

SUMMARY
T cells undergo metabolic reprogramming with major changes in cellular energy metabolism during activation. In patients with mitochondrial disease, clinical data were marked by frequent infections and immunodeficiency, prompting us to explore the consequences of oxidative phosphorylation dysfunction in T cells. Since cytochrome c oxidase (COX) is a critical regulator of OXPHOS, we created a mouse model with isolated dysfunction in T cells by targeting a gene, COX10, that produces mitochondrial disease in humans. COX dysfunction resulted in increased apoptosis following activation in vitro and immunodeficiency in vivo. Select T cell effector subsets were particularly affected; this could be traced to their bioenergetic requirements. In summary, the findings presented herein emphasize the role of COX particularly in T cells as a metabolic checkpoint for cell fate decisions following T cell activation, with heterogeneous effects in T cell subsets. In addition, our studies highlight the utility of translational models that recapitulate human mitochondrial disease for understanding immunometabolism.
Graphical abstract
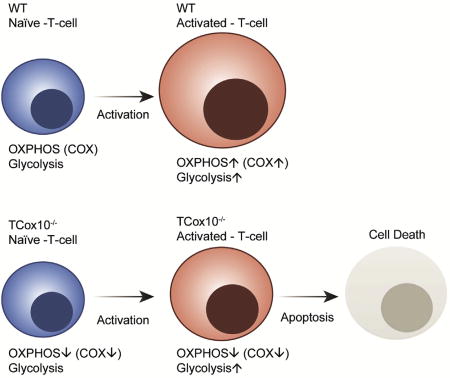
Mitochondrial diseases are disorders of oxidative phosphorylation. Using mitochondrial disease as a model system, Tarasenko et al. demonstrate that cytochrome c oxidase deficiency differentially affects T cell effector subsets based on their bioenergetic requirements. Mouse T cell COX deficiency produces an immunodeficiency similar to that of patients with mitochondrial disease.
INTRODUCTION
Although the majority of the immunometabolism literature on T cells has concentrated on the role of mitochondria in memory and regulatory subsets (Beier et al., 2015; Pearce et al., 2009, 2013; van der Windt et al., 2012, 2013), mitochondria are no less critical during T cell activation (Ron-Harel et al., 2016; Sena et al., 2013; van der Windt et al., 2013). In activated T cells, mitochondria localize to the immune synapse and play a role in the regulation of Ca2+ flux (Quintana et al., 2007; Schwindling et al., 2010). Despite adopting a glycolytic metabotype, oxidative phosphorylation (OXPHOS) is also increased during primary activation, albeit at a comparatively lower rate (Frauwirth et al., 2002; O’Rourke and Rider, 1989; Wang et al., 2011). In conjunction with supporting ATP production, individual OXPHOS complexes play unique roles in T cell activation. This question was first addressed for complex III in a seminal paper using a model of T cell intrinsic complex III dysfunction (T-Uqcrfs−/− mice). To date, there has not been a mitochondrial disease that has been demonstrated to be due to UQCRFS mutations. Despite this limitation, the T-Uqcrfs−/− model was critical in identifying the unique function of complex III in T cell activation: ROS production for NFAT signaling and the downstream production of IL-2 (Sena et al., 2013).
Cytochrome c oxidase (COX) is a multiprotein complex composed of 13 subunits, three of which are encoded in mitochondrial DNA (mtDNA), that is assembled with the help of 20 nuclear proteins (Fontanesi et al., 2006; Soto et al., 2012). COX10 is an assembly factor for complex IV, and deletion of COX10 in fibroblasts results in a significant reduction in COX subunits COX1, COX5b, COX6b, and COX4 (Diaz et al., 2006). COX deficiency due to COX10 mutations produces a heterogeneous mitochondrial disease in humans that is marked by Leigh disease, seizures, ataxia, hypotonia, muscle weakness, metabolic acidosis, proximal tubulopathy, and early death (Valnot et al., 2000; Antonicka et al., 2003) Tissue-specific COX10 deficiency in mice may result in hepatopathy, myopathy, and encephalopathy (Diaz, 2010; Diaz et al., 2012).
Due to its strategic positon in the respiratory chain as the rate-limiting complex, COX maintains tight control over OXPHOS flux and ATP production (Fukuda et al., 2007; Helling et al., 2012; Hüttemann et al., 2012; Semenza, 2011). As discussed above, OXPHOS components also have individual functions, such as ROS production—i.e., complexes I and III—that play a role in cellular activation and function. However, unlike complexes I and III, the unique function of COX is found in the mediation of apoptosis, ultimately determining cell fate (Diaz, 2010; Diaz et al., 2012; Schüll et al., 2015; Villani et al., 1998). While much is known about the function of COX in other cell types, the unique role played by COX in T cells remains unresolved.
Rare genetic diseases have contributed significantly to our understanding of human biology. Mitochondrial diseases (MD) are clinically heterogeneous disorders that can be inherited from mtDNA and nDNA. Even with the contributions of this dual genome, the majority of OXPHOS components are encoded in the nucleus, and as such are multisystemic. To characterize the role of COX in T cell function, we chose mitochondrial disease as a model system. Using a CD4-Cre recombinase, we targeted Cox10 to produce a model of T cell COX dysfunction (TCox10−/−). TCox10−/− mice displayed many similarities to patients with mitochondrial disease and immunodeficiency.
RESULTS AND DISCUSSION
Immunodeficiency in Patients with Mitochondrial Disease
Although mitochondrial disease (MD) is a multisystem disorder affecting tissues and organs with increased energetic requirements, immunodeficiency is generally under-recognized and is not part of the diagnostic criteria (Walker et al., 1996). A clinical case series suggested B cell dysfunction and enhanced susceptibility to sepsis and SIRS in patients with MD (Walker et al., 2014), prompting us to expand the immune phenotype of patients with MD. We began by conducting a retrospective chart review on 62 pediatric patients (38 female, 24 male; mean age, 7.4 years) with “probable” (n = 25) or “definite” (n = 37) MD (Figure 1A). The most common finding was recurrent or severe infections (89%) involving the respiratory tract: upper (47%) and lower (pneumonia, 40%) respiratory tract infections, otitis media (24%), and sinusitis (32%). Immunoglobulin therapy for recurrent infection was found in 21% of patients. The toll infection takes on patients with MD is well recognized by clinicians: up to 50% of cases result in life-threatening or neurode-generative sequelae (Edmonds et al., 2002).
Figure 1. Patients with Mitochondrial Disease Have Clinical Indicators of Immunodeficiency.

(A) Chart review of immune symptoms from patients with primary mitochondrial disease (N = 55).
(B) Clinical flow cytometry for naive (CD45RA+) and memory (CD45RO+) T cells in patients with various mitochondrial diseases (n = 10). Hashed lines indicate normal range of values.
(C) White blood cell counts during infection. Error bars denote SEM.
Since recurrent infections suggest compromised T cell immunity, we performed flow cytometry on ten pediatric patients with MD (3 female, 7 male). In the subjects surveyed, we found a preponderance of naive T cells (CD45RA+) and a paucity of memory T cells (CD45RO+) (Figure 1B). The enzymatic perturbations in OXPHOS ranged from select (complex I, complex I and III, or complex IV) to more generalized (global OXPHOS or mtDNA depletion) deficiencies, suggesting that multiple OXPHOS deficiencies may produce immunodeficiency. In addition to baseline abnormalities, immune cell dysfunction could also be provoked during episodes of stress. Two patients with complex III and complex I and III deficiencies by enzymology were admitted to hospital for an acute infectious illness. During their hospital course, both patients experienced leukopenia, which recovered with the resolution of their illness (Figure 1C). Overall, our clinical data serve to expand the immune phenotype of MD to include memory T cell deficits and leukopenia due to bioenergetic stress; they also suggest that T cells are affected by cell-intrinsic OXPHOS deficiency.
OXPHOS and COX Activity Are Enhanced during T Cell Activation
Activated and effector T cells undergo metabolic reprogramming that includes the adoption of a glycolytic (Warburg) metabotype: enhanced glycolysis with lactate production despite the availability of oxygen (Frauwirth et al., 2002; O’Rourke and Rider, 1989; Wang et al., 2011; MacIver et al., 2013; Vander Heiden et al., 2009). The glycolytic metabotype, although energy inefficient, is necessary for the synthesis of intermediates and macromolecules to support T cell proliferation. In general, metabotypes are defined by relative changes in glycolysis or OXPHOS. However, this definition may in some ways de-emphasize the relative contributions of the subdominant pathway. T cell activation also involves enhanced mitochondrial metabolism (Frauwirth et al., 2002; O’Rourke and Rider, 1989; Wang et al., 2011). In mouse wild-type (WT) T cells, after 24 hr of stimulation and following normalization for cell size, MitoGreen staining increased in activated T cells, indicating increased mitochondrial mass (Ron-Harel et al., 2016; Figure 2A, left). Mitochondrial membrane potential (Ψm) was also amplified in activated T cells secondary to augmented mitochondrial mass. Indeed, when normalized to mitochondrial mass, Ψm was unchanged (Figure 2A, right, bar graph). Profiling bioenergetics by extracellular flux analysis, we determined the extracellular acidification rate (ECAR), a proxy for glycolysis, and the oxygen consumption rate (OCR), a proxy for OXPHOS, in activated T cells. Consistent with previous reports (Ron-Harel et al., 2016; Sena et al., 2013; van der Windt et al., 2013), glycolysis outpaced OXPHOS upon T cell activation: glycolysis (ECAR) was upregulated ~13 × (p < 0.0001) (Figure 2B, left), while mitochondrial metabolism (OCR) was upregulated ~7 × (p < 0.0001) (Figure 2B, right).
Figure 2. Mitochondria Are Required in Activated Mouse Splenic T Cells.

WT T cells were stimulated with anti-CD3/CD28 for 24 hr.
(A) Mitochondrial content with MitoGreen (n = 3) and membrane potential ψm (TMRE, tetramethylrhodamine, ethyl ester, n = 3 per condition) and normalized membrane potential (bar graph).
(B) Extracellular flux analysis of naive and activated WT T cells (n = 4–5).
(C) Complex IV activity (n = 10 per condition).
(D) Cytochrome a content (n = 3 per condition).
(E) Cellular proliferation in glucose or galactose media (n = 3 per condition).
(F) Cell proliferation and apoptosis (inset) with potassium cyanide (KCN) (n = 4 per condition).
(G) Extracellular flux analysis in the presence of KCN (n = 4 per condition).
