of myelodysplastic and myeloproliferative diseases (MDS/MPN) (41), whereas targeted deletion of both genes induces lethal acute myeloid leukemia in mice (42). A recent study showed that overexpression of NR4A1 induced massive apoptotic cell death of aggressive lymphoma cell lines (43). In multiple myeloma, NR4A1-mimicking peptide induced Bcl-B–dependent apoptosis in multiple myeloma cells (34). These findings define NR4A1 as a candidate tumor suppressor in multiple myeloma. Using overexpression experiments, we confirmed the antioncogenic role of NR4A1 in multiple myeloma. Significantly, MYC is reportedly a direct target of NR4As (35). UNC1999 treatment profoundly suppressed MYC expression in multiple myeloma cells. As previously reported (44), bortezomib alone also down regulated MYC probably due to reduced E2F1, a transactivator of MYC (45), and enhanced UNC1999-induced suppression of MYC as evidenced by the marked suppression of MYC-related gene sets. Therefore, UNC1999 and bortezomib cooperatively repress the transcription of MYC, one of the most potent oncogenes in multiple myeloma, resulting in a remarkable synergistic effect.
Other notable UNC1999 targets included two novel candidate tumor suppressor genes in multiple myeloma: LTB and EGR1. LTB, a TNF family member (46), implicated in the NF-κB pathway (47), was reportedly inactivated in multiple myeloma patients (30). EGR1 encodes a protein that induces the expression of tumor suppressors such as TP53 (48) and PTEN (49). In multiple myeloma, recurrent mutations of EGR1 were reported (30). In addition, low expression of EGR1 strongly correlated with disease progression and knockdown of EGR1 in multiple myeloma cell lines conferred resistance to bortezomib (31). CDKN1C, which encodes the cyclin-dependent kinase inhibitor p57 (also known as kip2), is another direct target of UNC1999. Inactivation of CDKN1C by aberrant methylation of its promoter has been reported in several types of cancer (50). The detailed functions of these genes in the pathogenesis of multiple myeloma remain to be unveiled in future studies.
In conclusion, our findings demonstrate that targeting both EZH2 and EZH1, alone and in combination with proteasome inhibitors, could be a new therapeutic option for the treatment of PRC2-dependent cancers. Hopefully, this study will pave the way for clinical evaluation of this novel therapeutic approach.
Supplementary Material
Translational Relevance.
Although proteasome inhibitors have improved patient outcome in multiple myeloma, patients eventually develop drug resistance which needs to be overcome by novel combination strategies. In this study, we demonstrate that the combination of proteasome inhibitors with UNC1999, a dual inhibitor of EZH2 and EZH1, induces synergistic antimyeloma activity in vitro as well as in vivo. Importantly, EZH2 expression in primary multiple myeloma cells and cell lines is associated with bortezomib-resistance which is overcome by UNC1999. Bortezomib down regulates EZH2, thereby sensitizing EZH2-dependent tumors to EZH1 inhibition. UNC1999 derepresses the tumor suppressor NR4A1, leading to suppression of MYC which is enhanced by the combination with bortezomib. This combination is also synergistic in EZH2-overexpressing prostate cancer cells. Our study lays the preclinical framework for the broad clinical applications of proteasome inhibitors combined with dual inhibitors of EZH2 and EZH1 in PRC2-dependent cancers including multiple myeloma and prostate cancer.
Acknowledgments
The authors thank Drs. Y. Furukawa and A. Yoshimura for providing them with E2F1 and NR4A1 expression vectors, respectively. The authors thank Shorouq Mohammed Kamel Rizq for critical review of the article.
Grant Support
This work was supported in part by Grants-in-Aid for Scientific Research in Japan (#15H02544, #26860719, and #16K09839); Scientific Research on Innovative Areas “Stem Cell Aging and Disease” (#26115002) from MEXT, Japan; Next‐generation Cancer Research Strategy Promotion project (16cm0106516h0001) from Japan Agency for Medical Research and Development (AMED); and grants from the Uehara Memorial Foundation; Yasuda Medical Foundation; Mochida Memorial Foundation; Tokyo Biochemical Research Foundation; and Kanae Foundation for the promotion of Medical Science.
