

Abstract
Covalent histone modifications play an essential role in gene regulation and cellular specification required for multicellular organism development. Mono-ubiquitination of histone H2A (H2AUb1) is a reversible transcriptionally repressive mark. Exchange of histone H2A mono-ubiquitination and deubiquitination reflects the succession of transcriptional profiles during development required to produce cellular diversity from pluripotent cells. Germline pathogenic variants in components of the H2AUb1 regulatory axis are being identified as the genetic basis of congenital neurodevelopmental disorders. Here, we review the human genetics findings coalescing on molecular mechanisms that alter the genome-wide distribution of this histone modification required for development.
Keywords: Histone mono-ubiquitination, Polycomb repression, neurogenetics, neurodevelopment disorders
Chromatin modifications in brain development
Developmental decisions during lineage commitment are precisely coordinated at the genome level by gene expression programs that jointly activate or repress transcription [1]. Brain development requires the concurrent differentiation of neuronal cell types that must be organized into a complex organ [2, 3]. This process involves specification of pluripotent cells to ectoderm and neural precursors prior to terminal differentiation. Thus, brain development depends on precise temporal control of gene expression patterns, and disruption of transcriptional networks in brain development underlies neurodevelopmental disorders. It is not known how groups of genes are co-regulated during fate specification of neural lineages. Modification of histone marks is fundamentally important in specifying different cell types during embryonic development and pathogenic variants in genes that encode a broad-set of molecules that mediate chromatin modifications, including mono-ubiquitination of histone H2A lysine 119 (H2AUb1), are emerging as a prominent contribution to the genetic and molecular etiology of neurodevelopmental disorders [4–6].
DNA, wound around nucleosomes, is organized as chromatin fibers. Each nucleosome octamer is composed of two of each evolutionarily conserved histones H2A, H2B, H3 and H4 [7, 8]. The N-terminal tail of histones undergo numerous post-translational modifications (PTMs), including acetylation, ubiquitination, phosphorylation, or methylation of specific amino acid residues. Histone PTMs influence chromatin compaction, transcriptional regulation and interaction with protein complexes. Histone PTMs also correlate with active, repressive, and poised gene expression states. The genome-wide histone PTM landscape requires active maintenance to accommodate changing transcriptional profiles that allow for differentiation of diverse cell types and tissues from pluripotent stem cells during development [9].
Ubiquitin is a small molecule, which when covalently bound to substrates serves many biological functions. While poly-ubiquitination targets proteins for degradation via the 26S proteasome, monoubiquitination of histone H2A acts as a repressive chromatin modification. H2AUb1 has historically been associated with the E3 ubiquitin ligase activity of Polycomb repressive complex 1 (PRC1) and Polycomb transcriptional repression (Box 1), yet a variety of molecules that ubiquitinate or deubiquitinate H2A comprised of components of the H2AUb1 regulatory axis [10–13]. The components that decorate H2A with ubiquitin include canonical PRC1, variant PRC1, and TRIM37. Among the deubiquitinating components are the Polycomb repressive deubiquitinating complexes (PR-DUB) and USP16 [14, 15] (Figure 1, Key Figure). Dynamic exchange of this histone modification reflect the succession of transcriptional profile changes required to produce the cellular diversity from pluripotent cells during development.
Box 1. Polycomb Chromatin Modifiers.
Polycomb group (PcG) proteins function as chromatin-based transcriptional repressors and are essential for gene regulation during development. The functions of PcG complexes include modifying the genomic histone PTM landscape and establishing 3D chromatin topology and condensed chromatin at developmentally regulated loci [81]. First identified in Drosophila as important regulators of body plan specification, orthologous PcG systems were subsequently identified in vertebrates and also shown to be essential for developmental gene regulation [82–84]. Three PcG complexes, polycomb repressive complex 1 (PRC1), complex 2 (PRC2) and polycomb repressive deubiquitination complex (PR-DUB) have been identified and have various histone modifying activities (Figure. 1B) [64, 85]. PRC2 catalyzes trimethylation and dimethylation of histone H3 lysine 27 (H3K27me3/2) [86]. H3K27me3 plays a crucial role in the establishment of facultative heterochromatin throughout development. PRC1 catalyzes the mono-ubiquitination of H2A, while PR-DUB hydrolyzes ubiquitin from H2AUb1. The opposing functions of PRC1 and PR-DUB complex on histone H2AUb1 accentuates the important role for genome-wide H2AUb1 modification exchange in cell specification and development. In the hierarchical model, CBX recognizes the PRC2 H3K27me3 mark then recruits PRC1 to bivalent loci. However, when H3K27me3 levels were reduced in Eed knockout mESC, required for PRC2 stability, the levels and localization of H2AUb1 remained relatively unchanged implicating PRC2 independent H2A ubiquitination activity [76]. Furthermore, a series of experiments have demonstrated H2Aub1 recruiting PRC2 to genomic loci [87–89]. The mechanisms by which these PTM activities of PcG complexes are coordinated and recruited for transcriptional repression varies between organisms, across development and by tissue and is an important area of research elegantly addressed in recent reviews [11, 83, 85, 90]. A number of recent in vitro and in vivo studies have determined that the chromatin compaction functions of PRC1 and PRC2 can occur independent of their histone PTM activity [91–93]. Developmental repression of genomic loci by PRC1 is achieved by tethering compacted chromatin in nuclear foci that are modified during differentiation and development. Formation of these chromatin structures require chromatin tethering functions of canonical PRC1 complexes, but not H2AUb1 or variant PRC1 complexes.
Figure 1.
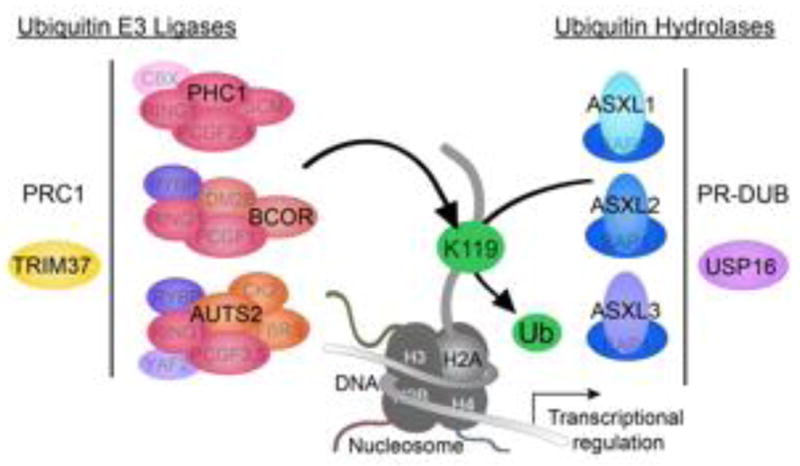
Histone H2AUb1 regulatory axis in neurodevelopmental disorders. Occurrence of genetic variants in the components that mediate H2AUb1 either independently or in association with Polycomb complexes. AUTS2, BCOR and PHC1 are components of different PRC1 complexes that mediate the ubiquitination of lysine 119 on H2A as highlighted with the green arrow. TRIM37 also functions as a ubiquitin E3 ligase but has not been shown to directly bind to a PRC1 complex. Along the right side, enzymes with H2AUb1 hydrolase activity are depicted. Individual ASXL family members form mutually exclusive PR-DUB complexes that mediate the deubiquitination of lysine 119 on H2A. The three PR-DUB complexes have overlapping and unique tissue and cell specificity. USP16 deubiquitinates H2AUb1 but does not interact with Polycomb complexes.