Experiments were performed three or more times with representative data shown. ECAR, extracellular acidification rate; OCR, oxygen consumption rate; TMPD, (tetramethyl-p-phenylenediamine). ***p < 0.001, ****p < 0.0001. Error bars denote SEM.
Since cytochrome c oxidase (COX) is the ultimate enzyme complex responsible for maintaining tight control over OXPHOS (Li et al., 2006), we next examined COX status. COX activity was determined using a complex-IV-specific electron donor, tetramethyl-p-phenylenediamine (TMPD). COX activity in activated T cells (24 hr) peaked at ~3.5 × (p < 0.001) above naive T cells (Figure 2C). Spectrophotometric measurement of heme a cytochrome showed a 50% increase in content, supporting enhanced activation of COX (Figure 2D). Despite the increase in enzyme activity, COX subunits were essentially unchanged, as demonstrated by immunoblot and proteomic analysis (Figure S1). Thus, when T cells become activated, oxidative capacity is increased without augmented synthesis of COX subunits.
A key component of the T cell immune response following activation is cellular proliferation. Cell proliferation proceeds robustly after about 48 hr in activated T cells. To explore the role of the mitochondria in supporting T cell proliferation, mouse splenic T cells were stimulated for 3 days in either glucose or galactose media. Galactose, as a carbon source, does not yield any net ATP by substrate-level phosphorylation, thus making the respiratory chain the sole source of ATP (Robinson et al., 1992). 3H-thymidine incorporation in cells cultured in galactose-containing media was reduced by 86% (p < 0.0001) (Figure 2E), suggesting that OXPHOS alone was insufficient to support T cell proliferation. However, it appears that glycolysis alone was also insufficient; T cells stimulated in the presence of the ionophore FCCP, a compound that dissipates the proton gradient in OXPHOS, showed diminished proliferation that was not due to apoptosis at the lowest dose (Figure S1C, inset). To examine the role of COX in supporting T cell proliferation, WT T cells activated as above were treated with potassium cyanide (KCN, dose range 0.1–2.5 mM), a complex IV inhibitor. Following 3 days of treatment, 2.5 mM KCN resulted in a precipitous drop in T cell proliferation with an increase in the apoptotic marker Annexin V (Figure 2F). This phenotype was observed when COX inhibition produced a ~50% decrease in OXPHOS, establishing a rudimentary threshold for survival and proliferation (Figure 2G). Similar to inhibition of COX, inhibition of the ATP synthase by oligomycin also resulted in depressed CD4+ T cell proliferation with enhanced apoptosis (Annexin+ PI+) (Figure S1D, inset). Our findings are consistent with previous reports citing loss of mitochondrial energy production causing cell death (Lartigue et al., 2009; Ricci et al., 2003). Taken as a whole, our data further support activated T cells’ dependence on both pathways and inability to function without intact OXPHOS (Ron-Harel et al., 2016; Sena et al., 2013; van der Windt et al., 2013). Although the role of OXPHOS in T cell activation is still being investigated, it has been suggested that mitochondrial ATP generation in naive T cells is necessary for enhanced glycolysis (van der Windt et al., 2013).
A Mouse Model of T Cell Cytochrome c Oxidase Dysfunction: TCox 10−/−
Based on the immune phenotype observed in MD patients (Walker et al., 2014; Figure 1) and our in vitro data demonstrating the importance of COX activity in activated T cells (Figure 2), we hypothesized that a genetic model of T cell COX dysfunction would reveal its unique role in T cell activation, proliferation, and cell-fate decisions. To target T cells specifically, we employed a CD4-Cre recombinase mouse model targeting Exon 6 of the nuclear gene Cox10 (TCox10−/−) (Figure S2A), a critical assembly factor for COX that is present in lymphocytes (Valnot et al., 2000). In general, TCox10−/− mice appeared healthy and attained a normal body weight (Figure S2B). Considering that the T cell co-receptor CD4 is expressed on all T cells during thymic development, all T cells are potentially affected by the CD4-Cre recombinase. We began by determining Exon 6 status in T cell populations isolated from lymphoid tissues using real time PCR of gDNA normalized to actin. Although the reported efficiency of the CD4-Cre recombinase is 95% (Schmidt-Supprian and Rajewsky, 2007), the number of copies of Exon 6 remained high. When compared to WT, thymic Exon 6 copy levels were at 22% in CD4+CD8+ (p < 0.001), 19% in CD4+ (p < 0.001), and 26% in CD8+ (p < 0.01) T cells (Figure S2C). In the spleen, Exon 6 copy levels were at 32% in CD3+ T cells (p < 0.01) (Figure S2D), and Cox10 mRNA levels (Figure S2E) were 29% of WT (p < 0.01). The deviations observed suggest reduced efficiency of the Cre recombinase or selective pressures on TCox10−/− naive T cells.
Consistent with the deletion of Exon 6 and the mRNA level of Cox10, COX enzyme activity was 22% of WT, while oxygen consumption (OCR) was 37% (p < 0.0001) of WT; this demonstrated that OXPHOS was significantly compromised (Figures 3A and 3B, left). To characterize the metabolic adaptations during T cell activation due to partial COX impairment, we profiled glycolysis using extracellular flux analysis and uniformly labeled 13C-glucose. After 24 hr of stimulation, T cells developed a glycolytic metabotype (ECAR) that was similar in WT and TCox10−/− (Figure 3B, right). When incubated with [U−13C] glucose, both WT- and TCox10−/−-activated T cells displayed similar m+3 isotopomeric patterns in dihydroxyacetone phosphate, 3-phosphoglycerate, phosphoenolpyruvate, pyruvate, lactate, and alanine, indicating that cells from both genotypes effectively took up and metabolized glucose via glycolysis (Figure S3). Despite these similar parameters, the citrate m+2 isotopomer was reduced by 37% (p < 0.05), suggesting that the transition from pyruvate to acetyl-CoA was downregulated and the tricarboxylic acid cycle (TCA) cycle was dysfunction (Table S1, Figure 3C). To probe for alterations in the TCA cycle, activated T cells were incubated with uniformly labeled 13C-glutamine. With [U−13C] glutamine entering the TCA cycle via α-ketoglutarate, m+4 labeling occurs on the first turn of the cycle, with additional labeling occurring on subsequent turns. At 24 hr of stimulation, TCox10−/−T cells exhibited an increase in fumarate (32%, p < 0.0001) and malate (17%, p < 0.0001) m+4 isotopomers, reflecting their formation from the first turn of the TCA cycle, but at the same time suggesting impaired turnover (Table S1, Figure 3D). Evidence for decreased TCA cycle turnover could also be seen in the reduced enrichment in other isotopomers as well as in increased m+4:m+2 ratios for citrate (p < 0.05), glutamate (p < 0.001), fumarate (p < 0.0001), and malate (p < 0.0001). Tumor cells with defective OXPHOS use glutamine-dependent reductive carboxylation as the source of citrate formation to support proliferation (Mullen et al., 2014). Glutamine-derived citrate provides acetyl-coenzyme A for lipid synthesis and four-carbon intermediates for TCA cycle anaplerosis. In TCox−/− T cells, reductive carboxylation does not appear to occur, as suggested by a lack of 13C-citrate m+5 isotopomer enrichment. Thus, our results indicate that COX dysfunction in T cells produces a significant impairment in OXPHOS and the TCA cycle due to reduced availability acetyl-CoA, and predicts compromised proliferation.
Figure 3. Mouse Model of T Cell Cytochrome c Oxidase Deficiency TCox10−/−.

(A) Cytochrome c oxidase (COX) enzyme activity (n = 5 per condition) normalized to citrate synthase (CS).
(B) OCR and ECAR.
(C) Citrate labeling following treatment with 13C-glucose (n = 3 per condition).
(D) TCA cycle flux following treatment with 13C-glutamine (n = 3 per condition). **p < 0.01, ***p < 0.001, ****p < 0.0001. Error bars denote SEM.
T Cell COX Dysfunction Results in Apoptosis In Vitro
Armed with a model of T-cell-specific dysfunction, we explored the role of COX in the early stages of T cell activation in vitro. Transmission electron microscopy (TEM) is a powerful tool employed in mitochondrial medicine for the morphologic examination of mitochondria (Vincent et al., 2016). We interrogated T cell mitochondrial morphology by TEM (Figure 4A) and produced mitochondrial masks to examine cristae morphology (Latorre-Pellicer et al., 2016). The mitochondria of naive WT T cells display rounded and elongated forms with thick cristae. TCox10−/− T cells also demonstrate heterogeneity in their shape; however, their cristae were less recognizable. In mitochondria from activated WT T cells, the mitochondria develop a rounded appearance and sharpening of the cristae. In contrast, activated TCox10−/− T cells failed to develop a rounded appearance, and the cristae were both abnormal in shape and disorganized. Other indicators of mitochondrial dysfunction, such as paracrystalline inclusions, angular cristae, onion-shaped cristae, and donut-shaped mitochondria, were not seen (Vincent et al., 2016; Latorre-Pellicer et al., 2016). Despite these morphologic findings suggesting mitochondrial dysfunction, early events following TCR engagement such as ERK phosphorylation (Figure S4A), calcium flux (Figure S4B), and ROS production (Figure S4C) were intact. TCox10−/− cells displayed normal levels of surface activation markers CD69 and CD25 at 24 hr; however, these were reduced at 72 hr compared to WT (Figures 4B and 4C). Along with the reduction in surface CD25 (IL-2R) at 72 hr, IL-2 production was attenuated by 62% (p < 0.05) (Figure 4D).
Figure 4. TCox10−/− T Cells Show Abnormal Activation In Vitro.

(A) Transmission electron microscopy of a representative sample with mitochondrial masks (n = 4–5 per condition).
(B) CD69 expression (n = 3 per condition).
(C) CD25 expression (n = 3 per condition).
(D) IL-2 mRNA expression (n = 3–5 per condition).
(E) Proliferation with 3H-thymidine (n = 6 per condition).
(F) Proliferation with CFSE (n = 3 per condition).
(G) Apoptosis in activated T cells (n = 3 per condition).
(H) Cox10 Exon 6 status in TCox10−/− cells sorted by division cycle (n = 3–4 per condition).
Experiments were performed three or more times with representative data shown. ****p < 0.0001. Error bars denote SEM.