Footnotes
Authors’ Contributions
Conception and design: O. Rizq, N. Mimura, A. Iwama
Development of methodology: O. Rizq, N. Mimura, C. Wang, A. Iwama
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): O. Rizq, N. Mimura, A. Saraya, S. Koide, Y. Kato, K. Aoyama, C. Wang, T. Chiba, J. Jin
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): O. Rizq, N. Mimura, M. Oshima, A. Saraya, C. Wang, T. Chiba, A. Iwama
Writing, review, and/or revision of the manuscript: O. Rizq, N. Mimura, A. Ma, J. Jin, C. Nakaseko, A. Iwama
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): O. Rizq, N. Mimura, A. Saraya, Y. Nakajima-Takagi, C. Wang, A. Ma, J. Jin, T. Iseki, C. Nakaseko, A. Iwama
Study supervision: N. Mimura, A. Iwama
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 2.Agger K, Cloos PAC, Christensen J, Pasini D, Rose S, Rappsilber J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–4. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 3.Laugesen A, Helin K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell. 2014;14:735–51. doi: 10.1016/j.stem.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 5.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346–57. doi: 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20:1329–39. doi: 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, MacNevin CJ, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324–34. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 11.Dimopoulos MA, Richardson PG, Moreau P, Anderson KC. Current treatment landscape for relapsed and/or refractory multiple myeloma. Nat Rev Clin Oncol. 2015;12:42–54. doi: 10.1038/nrclinonc.2014.200. [DOI] [PubMed] [Google Scholar]
- 12.Kalushkova A, Fryknäs M, Lemaire M, Fristedt C, Agarwal P, Eriksson M, et al. Polycomb target genes are silenced in multiple myeloma. PLoS One. 2010;5:e11483. doi: 10.1371/journal.pone.0011483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41:521–3. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005;24:6269–80. doi: 10.1038/sj.onc.1208771. [DOI] [PubMed] [Google Scholar]
- 15.Hernando H, Gelato KA, Lesche R, Beckmann G, Koehr S, Otto S, et al. EZH2 inhibition blocks multiple myeloma cell growth through upregulation of epithelial tumor suppressor genes. Mol Cancer Ther. 2016;15:287–98. doi: 10.1158/1535-7163.MCT-15-0486. [DOI] [PubMed] [Google Scholar]
- 16.Neo WH, Lim JF, Grumont R, Gerondakis S, Su IH. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J Biol Chem. 2014;289:31693–707. doi: 10.1074/jbc.M114.574517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal P, Alzrigat M, Párraga AA, Enroth S, Singh U, Ungerstedt J, et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget. 2016;7:6809–23. doi: 10.18632/oncotarget.6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mimura N, Hideshima T, Anderson KC. Novel therapeutic strategies for multiple myeloma. Exp Hematol. 2015;43:732–41. doi: 10.1016/j.exphem.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science. 2012;338:1465–9. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milano A, Iaffaioli RV, Caponigro F. The proteasome: a worthwhile target for the treatment of solid tumours? Eur J Cancer. 2007;43:1125–33. doi: 10.1016/j.ejca.2007.01.038. [DOI] [PubMed] [Google Scholar]
- 21.Voutsadakis IA, Papandreou CN. The ubiquitin-proteasome system in prostate cancer and its transition to castration resistance. Urol Oncol. 2012;30:752–61. doi: 10.1016/j.urolonc.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 22.Mimura N, Fulciniti M, Gorgun G, Tai YT, Cirstea D, Santo L, et al. Blockade of XBP1 splicing by inhibition of IRE1alpha is a promising therapeutic option in multiple myeloma. Blood. 2012;119:5772–81. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mimura N, Hideshima T, Shimomura T, Suzuki R, Ohguchi H, Rizq O, et al. Selective and potent Akt inhibition triggers anti-myeloma activities and enhances fatal endoplasmic reticulum stress induced by proteasome inhibition. Cancer Res. 2014;74:4458–69. doi: 10.1158/0008-5472.CAN-13-3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mochizuki-Kashio M, Aoyama K, Sashida G, Oshima M, Tomioka T, Muto T, et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1-dependent manner. Blood. 2015;126:1172–83. doi: 10.1182/blood-2015-03-634428. [DOI] [PubMed] [Google Scholar]
- 25.Chou T-C, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 26.Nara M, Teshima K, Watanabe A, Ito M, Iwamoto K, Kitabayashi A, et al. Bortezomib reduces the tumorigenicity of multiple myeloma via downregulation of upregulated targets in clonogenic side population cells. PLoS One. 2013;8:e56954. doi: 10.1371/journal.pone.0056954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRBE2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–35. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albero MP, Vaquer JM, Andreu EJ, Villanueva JJ, Franch L, Ivorra C, et al. Bortezomib decreases Rb phosphorylation and induces caspase-dependent apoptosis in Imatinib-sensitive and -resistant Bcr-Abl1-expressing cells. Oncogene. 2010;29:3276–86. doi: 10.1038/onc.2010.81. [DOI] [PubMed] [Google Scholar]