Large scale sequencing studies and widespread use of clinical exome sequencing are identifying inherited and de novo germline variants enriched in the H2AUb1 regulatory axis as important molecular pathology of syndromic neurodevelopmental disorders with features of autism and intellectual disability. Pathogenic human variants observed in Autism susceptibility candidate 2 (AUTS2), Polyhomeotic-like 1 (PHC1), Additional sex combs like family members (ASXl1,2,3), Tripartite motif-containing protein 37 (TRIM37), BCL6 corepressor (BCOR) and Ubiquitin-specific protease 16 (USP16) are components of the histone H2A ubiquitination regulatory axis. In this review, we will summarize the confluence of human neurogenetic findings around this important area of histone modification critical for brain development (Table 1).
Table 1.
Clinical features and model organism phenotypes for genes implicated in histone H2A ubiquitin remodeling and neurodevelopmental disorders.
| Gene/Disorder | Human Phenotypes | pLI* | Z score# | Model system/reported phenotypes | References |
|---|---|---|---|---|---|
| ASXL1/BOS | Developmental delays, ID, hypotonia, Hypoplastic/agenesis corpus callosum, delayed myelination, microcephaly, craniofacial dysmorphisms, feeding difficulties skeletal manifestations, VSD, ASD | 0.00 | 0.08 | Mouse/Craniofacial dysmorphism, skeletal transformations, myeloid dysplasia | [79, 96] |
| ASXL2/SPS | Delayed physcomotor development and speech, ID, ventriculomegaly, white matter volume loss, enlarged extra axial spaces, macrocephaly craniofacial dysmorphisms, feeding difficulties, skeletal manifestations, ASD | 0.99 | −0.01 | Mouse/Abnormal Heart morphology, skeletal transformations | [74] |
| ASXL3/BRS, Autism | Global psychomotor development delay, profound ID, microcephaly craniofacial dysmorphisms- feeding difficulties, skeletal manifestations | 1.00 | −0.94 | NA | [5] |
| USP16/Down Syndrome | ID, ventriculomegaly, Hypotonia, Skeletal manifestations, CHD, AVC | 0.01 | −0.89 | Mouse - Embryonic lethal < E7.5 Xenopus laevis-homeotic transformation |
[21, 62] |
| AUTS2/Autism | Delayed physcomotor development and speech, learning difficulties, microcephaly, ID, hypertonia, craniofacial dysmorphisms, feeding difficulties, skeletal manifestations | 1.00 | 3.09 | Mouse - developmental delay in sensorimotor, cognition and communication Zebrafish - craniofacial dysmorphism |
[4, 44] |
| PHC1/Microcephaly | Low-normal intelligence, microcephaly, craniofacial dysmorphisms- | 1.00 | 4.01 | Mouse - Congenital heart defects, homeotic transformations, craniofacial dysmorphisms | [39, 44] |
| TRIM37/Mulibrey Nanism | Large cerebral ventricles and cisternae, dysarthria, craniofacial dysmorphisms, dolichocephaly, skeletal manifestations, myocardial fibrosis, congestive heart failure, pericardial constriction, globular shaped heart | 0.17 | 1.42 | Mouse - cardiomyopathy, reproductive organ defects | [52] |
| BCOR/Syndromic Microphthalmia 2 | Mild ID, delayed motor development, hypoplastic or absent optic chiasm, spastic paraparesis, hypoplastic corpus callosum, microcephaly craniofacial dysmorphisms-Skeletal manifestations, ASD, VSD, Aortic valve stenosis, pulmonary valve stenosis, pentalogy of fallot, DORV, Dextrocardia, mitral valve prolapse, tricuspid valve inefficiency | 1.00 | 1.06 | Mouse - Heart defects, craniofacial dysmorphism, abnormal forebrain development, decreased cerebellar size | [59] |
ASD = Atrial septal defects, AVC = Atrioventricular canal, BOS = Bohring Opitz Syndrome, BRS = Bainbridge Ropers Syndrome, CHD = Congenital heart disease, SPS = Shashi-Pena Syndrome VSD = Ventricular septal defect
Both Zscore and pLI values have been obtained from the ExAC browser (http://exac.broadinstitute.org).
pLI is the probability of being loss-of-function (LoF) intolerant (pLI). The closer pLI is to one, the more LoF intolerant the gene appears to be.
for synonymous and missense Z score has been created. Z score is the deviation of observed counts from the expected number.
Positive Z scores indicate increased constraint (intolerance to variation) and therefore that the gene had fewer variants than expected. Negative Z scores are given to genes that had more variants than expected.
Mono-ubiquitinated histone H2A
Reversible H2AUb1 has diverse roles in a complex transcriptional milieu comprised of multiple chromatin modifications, DNA methylation, and chromatin remodeling molecules. H2AUb1 is enriched at bivalent promoters, characterized by the presence of both the activating H3K4me3 and repressive H3K27me3 histone PTMs [16–21]. Within ESCs, bivalent promoters are signature of developmentally important genes that establish cell identity [17, 22, 23]. H2AUb1 serves to prevent elongation of RNA polymerase machinery at bivalent loci, while RNA polymerase initiation remains unhindered [24, 25]. This poised state allows for dynamic transition between a transcriptionally active or repressed state required for normal development. Ubiquitinating and deubiquitinating components of the H2AUb1 regulatory axis reflect a similar genome-wide binding pattern implicating developmental modulation of this modification as an important regulatory mechanism [21, 26, 27]. Resolution of bivalent promoters at developmentally regulated genes towards active or fully repressed involves the concerted activity of various chromatin-modifying complexes [28, 29]. The emerging mechanisms suggest they can interact. The compounding effect among histone modifications due to alteration of a single histone modification can complicate interpreting the resulting molecular and transcriptional impacts.
The majority of mammalian gene promoters are encompassed by CpG islands, that correspond to contiguous non-methylated segments of the genome with a higher than average level of CpG dinucleotides [30]. DNA methylation of CpG islands acts as a stable and heritable repressive epigenetic mark essential for development and generating a diversity of cell fates from multipotent progenitor cells. H2AUb1 plays an important role in epigenetic establishment of mature cell fates by protecting CpG islands against DNA methylation required for constitutive repression of the corresponding genes [31, 32]. It still remains mechanistically unclear how developmental redistribution of H2AUb1 contributes to this diversity of transcriptional functions. Nevertheless, it underscores the diversity of clinical features and syndromic nature of inherited neurodevelopmental disorders that impact the H2AUb1 regulatory axis.
Polycomb H2A ubiquitination
PRC1 is an important source of H2A E3 ubiquitin ligase activity that has been the focus of substantial research. PRC1 is a multimeric complex with evolutionarily conserved function in transcriptional repression and non-enzymatic chromatin compaction. Polycomb group (PcG) proteins were originally described in Drosophila based on their regulation of Hox gene expression, that when dysregulated results in homeotic transformations, including additional sex combs, a structure present on the legs of male flies. Genetic screens of Drosophila mutants that exhibit this phenotype were important for identifying the composition of PcG complexes and mechanisms of transcriptional repression. PRC1 and PRC2 were the founding complexes that choreograph H2A monoubiquitination and H3 lysine 27 trimethylation (H3K27Me3) respectively required for transcriptional regulation (Box 1).