During evolution, mitochondria yielded many of their prokaryotic genes to the nucleus. Yet, at the same time, they acquired a critical role in governing cell proliferation and apoptosis (Antico Arciuch et al., 2012), ultimately regulating cell fate and function. Based on our stable isotope studies showing an impaired TCA cycle in TCox10−/− T cells, we hypothesized that proliferation would be profoundly affected. Stimulated TCox10−/− T cells had a ~50% reduction in proliferation at 3 days (p < 0.001) by 3H-thymidine incorporation (Figure 4E). The curtailment of proliferation could also be seen by carboxyfluorescein succinimidyl ester (CFSE) dilution. While WT T cells went through 4–5 division cycles in 3 days (Figure 4F, left), TCox10−/− T cells went through 2–3 divisions (Figure 4F, right). Attempts to overcome the proliferation (and IL-2) defect by IL-2 supplementation was not successful (Figure S4D).
Unlike T cells with isolated complex III dysfunction that are stagnant upon activation (Sena et al., 2013), TCox10−/− T cells exhibited a 1.6× increase in dead cells (Annexin V+ PI+) and a 2.5× increase in actively dying cells (Annexin V+) after 72 hr of stimulation (Figure 4G). The apoptotic phenotype observed was not due to increased FasL expression (data not shown). Although numerous OXPHOS deficiencies may be associated with enhanced apoptosis, the mechanism by which COX dysfunction leads to apoptotic cell death is beginning to be unraveled. COX-deficient fibroblasts (Cox10−/−) experiencing oxidative stress upregulated ceramide synthase (CerS6), leading to elevated C16:0 ceramide and causing cytochrome c release and accelerated rates of apoptosis (Schüll et al., 2015). Our cells did not appear to experience enhanced oxidative stress; however, ROS generation is necessary for T cell activation.
Due to the apoptotic phenotype, we surveyed the deletion status of TCox10−/− T cells by sorting cells by division cycle and performing PCR for Cox10 Exon 6, as above (Figure 4H). By the second to third cell division, the Exon 6 deletion approached 98%, suggesting that the Cre recombinase was active, leading to worsening COX function in activated TCox10−/− T cells. Although there were too few cells to perform extracellular flux analyses, previous work by our co-author (Diaz, 2010; Diaz et al., 2012) allows us to predict that COX activity is significantly impaired in these T cells.
COX Dysfunction Reveals CD8+ T Cell Vulnerability
In response to infection, T cell doubling time is 4–5 hr, generating >1012 cells in 1 week and generating a large metabolic demand. The scale of metabolic demand differs among various T cell subsets and may be related to their proliferative requirements. In contrast to CD4+ T cells, CD8+ T cells have greater proliferative potential and higher rates of apoptosis (Foulds et al., 2002). Given the involvement of COX in cell proliferation and apoptosis, we hypothesized that CD4+ and CD8+ T cells would be differentially influenced. In order to explore this hypothesis, we began by profiling proliferation and apoptosis in TCox10−/−CD4+ and CD8+ T cells. As predicted, TCox10−/− CD8+ T cells displayed significant proliferation defects at 3 days, while TCox10−/− CD4+ T cells were closer to WT (Figure 5A). At 24 hr, prior to the induction of proliferation, TCox10−/− CD8+ T cells displayed a ~2× increase in Annexin V staining (Figure S5A) and cleaved caspase 3b (Figure S5B) suggesting apoptosis (McIlwain et al., 2015). To confirm the involvement of apoptosis, we incubated T cells with zVAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone), a pan-caspase inhibitor that halts apoptosis. Following treatment with zVAD FMK for 3 days, the number of Annexin V+ PI+ cells was reduced in a dose-dependent manner for TCox10−/− CD4+ and CD8+ as well as WT CD8+ T cells (Figure S5C). WT CD4+T cells were unaffected by zVAD-FMK treatment. Small increments in proliferation (CFSE) were noted for TCox10−/− CD4+ (5 µM, green line, arrow) and CD8+ (1 µM, blue line, arrow) cells following zVAD treatment (Figure S5D). Despite the lack of a robust recovery of proliferation, CD4+ IL-2 production improved for both WT and TCox10−/− at the 1 µM dose (Figure S5E), suggesting partial phenotypic recovery.
Figure 5. TCox10−/− Effector T Cells Are Differentially Affected by COX Dysfunction.

(A) Proliferation by CFSE (n = 3 per condition).
(B) Metabotype by extracellular flux analysis (n = 6–9 per condition).
(C) Cell proliferation during 2-DG treatment (n = 3 per condition).
(D) Apoptosis during FG4592 treatment (n = 3 per condition).
(E and F) In vitro differentiation of (E) Th17 (n = 3 per condition) and (F) Treg (n = 3 per condition). Bar graphs (middle) summarize data. Proliferation and lineage-specific marker (right).
(G) Metabotype in activated Th17 and Treg cells (n = 5–9 per condition). 2-DG, 2-deoxyglucose; Th17, T helper 17; Treg, regulatory T cells. Experiments were performed three or more times with representative data shown. *p < 0.05, ****p < 0.0001. Error bars denote SEM.
COX Dysfunction Reveals Differences in Bioenergetic Requirements between CD4+ and CD8+ T Cells
Since TCox10−/− CD8+ T cells were more significantly affected by COX dysfunction, we hypothesized that the phenotypic characteristics observed were related to differences in bioenergetic requirements. To address this question, we performed in-depth profiling on CD4+ and CD8+ T cells that were stimulated for 24 hr. WT CD8+ T cells displayed a higher peak of Ψm when compared to WT CD4+ T cells (Figure S6A), suggesting subpopulations of cells with enhanced OXPHOS. TCox10−/− CD8+ T cells displayed even greater heterogeneity in the distribution of Jm than did WT (Figure S6A), supporting our findings describing apoptosis. Consistent with their proliferative needs, bioenergetic profiling of WT CD8+ T cells revealed glycolytic and OXPHOS requirements that surpassed CD4+ T cells: ECAR increased by 66% (p < 0.0001) and OCR increased by 74% (p < 0.0001) (Figure 5B). Despite this larger increase in bioenergetic requirements in CD8+ T cells, the glucose transporter Glut-1 (Figure S6B) and glucose transport by 2-NDBG (Figure S6C) were similar in both WT CD4+ and CD8+ T cells; this suggested that, while the substrate was equally available, the differences observed were driven by metabolic flux. Even though Glut-1 expression (Figure S6D) was decreased in both TCox10−/− CD4+ and CD8+ T cells, glucose transport (Figure S6E) mirrored WT. As expected, both TCox10−/− CD4+ (69% of WT, p < 0.0001) and CD8+ (28% of WT, p < 0.0001) T cells had much lower rates of OXPHOS (Figure 5B). Interestingly, TCox10−/− CD4+ T cells increased glycolysis by 26% (p < 0.0001) over WT, while TCox10−/− CD8+ T cells were somewhat lower than WT, although these findings were not significant (p > 0.05).
Unlike TCox10−/− CD4+ T cells, TCox10−/− CD8+ T cells were unable to upregulate glycolysis in the setting of OXPHOS dysfunction. Based on this finding, we hypothesized that TCox10−/− CD8+ T cells were operating at maximum for glycolysis (Cao et al., 2014) and perturbations would result in further inhibition of proliferation. To answer this question, we probed for glycolytic dependence in T cells for proliferation using 2-deoxyglucose (2-DG), an inhibitor of glycolysis. A dose-dependent response to 2-DG was seen in both WT CD4+ and CD8+ T cells. At the lowest dose of 2-DG (0.125 mM), proliferation was mildly depressed in TCox10−/− CD4+ T cells, while TCox10−/− CD8+ T cells were more significantly affected (Figure 5C) due to apoptosis (data not shown). Since TCox10−/−CD8+ T cells were completely suppressed by 2-DG at the lowest dose, we suggest that these cells have an increased demand on energetic resources and decreased metabolic flexibility.
We next asked whether we could overcome the apoptotic and proliferative phenotypes by enhancing glycolysis. Targeting the hypoxia response has been suggested as a therapy for MD (Jain et al., 2016). Furthermore, hypoxia-inducing factor-1a (HIF1a) regulates cytochrome oxidase subunits to optimize efficiency of respiration (Fukuda et al., 2007). FG4592 is a HIF1α-stabilizing factor that promotes glycolysis and improves survival of cells with OXPHOS dysfunction both in vitro and in vivo (Jain et al., 2016). After 24 hr of incubation with FG4592, enhanced glycolysis was seen in all treated cells (Figure S6F and S6G) but was more pronounced in TCox10−/− CD4+ (84% increase, p < 0.0001) and CD8+T cells (24% increase, p < 0.003). Unfortunately, this came at the expense of a further depression of OXPHOS: WT CD4+ and CD8+ T cells were decreased by 57% (p < 0.0001) and 48% (p < 0.0001), respectively, while TCox10−/− CD4+ and CD8+ T cells were depressed by 50% (p < 0.01) and 81% (p < 0.003), respectively. Proliferation by CFSE measured at 3 days showed a depression in WT CD8+ T cells with FG4592 treatment, highlighting the need for OXPHOS (Figure S6H). TCox10−/− CD4+ T cells also showed an increase in nonproliferating cells, while TCox10−/− CD8+ T cells remained essentially unchanged. In contrast to previously published models of OXPHOS dysfunction (Jain et al., 2016), FG4592 had the exact opposite effect on viability in CD8+ T cells with mitochondrial dysfunction. At 3 days, the number of cells undergoing apoptosis (Annexin+) increased by 67% in TCox10−/− CD8+ T cells (p < 0.05) (Figure 5D), while all other cell types, including TCox10−/− CD4+, were comparable to control. Unlike other in vivo and in vitro models of mitochondrial dysfunction (Jain et al., 2016), activation of the hypoxia response via FG4592 was ineffective in ameliorating the apoptotic effects of COX dysfunction. Besides revealing vulnerabilities in CD8+ T cells, our studies on metabolic reprogramming with FG4592 speak to the unique properties of T cells, including the interde-pendency between glycolysis and OXPHOS. The source of energy is more critical than simply increasing the overall bioenergetic currency.