- 29.Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109:3177–88. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- 30.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L, Wang S, Zhou Y, Wu X, Entin I, Epstein J, et al. Identification of early growth response protein 1 (EGR-1) as a novel target for JUN-induced apoptosis in multiple myeloma. Blood. 2009;115:61–70. doi: 10.1182/blood-2009-03-210526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X, Karuturi RKM, Sun F, Aau M, Yu K, Shao R, et al. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One. 2009;4:e5011. doi: 10.1371/journal.pone.0005011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–69. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 34.Luciano F, Krajewska M, Ortiz-Rubio P, Krajewski S, Zhai D, Faustin B, et al. Nur77 converts phenotype of Bcl-B, an antiapoptotic protein expressed in plasma cells and myeloma. Blood. 2007;109:3849–55. doi: 10.1182/blood-2006-11-056879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boudreaux SP, Ramirez-Herrick AM, Duren RP, Conneely OM. Genome-wide profiling reveals transcriptional repression of MYC as a core component of NR4A tumor suppression in acute myeloid leukemia. Oncogenesis. 2012;1:e19. doi: 10.1038/oncsis.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 37.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. Proteasome inhibition as a novel therapeutic target in human cancer. J Clin Oncol. 2005;23:630–9. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 38.Araki K, Nakajima Y, Eto K, Ikeda M-A. Distinct recruitment of E2F family members to specific E2F-binding sites mediates activation and repression of the E2F1 promoter. Oncogene. 2003;22:7632–41. doi: 10.1038/sj.onc.1206840. [DOI] [PubMed] [Google Scholar]
- 39.Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med. 2013;210:2627–39. doi: 10.1084/jem.20131144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohan HM, Aherne CM, Rogers AC, Baird AW, Winter DC, Murphy EP. Molecular pathways: the role of NR4A orphan nuclear receptors in cancer. Clin Cancer Res. 2012;18:3223–8. doi: 10.1158/1078-0432.CCR-11-2953. [DOI] [PubMed] [Google Scholar]
- 41.Ramirez-Herrick AM, Mullican SE, Sheehan AM, Conneely OM. Reduced NR4A gene dosage leads to mixed myelodysplastic/myeloproliferative neoplasms in mice. Blood. 2011;117:2681–90. doi: 10.1182/blood-2010-02-267906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mullican SE, Zhang S, Konopleva M, Ruvolo V, Andreeff M, Milbrandt J, et al. Abrogation of nuclear receptors Nr4a3 andNr4a1 leads to development of acute myeloid leukemia. Nat Med. 2007;13:730–5. doi: 10.1038/nm1579. [DOI] [PubMed] [Google Scholar]
- 43.Deutsch AJA, Rinner B, Wenzl K, Pichler M, Troppan K, Steinbauer E, et al. NR4A1-mediated apoptosis suppresses lymphomagenesis and is associated with a favorable cancer-specific survival in patients with aggressive B-cell lymphomas. Blood. 2014;123:2367–77. doi: 10.1182/blood-2013-08-518878. [DOI] [PubMed] [Google Scholar]
- 44.Zhao W-L, Liu Y-Y, Zhang Q-L, Wang L, Leboeuf C, Zhang Y-W, et al. PRDM1 is involved in chemoresistance of T-cell lymphoma and downregulated by the proteasome inhibitor. Blood. 2008;111:3867–71. doi: 10.1182/blood-2007-08-108654. [DOI] [PubMed] [Google Scholar]
- 45.Hiebert SW, Lipp M, Nevins JR. E1A-dependent trans-activation of the human MYC promoter is mediated by the E2F factor. Proc Natl Acad Sci U S A. 1989;86:3594–8. doi: 10.1073/pnas.86.10.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Browning JL, Ngam-ek A, Lawton P, DeMarinis J, Tizard R, Pingchang Chow E, et al. Lymphotoxin β, a novel member of the TNF family that forms a heteromeric complex with lymphotoxin on the cell surface. Cell. 1993;72:847–56. doi: 10.1016/0092-8674(93)90574-a. [DOI] [PubMed] [Google Scholar]
- 47.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng W-J, et al. Promiscuous mutations activate the noncanonical NF-kB pathway in multiple myeloma. Cancer Cell. 2007;12:131–44. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nair P, Muthukkumar S, Sells SF, Han S-S, Sukhatme VP, Rangnekar VM. Early growth response-1-dependent apoptosis is mediated by p53. J Biol Chem. 1997;272:20131–8. doi: 10.1074/jbc.272.32.20131. [DOI] [PubMed] [Google Scholar]
- 49.Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, et al. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol. 2001;3:1124–8. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 50.Kikuchi T, Toyota M, Itoh F, Suzuki H, Obata T, Yamamoto H, et al. Inactivation of p57KIP2 by regional promoter hypermethylation and histone deacetylation in human tumors. Oncogene. 2002;21:2741–9. doi: 10.1038/sj.onc.1205376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.