Work in invertebrates and vertebrates are uncovering differences in the mechanisms of PcG transcriptional repression and the composition of PRC1 between these organisms. In Drosophila, the dynamic functions of PRC1 and PRC2 are coordinated in a sequential manner at polycomb repressive elements (PREs) to mediate transcriptional repression and chromatin compaction, with PRC2 initiating this molecular cascade. In vertebrates PREs are not the functional genomic element that provides the platform for PcG repression and coordination of PRC1 and PRC2 activities exhibits more diversity. Likewise, the compositions of PRC1 complexes are more heterogeneous in vertebrates (Box 2), yet the E3 ubiquitin ligase and chromatin compaction activities are conserved. Interestingly, evidence that these two PRC1 activities are functionally distinct has precipitated a series of studies designed to isolate their functional impacts on development.
Box 2. PRC1- Canonical vs. Variant.
Canonical and variant PRC1 complexes are defined by the presence of the E3 ubiquitin ligase RING1A or RING1B, but differ in protein assembly of the larger multimeric complex. In depth discussion of PRC1 components and complexities have been reviewed elsewhere [10, 11]. Briefly, canonical PRC1 is comprised of RING1, PHC1, and CBX family members as well as PCGF2 or PCGF4 (Figure 3). Each family of core proteins consists of a number of paralogs, RING1 has two, PHC1 three and CBX five, increasing the diversity of potential canonical PRC1 complexes. Variant PRC1 can contain a combination of these proteins and several accessory binding partners including BCOR, KDM2B, E2F6, RYBP, and AUTS2 that uniquely specify variant PRC1 complexes (Figure I). Canonical and variant PRC1 are distinguished and labeled by which of the PCGF are incorporated [12, 94]. Expression of family members and PRC1 composition vary across cell types and developmental time points [72, 73]. CBX7 is the only homolog present in the PRC1 complexes within ESCs. However, upon differentiation CBX7 expression is reduced while CBX2 and CBX4 are increased demonstrating cell type specificity [73]. Several distinct PRC1 complexes can coexist within the same cell type but bind to and regulate distinct genomic loci [57, 76]. In ESCs, RYBP and CBX form mutually exclusive PRC1 complexes, which bind to coincident and unique targets [11, 12, 73]. CBX-PRC1 only targets are enriched for developmental and lineage specific genes, whereas RYBP-Prc1 localized to metabolic and M-phase associated genes [73]. Variant PRC1 accessory proteins can also provide unique functionality to the PRC1 complexes. For example, the H3K36me2 demethylase, KDM2, interacts with the variant BCOR complex and recruits the complex to CPG islands [32, 77, 78]. Beyond ubiquitination, PRC1 complexes repress gene activity by chromatin compaction [20, 33, 92]. Canonical PRC1 complexes can generate self-contained interaction domains at target sites, which can be lost upon differentiation [92]. Chromosomal compaction by PRC1 in this manner is independent of H2AUb1 and variant PRC1 proteins [33, 92]. It will be interesting to elucidate the biological role of PRC1 complex diversity as it pertains to transcriptional repression or chromatin structure.
Ring1A and Ring1B (Ring1A/B) are central E3 ubiquitin ligases of all PRC1 complexes. Complete deletion of Ring1A/B in mESCs leads to loss of both PRC1 complex stability, essential for compaction function, and H2AUb1 ligase activity. Consequently, Ring1A/B−/− mESCs spontaneously differentiate and de-repress many developmentally regulated genes [19]. Introduction of a catalytically inactive Ring1B I53A variant allowed for the H2AUb1 ligase activity of PRC1 to be evaluated independent of chromatin compaction [33, 34]. Unexpectedly, these genetic manipulations revealed that formation of a stable multimeric PRC1 complex could target developmental genes and compact chromatin independent of H2AUb1. This highlighted the dual functions of PRC1 and questioned the requirement for H2AUb1 in development. While Ring1B−/− mice are embryonic lethal by ~ E10.5, Ring1BI53A/I53A embryos complete gastrulation, but are unable to survive past ~E15.5 emphasizing an essential function for H2Aub1 later in development [34]. In Drosophila, an I48A mutation in Sex Combs Extra (Sce), the Ring1 homolog, ablates the E3 ligase enzymatic activity and results in decreased H2AUb1 levels. SceI48a/I48a maintain repression of many classical PCG target genes and show less severe phenotypes than Sce−/−, but SceI48a/I48a arrest development at the end of embryogenesis due to undefined causes [35]. Both model organisms demonstrate the vital requirement for proper regulation of H2AUb1 during development and emphasize that H2AUb1 can regulate non-canonical PCG target genes. The essential nature of H2Aub in development is further emphasized when viewed from the perspective of the H2AUb1 regulatory axis, representing a number of complexes and single molecules with a primary function of H2AUb1 modification. Likewise, recurrent pathogenic variants in genes that encode H2A monoubiquitination or deubiquitination components in neurodevelopmental disorders bolster this argument and will be discussed.
Histone H2A ubiquitination syndromes
PHC1 in Autosomal recessive primary microcephaly
Autosomal recessive primary microcephaly (MCPH) is clinically characterized as a small head circumference that reflects a correspondingly small brain. This genetically heterogeneous disorder is largely attributed to defects in neural progenitor cell (NPC) proliferation, neurogenesis and apoptosis, which reduce the size of the NPC pool available to build a brain. PHC1 is a core component of the canonical PRC1. Homozygous missense PHC1 variants were identified as a genetic basis for MCPH11 in a family with reported consanguinity (OMIM; 615414) [6]. This inherited rare variant in PHC1 is noteworthy based on the loss-of-function (LOF) intolerance and high constraint for homozygous variants calculated for this gene by Exome Aggregation Consortium (ExAC) [36] (Table 1). The clinical presentation of MCPH11 includes primary microcephaly, low-normal cognitive function and short stature. Functional analysis of this pathogenic variant in patient cells revealed reduced PHC1 expression with lower H2AUb1 levels genome-wide and impaired recruitment of PHC1 to loci of DNA damage and repair. These defects were also accompanied by increased expression of GMNN (Geminin), which was previously shown to cooperate with PcG transcriptional repression to achieve germinal differentiation, lineage commitment and early specification of neural cell fate [37]. Differential expression of GMNN was not linked to alterations in H2AUb1 at the GMNN locus, but is consistent with decreased progenitor proliferation and differentiation implicated in the pathogenic mechanisms of MCPH.
Phc1 null mice display diverse and severe phenotypes, including cephalic neural crest defect, microcephaly, abnormal facies, parathyroid and thymic hypoplasia together with skeletal and cardiac abnormalities [38–41]. These phenotypes are consistent with constitutive disruption of both canonical PRC1 chromatin folding and E3 ligase activities. In comparison to the Phc1 knockout phenotype, the MCPH11 phenotype suggests the PHC1 missense variants function as a hypomorphic allele. As the defective protein is expressed at normal levels, PRC1 formation, and thus chromatin compaction properties, may not be altered. We instead speculate that the MCPH11 phenotypes may be explained as a defect in PRC1 E3 ligase activity, which alone has been shown generate a milder phenotype in animal models. Interestingly, the MCPH11 PHC1 variant predominantly impacted neural development in affected individuals, potentially highlighting a sensitivity of the developing brain to dysregulation of H2AUb1 modification exchange.