Differential Effects of COX Dysfunction on Effector CD4+ T Cell Subsets
Based on our observation demonstrating the vulnerability of CD8+ T cell subsets, and the discrete metabolic programs adopted by effector and regulatory T cells (Gerriets et al., 2015; Michalek et al., 2011), we hypothesized that T helper (Th) subsets would also be differentially affected by COX dysfunction. Th1, Th2, and Th17 cells rely on high rates of glycolysis, while regulatory T cells (Tregs) are oxidative. In vitro differentiation of Th1, Th2, Th17, and Treg was performed on isolated splenic T cells from WT and TCox10−/− mice. In TCox10−/− T cells, in vitro polarization of Th1 and Th2 cells was significantly diminished. By flow cytometry, Th1 cells (IFNγ+) were decreased by 60% (p < 0.05) (Figure S7A, left and center). Th2 cells (IL-4+) were similarly afflicted (63% reduction, p < 0.05) (Figure S7B, left and center). Analogous to Th1 and Th2 T cells, TCox10−/− Th17 cells (IL-17+) were reduced by 70% (p < 0.0001) (Figure 5E, left and center). Evidently, WT Th1, Th2, and Th17 cells underwent several rounds of proliferation (CFSE) prior to differentiation in vitro, a condition that TCox10−/− T cells were unable to meet (Figures S7A, S7B and 5E far right). Tregs help maintain self-tolerance and suppress autoimmune disease (Rudensky, 2011). Unlike the trend seen in WT T cells, TCox10−/− Tregs (Foxp3+) displayed similar differentiation patterns when compared to WT (p = 0.31) (Figure 5F, left and center). This finding may be explained by the overlapping proliferation profiles (Figure 5F, right). Tregs, unlike other Th subsets, do not undergo extensive proliferation prior to differentiation; this suggests distinct mitochondrial bioenergetic requirements for the generation of effectors.
Th17 (glycolytic) and Treg (oxidative) cells are interchangeable by metabolic reprogramming (Gerriets et al., 2015). Considering that Th17 cells required rounds of proliferation prior to differentiation, in contrast to Tregs, we hypothesized that their requirements for proliferation were related to their metabotype. Using extracellular flux analysis, we found that WT Th17 cells had greater bioenergetic requirements than Tregs: WT Th17 exceeded WT Treg ECAR by ~30% (p < 0.0001) and OXPHOS by ~280% (p < 0.0001) (Figure 5G). In the setting of COX dysfunction, TCox10−/− Th17 showed a ~15% reduction in ECAR (p < 0.01) and a~27% reduction in OCR (p < 0.05), consistent with their reduced ability to form Th17 cells. Although Tregs have been reported to primarily use OXPHOS (Beier et al., 2015), WT and TCox10−/− Tregs had comparable requirements for OXPHOS (OCR). This was surprising to us due to the published literature regarding the role of OXPHOS in Treg function (Beier et al., 2015). Based on our results, we suggest that Tregs may have oscillating bioenergetic requirements depending on their stage of development.
T Cell COX Dysfunction Produces Abnormal T Cell Populations In Vivo
With evidence of abnormal in vitro T cell activation and differentiation, we next sought to characterize TCox10−/− T cell populations in vivo. In the thymus of TCox10−/− mice, total thymocyte number and percentage of CD4+CD8+ T cells were equivalent when compared with WT (Figure 6A, left, center left). While the percent of CD4+ T cells in TCox10−/− mice was slightly reduced (p < 0.05) (Figure 6A, center right), the percent of CD8+ T cells was comparable to WT mice (Figure 6A, right). In the spleen of TCox10−/− mice, CD4+ T cells were diminished by 32% (p < 0.05) (Figure 6B, left, center left). Consistent with our in vitro results demonstrating greater influence of COX dysfunction in CD8+ T cells, TCox10−/− splenic CD8+ T cells showed a 47% reduction (p < 0.01). Splenic B cell percentages (B220+) were unaffected, and myeloid cells (CD11b+) increased 2× (p < 0.01), potentially reflecting changes in relative cell number (Figure 6B).
Figure 6. T Cell Populations Are Altered in TCox10−/− Mice In Vivo.

(A) Thymic T cell populations (n = 3 per condition).
(B) Splenic cell populations (n = 3 per condition).
(C) CD45RB expression on splenic T cells (n = 3 per condition).
(D) Regulatory T cells (n = 3 per condition).
(E) Bone marrow transfer studies (n = 4 per condition).
Experiments were performed three or more times with representative data shown. *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars denote SEM.
As T cells become activated and eventually transition to memory, CD45RB downregulates from the cell surface. In WT mice, staining for CD45RB demonstrated two main populations: CD45RBhi represented naive T cells, while CD45RBlo represented activated or memory T cells (Figure 6C). In TCox10−/−, this relationship is not seen; there are primarily naive T cells (CD45RBhi) with significantly reduced activated and memory T cells (CD45RBlo). These data are consistent with our clinical findings (Figure 1B), in which patients with MD show elevated levels of naive T cells and a paucity of memory T cells. Despite the ability to differentiate in vitro (Figure 5F), 2-month-old TCox10−/− mice had 27% fewer Treg cells than WT (p < 0.05). These results support our assertion that Treg may have varying bioenergetic requirements that are dependent upon the stage of cell development, a point that bears further investigation.
Due to their diminished cell fitness in vitro and altered T cell populations in vivo, we hypothesized that TCox10−/− T cells would be unable to compete for immunologic niches in vivo to permit their survival. To address this question, we performed bone marrow chimera studies using CD45.1 as a lineage marker to examine the relative contribution of TCox10−/−bone marrow to CD4+ and CD8+ T cell populations (Figure 6E). At 3 months after transfer of equal numbers of WT and TCox10−/− T cells, TCox10−/− CD4+ T cells were decreased by 25% (p < 0.01) when normalized to B cells. Consistent with our in vitro findings, CD8+ cells were affected more by their COX dysfunction, showing a 55% reduction when compared to WT (p < 0.0001).
T Cell COX Dysfunction Results in Abnormal Vaccine Responses In Vivo
Adaptive immunity creates highly specific immunological memory in T cells and B cells after initial exposure to a pathogen. This priming produces an enhanced response to subsequent encounters resulting in either an abrogation or an attenuation of infection and is the basis of vaccination. To characterize the development of immune memory and protection, we conducted vaccination studies in TCox10−/− mice. As many vaccines are derived from protein antigens, we began by immunizing TCox10−/− and WT mice with 2,4,6-trinitrophenyl chicken gamma globulin (TNP-CGG) (Figure 7A). T-dependent primary immune responses to TNP (IgG1) in TCox10−/− mice were reduced to 22% of WT (p < 0.0001), while secondary immune responses were similarly affected at 12% of WT (p < 0.0001). To elicit T-dependent B cell germinal center (GC) formation and antibody response, mice were vaccinated with sheep red blood cells. SRBC specific IgG was present at 9% of normal in TCox10−/− (p < 0.05) (Figure 7B). The diminished production of SRBC IgG was due to a failure of GC formation. GC B cells (B220+, IgDlo, Fas+, GL7+, [Bgc]) were 44% of WT (p < 0.05) (Figure 7C). Although T follicular helper cell numbers (PD-1+, CXCR5+, Tfh) appeared to be unchanged due to COX dysfunction, they are unable to support GC formation, suggesting that these cells are not functional.
Figure 7. Abnormal Vaccine Responses in TCox10−/− Mice In Vivo.

(A) Antibody response to TNP-CGG (n = 11 of 13).
(B) Antibody response to sheep red blood cell (SRBC) immunization (n = 4 per condition).
(C) Germinal center formation (n = 3 per condition); bar graphs (right).
(D) Peripheral lymphocytes at baseline (top) and during H3N2 (X31) influenza infection (bottom) (n = 5 per condition).
(E) Viral titers following X31 influenza immunization and rechallenge (n = 3–6 per condition).
(F) Viral-specific T cells following X31 immunization and PR8 influenza challenge (n = 5 per condition).
(G) Viral titers following X31 immunization and PR8 influenza challenge (n = 4 per condition).
(H) Expansion of OT-I cells following challenge with X31-OVA (n = 5 per condition).
(I) Viral lung titer for NS1 (n = 5 per condition).
TNP-CGG, 2,4,6-trinitrophenyl chicken gamma globulin; neg ctrl, negative control; X31-OVA, X31 influenza virus containing ovalbumin; NS-1,influenza non-structural protein 1. Experiments (n = 3 per condition) were performed three or more times with representative data shown. *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars denote SEM.
Since patients with MD suffer from recurrent upper- and lower-respiratory infections (Figure 1), which may result in either death or long-term sequelae (Edmonds et al., 2002), we decided to evaluate viral immunity in TCox10−/− mice. Mice were infected with mouse-adapted H3N2 influenza A/X/31 (X31). At baseline, TCox10−/− mice showed normal peripheral WBC and lymphocyte counts (Figure 7D, top). After 5 days of influenza infection, lymphocyte counts (p < 0.05) and percentage (p < 0.001) dropped, mirroring our clinical data (Figure 1C). To characterize the memory immune response, secondary challenge was performed with X31virus (H3N2) at 28 days and mice were euthanized at 35 days to examine lung viral titers by PCR (Figure 7E). Unlike WT mice, TCox10−/− mice were unable to clear X31, as evidenced by significantly elevated lung viral titers (p < 0.01). As the immunologic response to X31 involves both T cells and B cells, we wanted to isolate the viral specific T cell response (CD4+ and CD8+) by performing a viral isotype switch experiment. Primary infection was performed using X31 virus followed 28 days later by infection with H1N1 influenza A/PR/8 (PR8, H1N1). In this experiment, CD4+ and CD8+ T cell responses are specific for viral core proteins, which are shared between the two viruses. At 35 days after infection—7 days after secondary challenge with PR8—the number of viral-specific T cells in the spleen was slightly lower (p < 0.05) (Figure 7F). In keeping with reduced viral-specific T cells, the lung viral titer in TCox10−/− mice was ~100 times greater than in WT mice (p < 0.01) (Figure 7G).
Because our isotype-switch experiment was a mixed CD4+ and CD8+ T cell response, we next sought to establish the consequences of COX dysfunction in CD8+ T cells for the clearance of influenza using an attenuated ovalbumin containing X31 influenza virus (X31-OVA). TCox10−/− and WT CD8+ OT-I T cells (CD45.2+) were transferred to a WT mouse (CD45.1+), which was subsequently infected with X31-OVA. After 7 days of infection, X31-OVA-specific TCox10−/− CD8+ T cells failed to expand as robustly as transferred WT T cells (p < 0.01) (Figure 7H). This attenuation of X31-OVA-specific T cells translated into an ~6× increase in lung viral titer (p < 0.001) in TCox10−/− (Figure 7I). Our findings are consistent with persistent influenza infection in TCox10−/− mice and suggest that COX dysfunction significantly affects T memory responses in vivo.