AUTS2 in Autism Spectrum and Neurodevelopmental Disorders
AUTS2 was originally identified as a candidate for neurodevelopmental disorders by resolution of a translocation-breakpoint in twins with autism spectrum disorder (ASD), developmental delay (DD), and epilepsy [42]. Following this initial description, AUTS2 structural variants and de novo dominant variants were identified by recent large-scale WES studies [43–47]. AUTS2 is a component of variant the PRC1 complex PRC1.5 (Box 1). PRC1.5 is sufficient for H2A ubiquitination in vivo and in vitro, a finding confirmed in the PCGF5 knockout mouse that disrupts the formation of PRC1.5 and is accompanied by a reduction of H2AUb1 levels [48]. In 293T-REx cells AUTS2 stably recruits casein kinase 2 (CK2) to PRC1.5. In an in vitro nucleosomal assay CK2 suppresses PRC1.5 H2A mono-ubiquitination activity through a RING1B phosphorylation event [4]. This reduction in H2AUb1 corresponds to transcriptional activation associated with the recruitment of P300. While direct experimental evidence is needed to confirm this molecular mechanism in developing brain, it highlights the importance of modulating H2AUb1 activity, in this scenario by RING1B phosphorylation, for proper neural development. Of note, heterozygous de novo mutations in EP300 (E1A binding protein p300), which encodes the histone H3 acetyltransferase P300, is a genetic basis for Rubinstein-Taybi syndrome (RSTS1 MIM: 180849), a neurodevelopmental disorder that shares clinical features AUTS2-ASD. AUTS2 associated chromatin biology exemplifies the complexity and interconnected nature of coordinated histone modifications, the molecular mechanisms of which will need to be elucidated to fully appreciate their role in brain development and neuropathology.
Homozygous Auts2 knockout mice exhibit reduced intrauterine and postnatal growth, altered motor control and reduced vocalizations relative to controls [4]. While these findings corroborate human genetic findings associated with AUTS2 and implicate an important role in neuropathology, the molecular function of AUTS2 in neural development and cortical neuron fate specification are yet to explored. AUTS2 exhibits highest expression in the developing cortex [49]. During cortical development, neural progenitor cells (NPCs) produce mature neuronal subtypes and glial cells in a defined temporal order. The timing of fate switches that determine the number of neuronal subtypes that will make up the composition of the six-layer cortex is governed by dynamic changes in NPC chromatin. Abnormal productions of cortical progeny have been shown to underlie ASD neuropathology. Uncovering the epigenomic role of AUTS2 and H2AUb1 dysregulation within the pathogenic PRC1.5 mechanism and more generally in normal development will be an important direction for future research.
TRIM37 in Mulibrey nanism
Mulibrey nanism (MUL; OMIM: 253250) is a rare autosomal recessive genetic disorder. The acronym “mu–li–br–ey” reflects the “muscle–liver–brain–eye” involvement in this syndrome. MUL is characterized by prenatal-onset growth retardation, short stature, craniofacial features, hepatomegaly, enlarged ventricles and dysarthria. Cardiac involvement is illustrated by progressive constrictive pericarditis, myocardial hypertrophy, and variable myocardial fibrosis. Pathogenic variants in TRIM37 are the genetic basis of MUL [50]. TRIM37 encodes a RING finger domain like the hallmark E3 ubiquitin ligases RING1A/B, but functions independent of the multimeric PRC1 complexes. Thus, TRIM37 does not exhibit chromatin compaction properties conveyed by PRC1 complexes, highlighting the critical role of H2AUb1 in development [51]. While the H2A ubiquitin ligase activity of TRIM37 has been experimentally demonstrated in vitro and in vivo, the molecular pathology associated with pathogenic TRIM37 variants has not been explored. Likewise, further studies are needed to confirm the dynamics of H2A ubiquitination in primary cells from individuals with MUL. The spectrum of phenotypes described for the Trim37−/− mouse model recapitulates many clinical features of MUL and thus provides a good model to study pathogenesis related to TRIM37 deficiency [52]. Despite the prominent neurological features of MUL, the role of TRIM37 in neural development has not been investigated.
BCL6 corepressor (BCOR) in Syndromic microphthalmia 2
Syndromic microphthalmia 2 (MCOPS2, MIM 300166) is a rare, X-linked disorder characterized by microphthalmia, congenital cataracts, dysmorphic facial features, congenital heart defects, malformed ears, dental, skeletal, renal and urogenital anomalies. Neurologically, MCOPS2 is associated with ID, microcephaly and structural brain abnormalities. Pathogenic variants in X-linked BCOR (BCL6 interacting corepressor) are the genetic basis of this disorder that presents with a broad phenotypic spectrum consistent with hemizygosity in males and random X inactivation in females [53, 54]. These phenotypes also encompass those associated with reported copy number variants of BCOR. Pathogenic variants in BCOR are associated with a significant reduction in genome-wide H2AUb1 levels in mesenchymal stem cells isolated from an individual with MCOPS2 compared to control, implicating H2AUb1 dysregulation in the molecular pathology [55].
BCOR is a transcriptional corepressor that was originally identified by its ability to interact with the site-specific transcriptional repressor BCL6 [56]. BCOR, KDM2B (lysine demethylase 2B) and PCGF1 are PcG-associated proteins that characterize the composition of variant PRC1.1 [12, 57]. PCGF1 knockdown experiments exhibit a dramatic reduction of H2AUb1, mirroring H2AUb1 deposition activity of PRC1.1 [12]. A role for KDM2B in the BCOR MCOPS2 molecular pathology has not been investigated, but KDM2B binds DNA and is critical for recruiting PRC1.1 E3 ligase activity to unmethylated CpG islands to protect against de novo methylation and constitutive repression (Figure 2) [31]. Such PRC1.1 functions would allow genome-wide epigenetic changes during development required to maintain the balance between proliferation and differentiation, and the subsequent emergence of cellular diversity defined by unique genome-wide DNA methylation patterns [58].
Figure 2.

Transcriptional regulation by the H2AUb1 regulatory axis. Schematic showing the monoubiquitination and deubiquitination of histone H2A at Bivalent and repressed loci. (A) Trim37 and PRC1 (Canonical or Variant) catalyze H2A monoubiquitination at unmethylated CpG islands of developmentally regulated genes that are enriched for H3K27me3 and H3K4me3 chromatin marks. In this bivalent state, RNA polymerase is initiated but unable to elongate, preventing RNA transcription. PR-DUB or USP16 deubiquitinate H2AUb, allowing activation of transcription and RNA pol II to elongate. (B) At repressed chromatin H2A monoubiquitination prevents the methylation of CpG Island. Removal of H2AUb1 by PR-DUB or USP16 allows for de novo DNA methylation and constitutive repression.
A role of H2AUb1 dysregulation has not been described for the neural developmental defects described for the Bcor conditional knockout mouse where BCOR is required for Sonic Hedgehog (SHH) signaling suppression by BCL6 in cerebellar development [59]. The epigenetic changes that mediate transcriptional repression of SHH signaling effectors in association with BCOR have not been elucidated, but recruitment of the histone deacetylation SIRT1 by BCOR to BCL6 target genes has been shown to be important. This again emphasizes a role for co-regulation of histone modifications in H2AUb1 associated transcriptional regulation. A similar SHH mechanism could contribute to the pathogenesis of medulloblastoma tumorigenesis associated with somatic variants in BCOR and cardiac laterality defects observed in individuals with MCOPS2 [60].
H2AUb1 deubiquitination syndromes
Ubiquitin-specific protease 16 (USP16) in Down’s Syndrome
Down Syndrome (DS; OMIM: 190685) results from full or partial trisomy of chromosome 21 and is characterized by reduced growth, facial dysmorphisms, ID, motor deficits and early onset Alzheimer’s disease. The constellations of DS phenotypes are attributed to the overexpression of a number of dosage-sensitive genes on chromosome 21 (Box 2). Among the genes that have been evaluated is Usp16 [61]. USP16 can independently remove ubiquitin moieties from histone H2A and exhibits high expression in stem cell populations. The Ts65Dn mouse model that has a third copy of approximately two-thirds of the murine genes homologous to genes on human chromosome 21, including Usp16, recapitulates several traits of the human disorder. Ts65Dn mice have 1.5-fold increase in Usp16 expression and a 40% decrease in H2AUb1. This molecular defect is proposed to account for decreased stem cell proliferation and increased senescence. Restoring Usp16 expression to disomy levels by reducing the USP16 dosage or reducing Usp16 expression by shRNA knockdown, rescues the stem cell proliferation and premature senescence phenotypes. These experiments suggest the third copy of Usp16 in Ts65Dn mice disrupt maintenance of stem cell multipotency contributing to DS pathology. A survey of tissues from individuals with DS confirmed reduced self-renewal of hematopoietic stem cells and premature senescence of mammary epithelial cells, NPCs, and fibroblasts [21, 62]. Molecularly, this highlights the role of histone H2AUb1 in pluripotency, differentiation and cell lineage commitment.
Usp16 knockout mice are early embryonic lethal and further implicate an essential role for USP16 in development and lineage commitment. Usp16 knockout mESCs exhibit elevated H2AUb1 unlike Ts65Dn mice, proliferate normally and maintain pluripotency. Upon differentiation high H2AUb1 levels persist at transcriptional start sites of many pluripotency and developmentally regulated genes. This molecular defect corresponds to persistent expression of pluripotency genes, delayed expression of germinal layer markers and an impediment to differentiation. A role for USP16 H2A deubiquitination in neural development has not been explored. We speculate that the epigenomic functions of USP16 may underlie the highly dynamic disruption of gene expression in the brains of individuals with DS spanning mid-fetal development to adulthood [63]. These transcriptomic findings implicate cell-autonomous deficits in oligodendrocyte differentiation and the production of neocortical myelin in DS pathogenesis, molecular and developmental biology consistent with UPS16 H2AUb1 hydrolase activity.
Developmental Disorders: Additional sex combs-like (ASXL1, ASXL2 and ASXL3)
ASXL1, ASXL2 and ASXL3 are mammalian homologs of Drosophila Additional sex combs (Asx) and share a conserved domain structure [64]. The paralogs interact with BRCA1 Associated Protein-1 (BAP1) in a mutually exclusive fashion to form three separate PR-DUB complexes (Figure 1). BAP1 provides the H2AUb1 ubiquitin hydrolase activity for the PR-DUBs represented by each ASXL family member. BAP1 is a critical tumor suppressor that has attracted medical interest in the past years since its loss leads to a variety of cancers due to somatic variants, but no pathogenic germline variants have been identified as the genetic basis of a developmental disorder to date [65–67]. Conversely, de novo heterozygous germline variants have been detected in all three family members by research and clinical sequencing efforts, for multi-organ developmental disorders with shared features of hypotonia, ID, craniofacial dysmorphisms, white matter loss and skeletal manifestations. Despite these overlapping features, each gene is associated with its own distinct neurodevelopmental syndrome that differ based on their constellation of partially penetrant clinical features, molecular pathology and associated cancer risk (Box 3). De novo variants in ASXL1 are the genetic basis of Bohring–Opitz syndrome (BOS; MIM 605039) [68, 69]. BOS is distinctive among ASXL family member disorders based on severe and highly penetrant skeletal abnormalities, facial nevus flammeus, microcephaly, persistent feeding difficulties and delays in speech and gross motor skills. ASXL2 is the genetic basis of Shashi-Pena syndrome (SPS; MIM 617190), which is the least severe of the ASXL associated disorders and distinctive because of mild neonatal feeding difficulties, complete penetrance of macrocephaly, partially penetrant facial nevus flammeus, variability in severity of ID, normal height and weight and bone density abnormalities [70]. Pathogenic ASXL3 variants have been discovered in Bainbridge Ropers syndrome (BRS; MIM 615485). BRS is characterized by persistent severe feeding difficulties, partially penetrant microcephaly, highly penetrant severe hypotonia, failure to meet developmental milestones including walking and highly penetrant nonverbal outcomes [5, 71]. Comprehensively, these findings suggest an important role for H2AUb1 modification exchange in development, with prominent and distinct neurological defects.
Box 3. Gene dosage in neurodevelopmental disorders.
Advanced genomic sequencing and clinical genetic testing tools have significantly added to the list of de novo variants contributing to neurological syndromes. Pathogenic de novo dominant heterozygous variants can act through dominant negative, toxic gain-of-function or LOF genetic mechanisms. A large proportion of heterozygous variants are predicted to result in LOF and haploinsufficiency, yet only a fraction are experimentally confirmed. What underlies the vulnerability to haploinsufficiency is unclear, however, it accentuates the importance of gene dosage and the pathogenicity that results from such changes of dosage-sensitive genes. Chromatin modifying genes are an important group of disease-associated genes that are contributing to the recent expansion of early-onset reproductively lethal neurodevelopmental disorders attributed to pathogenic de novo variants. The molecular mechanisms that underlie the phenotypic spectrum of dosage-sensitive genes have been largely unexplored. Through comprehensive genetic testing and research sequencing the SNVs and CNVs will be important to define dosage sensitive genes and the corresponding spectrum of phenotypic outcomes. Reciprocal dosage of Usp16 that results in opposing phenotypes is an example of the relationship of dosage and phenotypic outcomes for dosage sensitive genes. Located on chromosome 21, triplicate copies of USP16 are present in Down syndrome [61]. Reduced dosage of USP16 has not been described in human pathogenesis. Yet, early embryonic lethality of the Usp16 knockout suggests USP16 may be essential for development in humans as well. An evaluation of different cell types, including differentiation of mESCs towards a germinal fate showed that knockout of Usp16 results in increased H2AUb1 levels at key developmental regulators and persistent multipotency, while an extra functional copy of USP16 promotes H2AUb1 deubiquitination, progenitor proliferation, accelerated differentiation and senescence. For most cases of dosage the resulting phenotypic outcomes and proposed molecular mechanisms are more variable than this direct relationship between gene dosage and phenotype. For many of the neurodevelopmental disorder genes that encode components of the histone H2A ubiquitination regulatory axis, in depth molecular studies will be required to understand how a gene dosage alters tissue- and developmental- specific expression and how the altered amount of a single subunit of a multi-subunit protein complex leads to recognizable phenotypes. These two criteria are particularly relevant given the variability in subunit composition of PRC1.