Conclusions
To date, the majority of publications on the role of OXPHOS in T cells have relied on the use of chemical inhibitors to induce dysfunction, with some exceptions. Studying MD as a translational model system for understanding immune cell bioenergetics is a unique approach. Our findings presented herein emphasize the unique role of COX in T cell activation, differentiation, and function using a genetic model of human MD. Unlike complex III, which mediates T cell activation through the generation of secondary messengers (Sena et al., 2013), COX is involved in life-and-death decisions early in cell activation, marking it as a critical metabolic checkpoint. Although the ways in which mitochondria contribute to T cell activation and function are just beginning to be explored (Ron-Harel et al., 2016; Sena et al., 2013), it is clear that COX activity is essential and that inhibition promotes a modulation of metabolic flux and increased susceptibility to apoptosis, revealing the vulnerability of different T cell subsets.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
-
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients
Murine Model of TCox10−/−
-
METHOD DETAILS
Influenza Infection
BM Transfer
Enzyme Assay
Spectroscopic Cytochrome a Quantitation
Proteomics
Stable Isotopes
Flow Cytometry
Cell Isolation and Proliferation Assays
Transmission Electron Microscopy
Real-Time PCR
Immunoblot Studies
In Vitro Differentiation
OCR and ECAR Measurement
Immunization and Serum Analysis
QUANTIFICATION AND STATISTICAL ANALYSIS
DATA AND SOFTWARE AVAILABILITY
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter McGuire (peter.mcguire@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients
A retrospective chart review was performed on patients evaluated by a neurologist (M.K.K.) and an immunologist (S.P.) at the Mitochondrial Center of Excellence at the University of Texas, Houston. Patients were assigned a diagnosis of “Probable” or “Definite” mitochondrial disease based on the Walker criteria (Walker et al., 1996). Age, sex, and sample size is provided in the text. The studies were carried out under the Institutional Review Boards of the University of Texas, Houston and the National Human Genome Research Institute.
Murine Model of TCox10−/−
B6.129×1-Cox10tm1Ctm/J mice were kind gift of C. Morales and F. Diaz (University Miami) and were crossed with CD4-Cre transgenic mice (B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ) from The Jackson Laboratory. The resultant mice are referred to throughout the paper as TCox10−/−. For all experiments except where otherwise indicated, male and female mice, 8–12 weeks were used. Littermate controls were randomly assigned to experimental procedures. Mice were housed in a pathogen-free facility, caged individually had access to a 22% protein pellet based feed, (Bio-Serv) and autoclaved reverse osmosis water. Mice were kept in a temperature (22 + 2°C) and humidity (30%–70%) controlled environment with a 12 hr light cycle. All animal care and procedures were carried out according to the criteria outlined in the ‘Guide for the Care and Use of Laboratory Animals’ prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985) and were authorized by the Animal Care and Use Committees of the National Human Genome Research Institute.
METHOD DETAILS
Influenza Infection
Mouse adapted human influenza virus A/PR/8/34 (PR8), A/X/31 (X31) and X31 containing ovalbumin (X31-OVA) for infection were used. Mice (N = 10 / genotype) were exposed to aerosolized (Glas-Col) 500 TCID or PR8 in 10mL of saline. Details and time points of infection are outlined in the text. Expression of viral hemagglutinin (HA) in the lungs of infected mice was determined by real time PCR.
BM Transfer
WT (CD45.1) and TCox10−/− (CD45.2) bone marrow cells were mixed 1:1, and a total of 5 × 106 were injected intraorbitally into 8–12 week old irradiated B6 mice (950 rad). Mice (N = 3) were then euthanized at time points as defined in the text.
Enzyme Assay
Cytochrome c oxidase enzyme activity was determined in T cells stimulated for 24 hr with anti CD3 and CD28 using a method previously published (DiMauro et al., 1987). Briefly, frozen T cells pellets (~5 million cells) were suspended in 1 × phosphate buffered saline (Fisher Scientific, Waltham, MA) and sonicated 10 s on ice. Activity of cytochrome c oxidase was measured by following the oxidation of 1% reduced cytochrome C at 550 nm at 30°C. Reduced cytochrome C was prepared by adding 5ul/ml of 5% sodium hydrosulfite (Fisher Scientific) to a solution of cytochrome C from bovine heart (Sigma-Aldrich) in 0.01M KPO4 (Fisher Scientific) pH7.
Spectroscopic Cytochrome a Quantitation
Cytochrome a content in retinas was determined spectrophotometrically in a commercial spectrophotometer (Shimadzu 2700 UV-Vis; Shimadzu Corp., Kyoto, Japan). T cell pellets (N = 3) were resuspended in 0.1 mL of ice-cold buffer containing 280 mM sucrose, 10 mM HEPES, 1mM EDTA and 1 mM EGTA, adjusted to pH 7.1 with KOH. An aliquot of the same buffer (0.9 mL) containing 1% n-dodecyl βD-maltoside was added to the homogenate, followed by vortexing for 45 s. After centrifugation for 2 min at 16,000 g, the supernatant was transferred to a plastic cuvette. Potassium hexacyanoferrate(III) 0.1 mM was added to ensure full oxidation of the cytochromes, and five spectra in the wavelength range of 500 to 650 nm were collected and averaged. Then, 0.5 mM of potassium cyanide was added together with 10 mM sodium ascorbate (pH 7.4) to reduce cytochrome a, and five reduced spectra were collected and averaged. The oxidized spectrum was subtracted from the reduced spectrum, and the absorbance difference at 605 nm was calculated after manually tracing a baseline from 575 and 630 nm. An extinction coefficient of 10.8 mM−1 cm−1 was used to obtain cytochrome a concentration (Balaban et al., 1996).
Proteomics
Sample Preparation and Fractionation
Five naive and five stimulated T cell pellets were thawed and resuspended with 100 µL of lysis buffer containing 100 mM triethylammonium bicarbonate (TEAB) and 1% SDS. Each of the samples was loaded onto a QIAshredder spin column (QIAGEN) placed in a 2 mL collection tube, and spun for 2 min at maximum speed in a microcentrifuge to disrupt DNA and reduce viscosity. The flow-through for each sample was collected and protein was quantified using the Pierce detergent compatible Bradford assay kit (Thermo Fisher Scientific). A volume corresponding to 100 mg of protein was taken to 100 mL by adding the necessary volume of lysis buffer. Each sample was reduced by mixing with 5 µL of the 200 mM DTT and incubating at 55°C for 1 hr, and then alkylated by adding 5 µL of 375mM iodoacetamide and incubating for 30 min protected from light at room temperature. Protein in each sample was precipitated by adding 600 µL of pre-chilled acetone and storing overnight at −20°C. Samples were then centrifuged at 8000 × g for 10 min at 4°C, and acetone was carefully decanted without disturbing the white pellet, which was allowed to dry for 2–3 min. Each protein pellet was resuspended in 100 µL of a buffer containing 100 mM TEAB and 0.1% of Progenta anionic acid-labile surfactant I (AALSI, Protea Biosciences). Each sample was proteolyzed by adding 10 µL of 1.25 µg/µl sequencing grade modified trypsin (Promega) dissolved in 100 mM TEAB and incubated overnight at 37°C. Each sample was labeled with a different TMT label reagent (Thermo Fisher Scientific) by adding the contents of each label tube after dissolving with 41 µL of acetonitrile. The reaction was allowed to proceed for 1 hr at room temperature, and was quenched by adding 8 µL of 5% hydroxylamine to each sample and incubating for 15 min. AALSI was cleaved by adding 30 µL of 10% trifluoroacetic acid (TFA). All ten samples were combined samples in a new microcentrifuge tube, which was dried under vacuum until all acetonitrile was removed. The combined sample was desalted using an Oasis HLB column (Waters) and dried under vacuum. High pH reversed-phase liquid chromatography was performed on an offline Agilent 1200 series HPLC. The desalted peptides were resuspended in 0.1 mL 10 mM TEAB with 5% (v/v) acetonitrile. Peptides were loaded onto an Xbridge C18 HPLC column (Waters; 2.1 mm inner diameter × 100 mm, 5 µm particle size), and profiled with a linear gradient of 5%–40% buffer B (90% acetonitrile, 10 mM TEAB) over 60 min, at a flowrate of 0.15 ml/min. The chromatographic performance was monitored by sampling the eluate with a diode array detector (1200 series HPLC, Agilent) scanning between wavelengths of 200 and 400 nm. Fractions were collected at 1 min intervals followed by fraction concatenation. Twelve concatenated fractions were dried and resuspended in 0.01% formic acid, 5% acetonitrile. Approximately 500 ng of peptide mixture was loaded per liquid chromatography-mass spectrometry run.
Mass Spectrometry
All mass spectrometry experiments were performed on an Orbitrap Fusion coupled with a Ultimate 3000-nLC (Thermo Fisher Scientific). Peptides were separated on a EASY-Spray C18 column (Thermo Scientific; 75 µm × 25 cm inner diameter, 2 µm particle size and 100 Å pore size). Separation was achieved by first applying a quick 5%–7% gradient of acetonitrile +0.1% formic acid over 5 min at 300 nl(min−1), followed by 7%–25% acetonitrile + 0.1% formic acid over 65 min and ending at 25%–40% acetonitrile + 0.1% formic acid over 10 min. An electrospray voltage of 1.9 kV was applied to the eluent via the EASY-Spray column electrode. The Orbitrap Fusion was operated in positive ion data-dependent mode, using Synchronous Precursor Selection (SPS-MS3). Full scan MS1 was performed in the Orbitrap with a precursor selection range of 380–1,500 m/z at nominal resolution of 1.2 × 105. The AGC target and maximum accumulation time settings were set to 2 × 105 and 60 ms, respectively. MS2 was triggered by selecting the most intense precursor ions above an intensity threshold of 1 × 104 for collision induced dissociation (CID)-MS2 fragmentation with an AGC target and maximum accumulation time settings of 1 × 104 and 70 ms, respectively. Mass filtering was performed by the quadruple with 1.6 m/z transmission window, followed by CID fragmentation in the linear ion trap with 35% normalized collision energy in rapid scan mode. SPS was applied to co-select 10 fragment ions for HCD-MS3 analysis. SPS ions were all selected within the 400–2,000 m/z range, and were set to preclude selection of the precursor ion and TMTC ion series. The AGC target and maximum accumulation time were set to 2 × 104 and 120 ms (respectively) and parallelizable time option was selected. Co-selected precursors for SPS-MS3 underwent HCD fragmentation with 65% normalized collision energy, and were analyzed in the Orbitrap with nominal resolution of 3 × 104. The number of SPS- MS3 spectra acquired between full scans was restricted to a duty cycle of 3 s.