The Asxl1 and Asxl2 knockout mice exhibit both posterior and an anterior homeotic skeletal transformation, suggesting their role in Hox regulation is conserved in vertebrates [72–74]. Both genotypes were born at less than Mendelian ratio, demonstrated perinatal lethality with the null animals proportionally smaller than control littermates [73, 75]. Besides skeletal transformations, Asxl1−/− mice exhibit microphthalmia, craniofacial dysmorphisms and myelodysplasia but have not been described as microcephalic [33, 75–77]. Asxl2−/− mice showed enlarged hearts with interstitial fibrosis and cardiomyocyte disarray and do not exhibit macrocephaly [73, 78]. This discrepancy in brain size phenotypes between mice and humans is an interesting observation that warrants further analysis, as the role for PR-DUB function in brain development has not yet been described.
Between analysis of mouse models and primary cells established from affected individuals, dysregulation of H2AUb1 DUB activity is a key molecular defect of ASXL pathogenic variants [5]. However, molecular differences may also contribute to their phenotypic differences. In addition to H2AUb1, ASXL1 molecular pathology is also impacted by dysregulation of H3K27Me3 [79]. ASXL3 molecular pathology has been linked primarily to defects in H2AUb1 DUB activity and elevated genome-wide levels of H2AUb1 [5]. Pathogenic variants in ASXL2 have been implicated to function in a dominant-negative manner in the PR-DUB complex, a finding that will need to be confirmed [5]. Additionally, Asxl2−/− mice demonstrate elevated levels of H2AUb1 presumably due to an absence of PR-DUB activity [80]. Further investigating these pathogenic mechanisms in affected tissue will provide insight into the tissue-specific H2AUb1 modification exchange in fate specification of distinct cell lineages.
Concluding remarks
Convergence of neurogenetics findings on genes that encode components of the H2AUb1 regulatory axis provides genetic evidence for the important function of this histone PTM in normal development and pathogenesis. The diversity of syndromic features that define these neurodevelopmental disorders belies the dosage sensitivity and molecular complexity that accompanies developmental H2AUb1 exchange. Divergent phenotypes exhibited by pathogenic variants in genes that encode components of overlapping protein complexes with H2AUb1 modifying activity also suggests tissue- and developmental- specific expression. These genetic observations will be critical for designing experimental approaches to understand the fundamental epigenetic mechanisms of H2AUb1. More mechanistic studies will be needed to translate these genetic perturbations into a better understanding of the polycomb histone modifications required for the generation and maintenance of cellular diversity of different organ systems, including the brain (see outstanding questions). These genetic findings provide a strong foundation on which to base these future research goals.
Outstanding Questions.
Sequencing initiatives and clinical genetic testing are uncovering an array of alleles that will be invaluable for dissecting the molecular mechanisms of developmental H2AUb1 remodeling. Particularly relevant to this endeavor will be the pathogenic mechanisms of individual protein domains disrupted by pathogenic missense variants that do not disrupt expression.
Are the chromatin condensation and folding functions of PRC1 completely independent of H2AUb1 in all tissues and what are the roles for each in neurodevelopment?
What is the role of H2AUb1 exchange kinetics in neurodevelopment?
Are molecular mechanisms shared by pathogenic germline and somatic variants? Is there a common set of dysregulated genes that drive pathogenicity?
What are the molecular mechanisms of gene dosage for individual subunits of large multimeric PRC1s?
Histone H2A E3 ligases are often in association with proteins that perform an array of histone PTMs in addition to H2A ubiquitination (CK2/P300 and PRC1.5; KDM2B and PRC1.1; TRIM37 and PRC2). How are the constellation of histone PTMs in each of these cases integrated to determine developmental transcriptional profiles?
What is the developmental choreography of histone H2A ubiquitination relative to deubiquitination? Is this dynamic remodeling critical for cell fate determination? What neuronal cell types are disrupted in neurodevelopmental disorders associated with pathogenic variants in components of the H2AUb1 regulatory axis?
Figure 3.
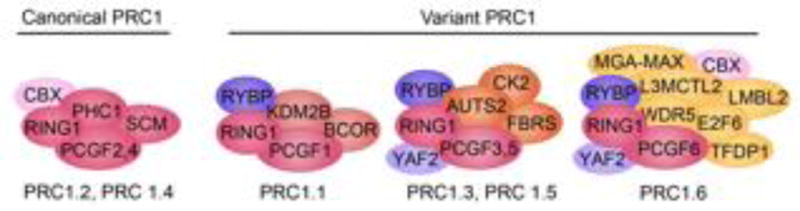
Components of canonical and variant Polycomb repressive complex I. There are two categories of PRC1 complexes, canonical and variant, which are distinguished by the mutually exclusive incorporation of one of six PCGF paralogs. This family of complexes is biochemically defined by the presence of RING1A/B. Components of canonical PRC1 include RING1A/B, PHC1, and CBX family members and are labeled as either PRC1.2 or PRC1.4 based on incorporation of PCGF2 or PCGF4 respectively. RYBP/YAF2, PCGF and RING1A/B are shared core components of variant PRC1, that are characterized by accessory binding partners, including BCOR, KDM2B, E2F6, RYBP, and AUTS2, and labeled according to the PCGF1, PCGF3, PCGF5 or PCGF6 subunits (PRC1.1, PRC1.3, PRC1.5 and PRC1.6).
Box 4. Coincidence of variants in cancers and neurodevelopmental disorders.
A decidedly unexpected finding from comprehensive analysis of data from large scale sequencing efforts and routine clinical genome-wide genetic tests was the coincidence of variants in chromatin modifying genes that are attributed to both neurodevelopmental disorders and cancer. Similar to germline variants in components of the histone H2AUb1 regulatory axis detected for neurodevelopmental disorders, somatic variants are being identified as potential drivers of a variety of tissue malignancies. Individually mutated genes can be preferentially associated with individual tumor types, as well as overlapping contributions to a broad range of cancers including neural, epithelial and hematopoietic malignancies. The ASXL family members are an example of this complex interconnectedness. Pathogenic germline variants in ASXL1, ASXL2 and ASXL3 are each associated with distinct neurodevelopmental disorders that share some overlapping phenotypes. Likewise, somatic variants in ASXL1, ASXL2 and ASXL3 are detected in many malignancies, but each is also associated prominently with AML, breast cancer and melanoma respectively [95, 96]. These cases highlight the importance of tissue- and developmental- specific expression to pathogenesis and suggest shared molecular mechanisms conveyed by germline and somatic variants in the same gene. An outstanding question is how variants in different subunits of the same complex can generate divergent phenotypes and susceptibility to tumorigenesis. Thus, the context of genetic variations, including timing, genetic background and cell type, may contribute to the differing disease phenotypes, despite similar protein defects. The cell and tissue choices for investigating the molecular mechanisms of pathogenesis will be critically important to uncover the pathogenic mechanism of neurodevelopmental defects and generation of cancer.
Trends.
Human neurogenetic findings highlight a previously undescribed role for developmental remodeling of histone H2A mono-ubiquitination in early brain development.
Pathogenic variants that disrupt components of the H2AUb1 regulatory axis accentuates the important role for ubiquitin exchange at Histone H2A Lysine 119 in cell specification and development.
The diversity of syndromic features that define neurodevelopmental disorders belies the dosage sensitivity and molecular complexity that accompanies H2AUb1 exchange.