Data Processing
Raw data files were processed using Proteome Discoverer (v2.1, Thermo Fisher Scientific), using three embedded search nodes; Mascot (v2.5.1, Matrix Science), Sequest HT (Thermo Fisher Scientific) and MS Amanda (v2.1.5, Bioinformatics Research Group) search algorithms. All searches were performed against SwissProt human database (May 2016, 20,202 sequences), with carbamidomethylation of cysteine, and TMT 10-plex modification of lysine and peptide N termini set as static modifications and oxidation of methionine as dynamic. For SPS-MS3 the precursor and fragment ion tolerances of 12 ppm and 0.6 Da were applied, respectively. Up to two-missed tryptic cleavages were permitted. Percolator algorithm was used to calculate the false discovery rate (FDR) of peptide spectrum matches, set to a q-value of 0.05.
Peptide quantification and normalization was also performed by Proteome Discoverer v2.1 by calculating the reporter peak intensity based on the sum of centroid ions within 20 ppm window around the expected m/z for each of the 10 TMT reporter ions. Quantification was performed at the MS3 level where the median of all quantifiable PSMs for each protein group was used for protein ratios, and only those PSMs in which all 10 tags were detected were considered. Normalization was performed on total peptide amount: after aggregating all abundance values per quantification channel, the node normalizes the abundance values of each channel with a constant channel-specific factor so that all channels have the same total peptide abundance at the end.
Stable Isotopes
Mouse T cells were stimulated for 24 hr with plate-bound anti-CD3/CD28. Labeling experiments were performed essentially as described previously (Cheng et al., 2011; Mullen et al., 2012). All labeling experiments were performed with 1 million cells/mL cultured in RPMI containing 11 mM glucose and 2 mM glutamine, with one nutrient or the other replaced by a uniformly 13C-labeled analog (i.e., [U−13C] glucose or [U−13C] glutamine; Cambridge Isotope Laboratories). Cells were rinsed in phosphate-buffered saline, then replenished with labeling medium at time 0. Culture proceeded for 24 hr, then the cells were briefly rinsed in cold saline, pelleted, and lysed in cold 50% methanol. The lysates were subjected to at least three freeze-thaw cycles, then centrifuged to remove debris. The supernatants were evaporated to dryness methoximated and derivatized by tert-butyl dimethylsilylation. One µL of the derivatized material was injected onto an Agilent 6970 gas chromatograph equipped with a fused silica capillary GC column (30 m length, 0.25 mm diameter) and networked to either an Agilent 5973 or a 5975 Mass Selective Detector. Retention times of all metabolites of interest were validated using pure standards. The measured distribution of mass isotopomers was corrected for natural abundance of 13C (Des Rosiers et al., 1994).
Flow Cytometry
Single-cell suspension of tissues were prepared. Anti- CD4, CD8, CD44, B220, CD69, CD247, IgM, CXCR5, PD-1, Foxp3 antibody were purchased from BDBiosciences or ebioscience. Data were acquired on a FACSCalibur flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star). Labeled tetramers (NIH tetramer core facility) were used to identify viral specific T cells. MitoTracker Green (Molecular Probes) and TMRE (Abcam) were used according to manufacturer instructions. Apoptosis analysis was measured by Annexin V staining (ebioscience). Substrate cleavage by caspase3 was measured with caspase3 substrate PhiPhilus-G1D2 (OncoImmunin) according to the manufacturer instructions. Cells were loaded with 5 µM CFSE (ThermoFisher Scientific) and proliferation were estimated on day 3 by FACS. For calcium influx assays, T cells were loaded with Fluo4-NW (Molecular Probes, Eugene, OR) according to the manufacturer’s instructions. Cells were stimulated with biotinylated anti CD3 and crosslinked with streptavidin (Sigma-Aldrich). All assays were performed with N = 3 or more per condition and repeated 3 times.
Cell Isolation and Proliferation Assays
Pan T cells, CD8+ T and CD4+ cells were enriched using isolation kits (Miltenyi Biotec). Purity of T cells was > 95% in all cases. T cells were stimulated with plate-bound anti-CD3 (5 µg/ml) and anti-CD28 (0.5 µg/ml) in glucose or galactose containing media (Sigma-Aldrich) with various concentrations of FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) (Sigma-Aidrich) or Z-VAD-FMK (pan caspase inhibitor) (R&D systems, Minneapolis, MN).T cell proliferation was assessed by [3H]TdR incorporation in the last 8 hr of culture. Cell were treated with 2-deoxyglucose any varying concentrations and FG4592 at 50 µM as described in the results. All assays were performed with N = 3 or more per condition and repeated 3 times.
Transmission Electron Microscopy
T cells were fixed for 48 hr at 4°C in 2% glutaraldehyde and 1% paraformaldehyde in 0.1M cacodylate buffer (pH 7.4) and washed with cacodylate buffer three times. The T cells were fixed with 1% OsO4 for two hours, washed again with 0.1M cacodylate buffer three times, washed with water and placed in 1% uranyl acetate for 1 hr. The pellets were subsequently serially dehydrated in ethanol and propylene oxide and embedded in EMBed 812 resin (Electron Microscopy Sciences, Hatfield, PA, USA). Thin sections, approx. 80 nm, were obtained by utilizing the Leica ultracut-UCT ultramicrotome (Leica, Deerfield, IL, USA) and placed onto 300 mesh copper grids and stained with saturated uranyl acetate in 50% methanol and then with lead citrate. The grids were viewed in the JEM-1200EXII electron microscope (JEOL Ltd, Tokyo, Japan) at 80kV and images were recorded on the XR611M, mid mounted, 10.5Mpixel, CCD camera (Advanced Microscopy Techniques Corp, Danvers, MA, USA). Masks for representative mitochondria to demonstrate cristae morphology were created by adjusting the brightness and contrast of images.
Real-Time PCR
RNA was extracted from the tissues using Pure link RNA mini kit (Thermo Fisher Scientific) and was reverse transcribed to cDNA (iScript, Bio-Rad) according to the manufacturer’s instructions. Reactions were cycled and quantitated with an ABI 7500 Fast Real Time PCR System (Applied Biosystems). All assays were performed with N = 3 or more per condition and repeated 3 times.
Immunoblot Studies
For western blot analysis, approximately 30 mg of protein was loaded on either 10% (for pERK/ERK) or 4%–20% (for MTCO1, pPDH/ PDH) Tris-glycine polyacrylamide gels. The gels were run at 150 V for approximately 1.5 hr. The gels were transferred to either nitrocellulose (for pERK/ERK blots) or polyvinylidene difluoride membrane (for MTCO1, pPDH/PDH blots) using the iBlot Dry Blotting System (Life Technologies, Grand Island, NY). The membranes were incubated overnight at 4°C in blocking buffer (Li-Cor, Lincoln, NE). The membranes were probed with the following primary antibodies: MTCO1, phosphor-ERK (Cell Signaling, Danvers, MA), total ERK (Cell Signaling, Danvers, MA), PDH and phosphor-PDH (Abcam, San Francisco, CA) and b-actin (Sigma-Aldrich, St. Louis, MO). The membranes were washed three times for 10 min each with TBS containing 0.1% Tween 20, followed by incubation with indicated IRDye secondary antibodies (Li-Cor, Lincoln, NE). Image analyses were performed using an Odyssey Imager (Li-Cor, Lincoln, NE).
In Vitro Differentiation
Naive (CD4+ (PerCP/Cy5.5, clone RM4–5) CD44low (APC, clone IM7) CD62Lhi (eFluor 450, clone MEL-14) CD25neg (PE, clone PC61.5) T cells) were purified by cell sorting. Purity was greater than 99% purity. Sorted naive CD4+ T cells (2 × 105) were co-cultured at a ratio of 1:5 with mitomycin-treated T-depleted splenocytes as APCs in 48-well plates under various differentiation conditions for 3 days. Th1 conditions included 40 ng/ml IL-12 and anti-IL-4. Th2 conditions included 1 µg/ml anti-CD3, 3 µg/ml anti-CD28, 20 ng/ml IL-4 and 10 µg/ml anti-IL-12. Th17 used 20 ng/ml of IL-6, 5 ng/ml of TGF-β1, anti-IL-4, anti-IFN-γ, and anti-IL-12. Treg used 100 U/ml hIL-2, 5 ng/ml TGF-β1 with 0.1, 1, or 5 µg anti-CD3, and 10 µg/ml of each anti-IL-4, anti-IFN-γ, and anti-IL-12 antibodies. Antibodies were purchased from BioXcell unless otherwise indicated. All assays were performed with N = 3 or more per condition and repeated 3 times.
OCR and ECAR Measurement
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse XF24 analyzer (Seahorse Bioscience). T cells from mice activated for 24 hr with anti CD3 and anti CD28 were attached with Cell-Tak (Corning) according to manufacturer’s instructions at concentration 0.5 million cells/well in Seahorse BASE media with proprietary additives. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were determined using the Mitostress kit (Seahorse Biosciences) according to the manufacturer’s standard protocol. OCR and ECAR were calculated and recorded by the Seahorse XF-24 software. Complex IV activity (COX) was measured according to published methods (Salabei et al., 2014) using tetramethyl-p-phenylenediamine (TMPD) as an electron donor that is specific for complex IV with OCR as the readout. All assays were performed with N = 3 or more per condition and repeated 3 times.
Immunization and Serum Analysis
Mice were immunized with 50 µg of TNP-CGG in Imject Alum (Pierce Chemical) and re-immunized with TNP-CGG alone in 24 days. Sera were tested by ELISA for TNP reactivity. Briefly, plates were coated with TNP-BSA or (10 µg/ml; Biosearch Technologies), and bound immunoglobulins were detected by alkaline phosphotase-conjugated detection antibodies to specific mouse isotypes (Southern Biotechnology Associates). Mice were also immunized with sheep RBC and Tfh and GC B cell formation were evaluated on day 9 by FACS. All assays were performed with N = 3 or more per condition and repeated 3 times.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using Prism (Graphpad Software). Summary statistics were generated for all data. Unpaired Student’s t test was used for comparing two groups where the populations followed a normal distribution, similar variance, and were sampled independently. Nonparametric Mann-Whitney test was used for viral titers (Figure 7), where the data were not normally distributed. P value of < 0.05 was considered to be statistically significant. N signifies number tested, while error bars are standard error of the mean.
DATA AND SOFTWARE AVAILABILITY
The accession number for the stable isotope data reported in this paper is [Mendeley]: http://dx.doi.org/10.17632/s7f6r3x94n.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse T and B activation Antigen (clone GL7) FITC | BD Bioscience | Cat#553666 |
| Anti-Mouse IgD (clone 11-26c.2a) APC | BD Bioscience | Cat#560868 |
| Anti-Mouse CD3 (clone 145-2C11) | BD Bioscience | Cat# 553058 |
| Anti-Mouse CD45RB (clone 16A) | BD Bioscience | Cat# 553101 |
| Anti-Mouse CD45.1 (clone A20) APC | BD Bioscience | Cat# 561873 |
| Anti-Mouse CD3 (clone 145-2C11) biotin | BD Bioscience | Cat# 553060 |
| IL-9 Monoclonal AB (RM9A4) eFluor 660 | BD Bioscience | Cat#50-8091-82 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit mAb | Cell Signaling | Cat# 4370 |
| p44/42 MAPK (Erk1/2) (3A7) Mouse mAb | Cell Signaling | Cat# 9107 |
| Anti-Mouse CD45.2 (clone104) FITC | Ebioscience | Cat# 11-0454-81 |
| Anti-Mouse CD4 (clone RM4-5) PerCP-Cy5.5 | Ebioscience | Cat# 45-0042-82 |
| Anti-Mouse CD62L (clone MEL-14) eFluor 450 | Ebioscience | Cat# 48-0621-82 |
| Anti-Mouse CD8a (clone 53-6.7) | Ebioscience | Cat# 17-0081-82 |
| Anti-Mouse CD44 (clone IM7) PE | Ebioscience | Cat# 12-0441-82 |
| Anti-Mouse CD44 (clone IM7) APC | Ebioscience | Cat# 17-0441-82 |
| Anti-Mouse CD25 (clone PC61.5) PE | Ebioscience | Cat# 12-0251-82 |
| Anti-Mouse CD45R(B220) (clone RA3-6B2) | Ebioscience | Cat# 17-0452-82 |
| Anti-Mouse CD185 (CXCR5) (clone SPRCL5) PE | Ebioscience | Cat# 12-7185-82 |
| Anti-Mouse IgM (clone II/41) APC | Ebioscience | Cat#17-5790-82 |
| Anti-Mouse CD95 (clone APO-1/Fas) | Ebioscience | Cat#12-0951-81 |
| Anti-Mouse PD-1 | Ebioscience | Cat#11-9985-82 |
| Anti-Mouse C185(CXCR5) (clone SPRCL5) PE | Ebioscience | Cat#12-7185-82 |
| Anti mouse CD25 (clone PC61.5) Alexa Fluor | Ebioscience | Cat# 53-0251-82 |
| Anti-Mouse PD-1 (clone J43) FITC | Ebioscience | Cat# 11-9985-81 |
| Anti-Mouse CD28 Functional Grade Purified (clone37.51) | Ebioscience | Cat# 16-0281-82 |
| IFN gamma Monoclonal AB (XMG1.2) PerCp-Cyanine5.5 | Ebioscience | Cat#45-7311-82 |
| IL-4 Monoclonal AB (clone BVD6-24G2) FITC | Ebioscience | Cat#11-7042-82 |
| IL-17A Monoclonal AB (ebio17B7) PE | Ebioscience | Cat#12-7177-81 |
| FOXP3 Monoclonal AB (FJK-16 s) eFluor 450 | Ebioscience | Cat#48-5773-82 |
| Anti IL-4 (clone 11B11) | ebioscience | Cat#16-7041-81 |
| Anti-IL-12 (clone C17.8) | ebioscience | Cat#16-7123-81 |
| IRDye® 800CW Goat anti-Rabbit IgG (H + L) | Li-Cor | Cat# 925-32211 |
| IRDye® 680LT Goat anti-Mouse IgG (H + L), 0.1 mg | Li-Cor | Cat# 925-68020 |
| Anti-Mouse CD28 (clone 37.51) | Thermo Fisher Scientific | Cat#MA1-10172 |
| Anti-IFN-g (clone XMG1.2) | Thermo Fisher Scientific | Cat#MM700 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TMRE Mitochondrial Membrane Potential Assay kit | Abcam | Cat# ab113852 |
| Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) | Abcam | Cat#ab120081 |
| TNP-BSA | Biosearch Tech | Cat#T5050-10 |
| Sucrose | Calbiochem | Cat#573113 |
| KH2PO4 | Calbiochem | Cat#529568 |
| [U-13C]glucose | Cambridge Isotope Laboratories | Cat# CLM1396 |
| [U-13C]glutamine | Cambridge Isotope Laboratories | Cat# CLM-1822 |
| FG4592 | Cayman Chemical | Cat#15294 |
| Sheep red blood cells, Alsevers | Colorado Serum Company | Cat#31112-60ml |
| Cell-Tak | Corning | Cat#354240 |
| HEPES | Fisher Scientific | Cat# BP310-500 |
| HEPES | Fisher Scientific | Cat# BP310-500 |
| Sodium hydrosulfite | FisherScientific | Cat#S310-100 g |
| Potassium cyanide | J.T. Baker | Cat# 3080-01 |
| Sucrose | MP Biomedicals | Cat# 021528490 |
| IL-4 cytokine, mouse recombinant | PeproTech | Cat#214-14 |
| IL-6 cytokine, mouse recombinant | PeproTech | Cat#216-16 |
| TGFb1 cytokine, mouse | PeproTech | Cat#100-21 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5111 |
| Progenta Acid Labile Surfactant (AALS I) | Protea | Cat# ALS-100-1 |
| Recombinant Mouse TL1A | R&D | Cat#1896TL |
| Z-VAD-FMK | R&D | Cat#FMK001 |
| XF calibrant solution | Seahorse Bioscience | Cat#100840-000 |
| Seahorse XF Base Medium | Seahorse Bioscience | Cat#102353-100 |
| Seahorse XFe96 FluxPak mini | Seahorse Bioscience | 102601-100 |
| XF calibrant solution | Seahorse Bioscience | Cat#100840-000 |
| Cytochrome C from Bovine heart | Sigma Aldrich | Cat# C3131 |
| Oligomycin | Sigma Aldrich | Cat#75351-5mg |
| Glucose | Sigma Aldrich | Cat#D8270-100 g |
| Galactose | Sigma Aldrich | Cat#G0750-100 g |
| Rotenon | Sigma Aldrich | Cat#R8875-1g |
| Antimycin(AA) | Sigma Aldrich | Cat#A8674-100mg |
| 2-Deoxy-D-glucose | Sigma Aldrich | Cat#D8375-5g |
| NNNN Tetramethyl-p-phenylenediamine (TMPD) | Sigma Aldrich | Cat# T7394 |
| L-ascorbic acid | Sigma Aldrich | Cat# A5960 |
| Saponin | Sigma Aldrich | Cat#47036 |
| MgCl2 | Sigma Aldrich | Cat#M8266 |
| Protease inhibitor | Sigma Aldrich | Cat#P8340 |
| PMA | Sigma Aldrich | Cat#P1585-1MG |
| Ionomycin calcium salt from Streptomyces conglobatus | Sigma Aldrich | Cat#I0634-1MG |
| Methoxyamine | Sigma Aldrich | Cat#226904 |
| Pyridine | Sigma Aldrich | Cat# 270407 |
| N-tert-Butyldimethylsilyl-N-methyltrifluoroacetamide | Sigma Aldrich | Cat# 394882 |
| Potassium hexacyanoferrate(III) | Sigma-Aldrich | Cat# P8131 |
| Sodium dodecyl sulfate (SDS) | Sigma-Aldrich | Cat# 436143 |
| Triethylammonium bicarbonate (TEAB) | Sigma-Aldrich | Cat# 90360 |
| Potassium hexacyanoferrate(III) | Sigma-Aldrich | Cat# P8131 |
| Acetone | Sigma-Aldrich | Cat# 34850 |
| Ethylene glycol-bis(2-aminoethylether)-N,N, N′,N′-tetraacetic acid (EGTA) | Sigma-Aldrich | Cat# E4378 |
| Ethylenediaminetetraacetic acid dipotassium salt dihydrate (EDTA) | Sigma-Aldrich | Cat# ED2P |
| L-ascorbic acid | Sigma-Aldrich | Cat# A5960 |
| n-Dodecyl β-D-maltoside | Sigma-Aldrich | Cat# D4641 |
| Acetonitrile LC/MS Grade | Thermo Fisher Scientific | Cat# TS-51101 |
| Dithiothreitol (DTT) | Thermo Fisher Scientific | Cat# 20291 |
| Formic Acid LC-MS Grade | Thermo Fisher Scientific | Cat# 85178 |
| Hydroxylamine 50% | Thermo Fisher Scientific | Cat# 90115 |
| Iodoacetamide | Thermo Fisher Scientific | Cat# 90034 |
| Trifluoroacetic acid (TFA) | Thermo Fisher Scientific | Cat# 28901 |
| Water LC-MS Grade | Thermo Fisher Scientific | Cat# TS-51140 |
| Phosphatase inhibitor | Thermo Fisher Scientific | Cat#1862495 |
| B actin probe Mm00607939_ s1 one exon | Thermo Fisher Scientific | Cat#4331182 |
| Critical Commercial Assays | ||
| iScript cDNA syntesis kit | Bio-Rad | Cat#1708890 |
| Annexin V apoptosis Detection kit FITC | Ebioscience | Cat# 88-8005-74 |
| Anti-Mouse/Rat Foxp3 Staining Set PE | Ebioscience | Cat# 72-5775-40 |
| Pan T cell isolation kit | Miltenyi Biotec | Cat# 130-095-130 |
| CD4+ cell isolation kit | Miltenyi Biotec | Cat# 130-104-454 |
| CD8+ isolation kit | Miltenyi Biotec | Cat# 130-104-075 |
| Caspase 3 substrate reagent kit | Oncoimmunin | Cat# A304RIG |
| Pure link RNA kit | Thermo Fisher Scientific | Cat# 12183025 |
| SBA clonotyping System | Southern Biotech | Cat# 5300-04B |
| Detergent Compatible Bradford Assay Kit | Thermo Fisher Scientific | Cat# 23246 |
| TMT10plex Isobaric Label Reagent Kit | Thermo Fisher Scientific | Cat# 90110 |
| QIAshredder Cell-lysate homogenizer Kit | QIAGEN | Cat# 79654 |
| MitoTracker Green FM | Thermo Fisher Scientific | Cat# M7514 |
| CellTrace CFSE Cell Proliferation kit | Thermo Fisher Scientific | Cat# C34554 |
| Fluo4-NW Calcium assay kit | Thermo Fisher Scientific | Cat#F36206 |
| Deposited Data | ||
| Stable isotope data | Mendeley | http://dx.doi.org/10.17632/s7f6r3x94n.1 |
| Experimental Models: Organisms/Strains | ||
| Cox10fl/fl | Dr. Francisca Diaz (U. of Miami) | |
| Oligonucleotides | ||
| PR8 NP For | IDT DNA | CAGCCTAATCAGACCAAATG |
| PR8 NP Rev | IDT DNA | TACCTGGTTCTCAGTTCAAG |
| Cox10 ex6 For | IDT DNA | AGGGTCAGCATCACCAATAC |
| Cox10 ex6 rev | IDT DNA | GGAGACACTTACCAGCATCAA |
| Software and Algorithms | ||
| Proteome Discoverer v2.1 | ThermoFisher Scientific | Cat# IQLAAEGABSFAKJMAUH |
| Mascot v2.5.1 | Perkins et al., 1999 | http://www.matrixscience.com |
| Sequest HT | Eng et al., 1994 | https://www.thermofisher.com/order/catalog/product/IQLAAEGABSFAKJMAUH |
| MS Amanda v2.1.5 | Dorfer et al., 2014 | http://ms.imp.ac.at/?goto=msamanda |
| Percolator | Käll et al., 2007 | http://percolator.ms |
| Swiss-Prot | Bairoch and Apweiler, 1999 | http://www.uniprot.org |
| Proteome Discoverer v2.1 | ThermoFisher Scientific | Cat# IQLAAEGABSFAKJMAUH |
| Other | ||
| Oasis HLB column | Waters | Cat# 186003908 |
| Xbridge C18 column | Waters | Cat# 186003109 |
| Easy-Spray C18 column | Thermo Fisher Scientific | Cat# ES802 |
Highlights.