Careful analysis of Human genetic findings guides the dissection of molecular defects in epigenetic mechanisms critical for development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davidson EH. Emerging properties of animal gene regulatory networks. Nature. 2010;468(7326):911–20. doi: 10.1038/nature09645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azevedo FA, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009;513(5):532–41. doi: 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
- 3.Greig LC, et al. Molecular logic of neocortical projection neuron specification, development and diversity. Nat Rev Neurosci. 2013;14(11):755–69. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao Z, et al. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature. 2014;516(7531):349–54. doi: 10.1038/nature13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srivastava A, et al. De novo dominant ASXL3 mutations alter H2A deubiquitination and transcription in Bainbridge-Ropers syndrome. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad S, et al. Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum Mol Genet. 2013;22(11):2200–13. doi: 10.1093/hmg/ddt072. [DOI] [PubMed] [Google Scholar]
- 7.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184(4139):868–71. doi: 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 8.Thomas JO, Kornberg RD. An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci U S A. 1975;72(7):2626–30. doi: 10.1073/pnas.72.7.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tee WW, Reinberg D. Chromatin features and the epigenetic regulation of pluripotency states in ESCs. Development. 2014;141(12):2376–90. doi: 10.1242/dev.096982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gil J, O’Loghlen A. PRC1 complex diversity: where is it taking us? Trends Cell Biol. 2014;24(11):632–41. doi: 10.1016/j.tcb.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20(10):1147–55. doi: 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- 12.Gao Z, et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell. 2012;45(3):344–56. doi: 10.1016/j.molcel.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidal M, Starowicz K. Polycomb complexes PRC1 and their function in hematopoiesis. Exp Hematol. 2017;48:12–31. doi: 10.1016/j.exphem.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Vissers JH, et al. The many faces of ubiquitinated histone H2A: insights from the DUBs. Cell Div. 2008;3:8. doi: 10.1186/1747-1028-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He M, et al. The emerging role of deubiquitinating enzymes in genomic integrity, diseases, and therapeutics. Cell Biosci. 2016;6:62. doi: 10.1186/s13578-016-0127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Endoh M, et al. Histone H2A mono-ubiquitination is a crucial step to mediate PRC1-dependent repression of developmental genes to maintain ES cell identity. PLoS Genet. 2012;8(7):e1002774. doi: 10.1371/journal.pgen.1002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 18.Endoh M, et al. Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development. 2008;135(8):1513–24. doi: 10.1242/dev.014340. [DOI] [PubMed] [Google Scholar]
- 19.Brookes E, et al. Polycomb associates genome-wide with a specific RNA polymerase II variant, and regulates metabolic genes in ESCs. Cell Stem Cell. 2012;10(2):157–70. doi: 10.1016/j.stem.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoenfelder S, et al. Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet. 2015;47(10):1179–86. doi: 10.1038/ng.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang W, et al. The histone H2A deubiquitinase Usp16 regulates embryonic stem cell gene expression and lineage commitment. Nat Commun. 2014;5:3818. doi: 10.1038/ncomms4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–60. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441(7091):349–53. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 24.Stock JK, et al. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9(12):1428–35. doi: 10.1038/ncb1663. [DOI] [PubMed] [Google Scholar]
- 25.Zhou W, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29(1):69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ku M, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4(10):e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahtoe DD, et al. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat Commun. 2016;7:10292. doi: 10.1038/ncomms10292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voigt P, et al. A double take on bivalent promoters. Genes Dev. 2013;27(12):1318–38. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science. 2016;352(6290):aad9780. doi: 10.1126/science.aad9780. [DOI] [PubMed] [Google Scholar]
- 30.Blackledge NP, Klose R. CpG island chromatin: a platform for gene regulation. Epigenetics. 2011;6(2):147–52. doi: 10.4161/epi.6.2.13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boulard M, et al. FBXL10 protects Polycomb-bound genes from hypermethylation. Nat Genet. 2015;47(5):479–85. doi: 10.1038/ng.3272. [DOI] [PubMed] [Google Scholar]
- 32.Wu X, et al. Fbxl10/Kdm2b recruits polycomb repressive complex 1 to CpG islands and regulates H2A ubiquitylation. Mol Cell. 2013;49(6):1134–46. doi: 10.1016/j.molcel.2013.01.016. [DOI] [PubMed] [Google Scholar]
- 33.Eskeland R, et al. Ring1B compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol Cell. 2010;38(3):452–64. doi: 10.1016/j.molcel.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Illingworth RS, et al. The E3 ubiquitin ligase activity of RING1B is not essential for early mouse development. Genes Dev. 2015;29(18):1897–902. doi: 10.1101/gad.268151.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutiérrez L, et al. The role of the histone H2A ubiquitinase Sce in Polycomb repression. Development. 2012;139(1):117–27. doi: 10.1242/dev.074450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sankar S, et al. Gene regulatory networks in neural cell fate acquisition from genome-wide chromatin association of Geminin and Zic1. Sci Rep. 2016;6:37412. doi: 10.1038/srep37412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohta H, et al. Polycomb group gene rae28 is required for sustaining activity of hematopoietic stem cells. J Exp Med. 2002;195(6):759–70. doi: 10.1084/jem.20011911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takihara Y, et al. Targeted disruption of the mouse homologue of the Drosophila polyhomeotic gene leads to altered anteroposterior patterning and neural crest defects. Development. 1997;124(19):3673–82. doi: 10.1242/dev.124.19.3673. [DOI] [PubMed] [Google Scholar]
- 40.Tokimasa S, et al. Lack of the Polycomb-group gene rae28 causes maturation arrest at the early B-cell developmental stage. Exp Hematol. 2001;29(1):93–103. doi: 10.1016/s0301-472x(00)00620-2. [DOI] [PubMed] [Google Scholar]
- 41.Nomura M, et al. Isolation and characterization of retinoic acid-inducible cDNA clones in F9 cells: one of the early inducible clones encodes a novel protein sharing several highly homologous regions with a Drosophila polyhomeotic protein. Differentiation. 1994;57(1):39–50. doi: 10.1046/j.1432-0436.1994.5710039.x. [DOI] [PubMed] [Google Scholar]
- 42.Sultana R, et al. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics. 2002;80(2):129–34. doi: 10.1006/geno.2002.6810. [DOI] [PubMed] [Google Scholar]
- 43.Talkowski ME, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149(3):525–37. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beunders G, et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet. 2013;92(2):210–20. doi: 10.1016/j.ajhg.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagamani SC, et al. Detection of copy-number variation in AUTS2 gene by targeted exonic array CGH in patients with developmental delay and autistic spectrum disorders. Eur J Hum Genet. 2013;21(3):343–6. doi: 10.1038/ejhg.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oksenberg N, Ahituv N. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 2013;29(10):600–8. doi: 10.1016/j.tig.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jolley A, et al. De novo intragenic deletion of the autism susceptibility candidate 2 (AUTS2) gene in a patient with developmental delay: a case report and literature review. Am J Med Genet A. 2013;161A(6):1508–12. doi: 10.1002/ajmg.a.35922. [DOI] [PubMed] [Google Scholar]
- 48.Si S, et al. Loss of Pcgf5 Affects Global H2A Monoubiquitination but Not the Function of Hematopoietic Stem and Progenitor Cells. PLoS One. 2016;11(5):e0154561. doi: 10.1371/journal.pone.0154561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bedogni F, et al. Autism susceptibility candidate 2 (Auts2) encodes a nuclear protein expressed in developing brain regions implicated in autism neuropathology. Gene Expr Patterns. 2010;10(1):9–15. doi: 10.1016/j.gep.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Avela K, et al. Gene encoding a new RING-B-box-Coiled-coil protein is mutated in mulibrey nanism. Nat Genet. 2000;25(3):298–301. doi: 10.1038/77053. [DOI] [PubMed] [Google Scholar]
- 51.Bhatnagar S, et al. TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature. 2014;516(7529):116–20. doi: 10.1038/nature13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kettunen KM, et al. Trim37-deficient mice recapitulate several features of the multi-organ disorder Mulibrey nanism. Biol Open. 2016;5(5):584–95. doi: 10.1242/bio.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilton E, et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. Eur J Hum Genet. 2009;17(10):1325–35. doi: 10.1038/ejhg.2009.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ng D, et al. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat Genet. 2004;36(4):411–6. doi: 10.1038/ng1321. [DOI] [PubMed] [Google Scholar]
- 55.Fan Z, et al. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat Cell Biol. 2009;11(8):1002–9. doi: 10.1038/ncb1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sánchez C, et al. Proteomics analysis of Ring1B/Rnf2 interactors identifies a novel complex with the Fbxl10/Jhdm1B histone demethylase and the Bcl6 interacting corepressor. Mol Cell Proteomics. 2007;6(5):820–34. doi: 10.1074/mcp.M600275-MCP200. [DOI] [PubMed] [Google Scholar]
- 57.Gearhart MD, et al. Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol Cell Biol. 2006;26(18):6880–9. doi: 10.1128/MCB.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mo A, et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron. 2015;86(6):1369–84. doi: 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tiberi L, et al. A BCL6/BCOR/SIRT1 complex triggers neurogenesis and suppresses medulloblastoma by repressing Sonic Hedgehog signaling. Cancer Cell. 2014;26(6):797–812. doi: 10.1016/j.ccell.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 60.Pennekamp P, et al. Situs inversus and ciliary abnormalities: 20 years later, what is the connection? Cilia. 2015;4(1):1. doi: 10.1186/s13630-014-0010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adorno M, et al. Usp16 contributes to somatic stem-cell defects in Down’s syndrome. Nature. 2013;501(7467):380–4. doi: 10.1038/nature12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Joo HY, et al. Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature. 2007;449(7165):1068–72. doi: 10.1038/nature06256. [DOI] [PubMed] [Google Scholar]
- 63.Olmos-Serrano JL, et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron. 2016;89(6):1208–22. doi: 10.1016/j.neuron.2016.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scheuermann JC, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–7. doi: 10.1038/nature08966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harbour JW, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–3. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wiesner T, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43(10):1018–21. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Testa JR, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43(10):1022–5. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoischen A, et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011;43(8):729–31. doi: 10.1038/ng.868. [DOI] [PubMed] [Google Scholar]
- 69.Dangiolo SB, et al. Bohring-Opitz syndrome (BOS) with a new ASXL1 pathogenic variant: Review of the most prevalent molecular and phenotypic features of the syndrome. Am J Med Genet A. 2015;167A(12):3161–6. doi: 10.1002/ajmg.a.37342. [DOI] [PubMed] [Google Scholar]
- 70.Shashi V, et al. De Novo Truncating Variants in ASXL2 Are Associated with a Unique and Recognizable Clinical Phenotype. Am J Hum Genet. 2017;100(1):179. doi: 10.1016/j.ajhg.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bainbridge MN, et al. De novo truncating mutations in ASXL3 are associated with a novel clinical phenotype with similarities to Bohring-Opitz syndrome. Genome Med. 2013;5(2):11. doi: 10.1186/gm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kloet SL, et al. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat Struct Mol Biol. 2016;23(7):682–90. doi: 10.1038/nsmb.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morey L, et al. Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell. 2012;10(1):47–62. doi: 10.1016/j.stem.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 74.Baskind HA, et al. Functional conservation of Asxl2, a murine homolog for the Drosophila enhancer of trithorax and polycomb group gene Asx. PLoS One. 2009;4(3):e4750. doi: 10.1371/journal.pone.0004750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Endoh M, et al. PCGF6-PRC1 suppresses premature differentiation of mouse embryonic stem cells by regulating germ cell-related genes. Elife. 2017:6. doi: 10.7554/eLife.21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tavares L, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148(4):664–78. doi: 10.1016/j.cell.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He J, et al. Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat Cell Biol. 2013;15(4):373–84. doi: 10.1038/ncb2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wong SJ, et al. KDM2B Recruitment of the Polycomb Group Complex, PRC1.1, Requires Cooperation between PCGF1 and BCORL1. Structure. 2016;24(10):1795–1801. doi: 10.1016/j.str.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abdel-Wahab O, et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210(12):2641–59. doi: 10.1084/jem.20131141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lai HL, Wang QT. Additional sex combs-like 2 is required for polycomb repressive complex 2 binding at select targets. PLoS One. 2013;8(9):e73983. doi: 10.1371/journal.pone.0073983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bantignies F, Cavalli G. Polycomb group proteins: repression in 3D. Trends Genet. 2011;27(11):454–64. doi: 10.1016/j.tig.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 82.Lewis EB. A gene complex controlling segmentation in Drosophila. Nature. 1978;276(5688):565–70. doi: 10.1038/276565a0. [DOI] [PubMed] [Google Scholar]
- 83.Schwartz YB, Pirrotta V. A new world of Polycombs: unexpected partnerships and emerging functions. Nat Rev Genet. 2013;14(12):853–64. doi: 10.1038/nrg3603. [DOI] [PubMed] [Google Scholar]
- 84.Duncan IM. Polycomblike: a gene that appears to be required for the normal expression of the bithorax and antennapedia gene complexes of Drosophila melanogaster. Genetics. 1982;102(1):49–70. doi: 10.1093/genetics/102.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aranda S, et al. Regulation of gene transcription by Polycomb proteins. Sci Adv. 2015;1(11):e1500737. doi: 10.1126/sciadv.1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Conway E, et al. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol. 2015;37:42–8. doi: 10.1016/j.ceb.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 87.Cooper S, et al. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 2014;7(5):1456–70. doi: 10.1016/j.celrep.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Blackledge NP, et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 2014;157(6):1445–59. doi: 10.1016/j.cell.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kalb R, et al. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol. 2014;21(6):569–71. doi: 10.1038/nsmb.2833. [DOI] [PubMed] [Google Scholar]
- 90.Entrevan M, et al. Regulation of Genome Architecture and Function by Polycomb Proteins. Trends Cell Biol. 2016;26(7):511–25. doi: 10.1016/j.tcb.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 91.Cruz-Molina S, et al. PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell. 2017 doi: 10.1016/j.stem.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 92.Kundu S, et al. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol Cell. 2017;65(3):432–446. e5. doi: 10.1016/j.molcel.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lau MS, et al. Mutation of a nucleosome compaction region disrupts Polycomb-mediated axial patterning. Science. 2017;355(6329):1081–1084. doi: 10.1126/science.aah5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hauri S, et al. A High-Density Map for Navigating the Human Polycomb Complexome. Cell Rep. 2016;17(2):583–595. doi: 10.1016/j.celrep.2016.08.096. [DOI] [PubMed] [Google Scholar]
- 95.Katoh M. Functional and cancer genomics of ASXL family members. Br J Cancer. 2013;109(2):299–306. doi: 10.1038/bjc.2013.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J, et al. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood. 2014;123(4):541–53. doi: 10.1182/blood-2013-05-500272. [DOI] [PMC free article] [PubMed] [Google Scholar]