Patients with mitochondrial disease have an underappreciated immune phenotype
COX regulates activation and proliferation in T cells through apoptosis
Effector T cell subsets are differentially affected by COX deficiency
Mouse T cell COX deficiency produces immunodeficiency in vivo
Acknowledgments
The authors would like to thank the Animal Core facility at NHGRI for support. This work was supported by the intramural research program of the National Institutes of Health (HG200381-03). Special thanks to Dr. Maryna Eichelberger for growing the influenza virus and to Dr. Pamela Schwartzberg for her editorial comments and suggestions. R.J.D. serves as an advisor for Agios Pharmaceuticals. M.K.K. receives research funding from Stealth BioTherapeutics, Reata Pharmaceuticals, and BioElectron.
Footnotes
Supplemental Information includes seven figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2017.05.007.
AUTHOR CONTRIBUTIONS
Conceptualization, T.N.T., S.E.P., and P.J.M.; Methodology, analysis, and investigation, T.N.T., S.E.P., M.K.K., J.G.-R., S.M.K., P.M.Z., E.B., J.S., R.J.D., R.C., R.S.B., S.D., and P.J.M.; Resources, T.N.T., S.E.P., M.K.K., J.G.-R., F.D., S.M.K., P.M.Z., E.B., J.S., R.J.D., R.C., R.S.B., S.D., and P.J.M.; Original draft, P.J.M.; Reviewing and editing, T.N.T., S.E.P., M.K.K., F.D., J.G.-R., S.M.K., E.B., J.S., R.J.D., R.C., R.S.B., S.D., and P.J.M.; Supervision, P.J.M.; Project administration, T.N.T. and P.J.M.; Funding acquisition, P.J.M.
References
- Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 2012;16:1150–1180. doi: 10.1089/ars.2011.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, Shoubridge EA. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet. 2003;12:2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 1999;27:49–54. doi: 10.1093/nar/27.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban RS, Mootha VK, Arai A. Spectroscopic determination of cytochrome c oxidase content in tissues containing myoglobin or hemoglobin. Anal. Biochem. 1996;237:274–278. doi: 10.1006/abio.1996.0239. [DOI] [PubMed] [Google Scholar]
- Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J. 2015;29:2315–2326. doi: 10.1096/fj.14-268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Rathmell JC, Macintyre AN. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS ONE. 2014;9:e104104. doi: 10.1371/journal.pone.0104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Matés JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Des Rosiers C, Fernandez CA, David F, Brunengraber H. Reversibility of the mitochondrial isocitrate dehydrogenase reaction in the perfused rat liver. Evidence from isotopomer analysis of citric acid cycle intermediates. J. Biol. Chem. 1994;269:27179–27182. [PubMed] [Google Scholar]
- Diaz F. Cytochrome c oxidase deficiency: patients and animal models. Biochim. Biophys. Acta. 2010;1802:100–110. doi: 10.1016/j.bbadis.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Diaz F, Fukui H, Garcia S, Moraes CT. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell. Biol. 2006;26:4872–4881. doi: 10.1128/MCB.01767-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F, Garcia S, Padgett KR, Moraes CT. A defect in the mitochondrial complex III, but not complex IV, triggers early ROS-dependent damage in defined brain regions. Hum. Mol. Genet. 2012;21:5066–5077. doi: 10.1093/hmg/dds350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Servidei S, Zeviani M, DiRocco M, DeVivo DC, DiDonato S, Uziel G, Berry K, Hoganson G, Johnsen SD, Johnsen PC. Cytochrome c oxidase deficiency in Leigh syndrome. Ann. Neurol. 1987;22:498–506. doi: 10.1002/ana.410220409. [DOI] [PubMed] [Google Scholar]
- Dorfer V, Pichler P, Stranzl T, Stadlmann J, Taus T, Winkler S, Mechtler K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014;13:3679–3684. doi: 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds JL, Kirse DJ, Kearns D, Deutsch R, Spruijt L, Naviaux RK. The otolaryngological manifestations of mitochondrial disease and the risk of neurodegeneration with infection. Arch. Otolaryngol. Head Neck Surg. 2002;128:355–362. doi: 10.1001/archotol.128.4.355. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Fontanesi F, Soto IC, Horn D, Barrientos A. Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am. J. Physiol. Cell Physiol. 2006;291:C1129–C1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J. Immunol. 2002;168:1528–1532. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helling S, Hüttemann M, Ramzan R, Kim SH, Lee I, Müller T, Langenfeld E, Meyer HE, Kadenbach B, Vogt S, Marcus K. Multiple phosphorylations of cytochrome c oxidase and their functions. Proteomics. 2012;12:950–959. doi: 10.1002/pmic.201100618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttemann M, Lee I, Grossman LI, Doan JW, Sanderson TH. Phosphorylation of mammalian cytochrome c and cytochrome c oxidase in the regulation of cell destiny: respiration, apoptosis, and human disease. Adv. Exp. Med. Biol. 2012;748:237–264. doi: 10.1007/978-1-4614-3573-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain IH, Zazzeron L, Goli R, Alexa K, Schatzman-Bone S, Dhillon H, Goldberger O, Peng J, Shalem O, Sanjana NE, et al. Hypoxia as a therapy for mitochondrial disease. Science. 2016;352:54–61. doi: 10.1126/science.aad9642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol. Biol. Cell. 2009;20:4871–4884. doi: 10.1091/mbc.E09-07-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sánchez-Cabo F, Torroja C, Acín-Pérez R, Calvo E, Aix E, González-Guerra A, Logan A, et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535:561–565. doi: 10.1038/nature18618. [DOI] [PubMed] [Google Scholar]
- Li Y, Park JS, Deng JH, Bai Y. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J. Bioenerg. Biomembr. 2006;38:283–291. doi: 10.1007/s10863-006-9052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015;7:a026716. doi: 10.1101/cshperspect.a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7:1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke AM, Rider CC. Glucose, glutamine and ketone body utilisation by resting and concanavalin A activated rat splenic lymphocytes. Biochim. Biophys. Acta. 1989;1010:342–345. doi: 10.1016/0167-4889(89)90059-1. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Quintana A, Schwindling C, Wenning AS, Becherer U, Rettig J, Schwarz EC, Hoth M. T cell activation requires mitochondrial translocation to the immunological synapse. Proc. Natl. Acad. Sci. USA. 2007;104:14418–14423. doi: 10.1073/pnas.0703126104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci JE, Gottlieb RA, Green DR. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 2003;160:65–75. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, Wallace DC. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer M, Spinelli JB, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 2016;24:104–117. doi: 10.1016/j.cmet.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudensky AY. Regulatory T cells and Foxp3. Immunol. Rev. 2011;241:260–268. doi: 10.1111/j.1600-065X.2011.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat. Immunol. 2007;8:665–668. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- Schüll S, Günther SD, Brodesser S, Seeger JM, Tosetti B, Wiegmann K, Pongratz C, Diaz F, Witt A, Andree M, et al. Cytochrome c oxidase deficiency accelerates mitochondrial apoptosis by activating ceramide synthase 6. Cell Death Dis. 2015;6:e1691. doi: 10.1038/cddis.2015.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindling C, Quintana A, Krause E, Hoth M. Mitochondria positioning controls local calcium influx in T cells. J. Immunol. 2010;184:184–190. doi: 10.4049/jimmunol.0902872. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta. 2011;1813:1263–1268. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto IC, Fontanesi F, Liu J, Barrientos A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta. 2012;1817:883–897. doi: 10.1016/j.bbabio.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valnot I, von Kleist-Retzow JC, Barrientos A, Gorbatyuk M, Taanman JW, Mehaye B, Rustin P, Tzagoloff A, Munnich A, Rötig A. A mutation in the human heme A: farnesyltransferase gene (COX10 ) causes cytochrome c oxidase deficiency. Hum. Mol. Genet. 2000;9:1245–1249. doi: 10.1093/hmg/9.8.1245. [DOI] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, Chang CH, Smith AM, Ai T, Faubert B, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani G, Greco M, Papa S, Attardi G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J. Biol. Chem. 1998;273:31829–31836. doi: 10.1074/jbc.273.48.31829. [DOI] [PubMed] [Google Scholar]
- Vincent AE, Ng YS, White K, Davey T, Mannella C, Falkous G, Feeney C, Schaefer AM, McFarland R, Gorman GS, et al. The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci. Rep. 2016;6:30610. doi: 10.1038/srep30610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker UA, Collins S, Byrne E. Respiratory chain encephalomyopathies: a diagnostic classification. Eur. Neurol. 1996;36:260–267. doi: 10.1159/000117269. [DOI] [PubMed] [Google Scholar]
- Walker MA, Slate N, Alejos A, Volpi S, Iyengar RS, Sweetser D, Sims KB, Walter JE. Predisposition to infection and SIRS in mitochondrial disorders: 8 years’ experience in an academic center. J. Allergy Clin. Immunol. Pract. 2014;2:465–468. 468.e1. doi: 10.1016/j.jaip.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.