

Abstract
DNA replication stress promotes genome instability in cancer. However, the contribution of the replication stress response to the development of malignancies remains unresolved. The DNA replication stress response protein SMARCAL1 stabilizes DNA replication forks and prevents replication fork collapse, a cause of DNA breaks and apoptosis. While the fork regression/remodeling functions of SMARCAL1 have been investigated, its in vivo functions in replication stress and cancer are unclear. Using a gamma radiation (IR)-induced replication stress T-cell lymphoma mouse model, we observed a significant inhibition of lymphomagenesis in mice lacking one or both alleles of Smarcal1. Notably, a quarter of the Smarcal1-deficient mice did not develop tumors. Moreover, hematopoietic stem/progenitor cells (HSPCs) and developing thymocytes in Smarcal1-deficient mice showed increased DNA damage and apoptosis during the proliferation burst following IR and an impaired ability to repopulate the thymus after IR. Additionally, mice lacking Smarcal1 showed significant HSPC defects when challenged to respond to other replication stress stimuli. Thus, our data reveal the critical function of the DNA replication stress response and specifically, Smarcal1 in hematopoietic cell survival and tumor development. Our results also provide important insight into the immunodeficiency observed in individuals with mutations in SMARCAL1 by suggesting that it is an HSPC defect.
Keywords: lymphoma, DNA replication stress, Smarcal1, DNA damage, apoptosis
Introduction
Genome instability is a hallmark of human cancer and contributes to tumor initiation and progression.1 A poorly understood contributor to genome instability is DNA replication stress, which refers to processes that induce replication fork stalling and/or collapse and impede DNA synthesis.2 DNA damage is accrued through the processing of stalled forks into double-stranded DNA breaks, and incomplete DNA replication can result in deletions and chromosomal abnormalities.2,3 Replication stress has been identified in both pre-malignant and cancerous lesions and is associated with tumor development, progression, and evolution.4,5 Replication-associated DNA damage is thought to induce selective pressure to inactivate tumor suppressive programs in pre-malignant cells and provide a source of mutation within tumor cells.6 However, in response to replication-associated DNA damage, cells activate a DNA damage response to facilitate the completion of DNA replication and repair damaged DNA to minimize the threat to the genome.3,7 While replication stress has been linked to tumorigenesis, the contribution and function of specific replication stress response proteins in tumor development remain unknown.
Mammalian cells express several proteins that repair and restart stalled replication forks and promote genome stability during replication stress. One such protein, SMARCAL1, binds forked DNA structures.8 It is recruited to stalled replication forks, through an interaction with the single-stranded (ss) DNA-binding protein replication protein A (RPA). There, SMARCAL1 promotes fork stabilization and repair by catalyzing the annealing of RPA-coated ssDNA to remodel stalled replication forks.9–16 Bi-allelic mutations in SMARCAL1 cause the pleiotropic disorder Schimke Immuno-osseous Dysplasia (SIOD), which is characterized by immunodeficiency, spondyloepiphyseal dysplasia, facial dysmorphism, and progressive nephropathy.17 While the biochemical function of SMARCAL1 at replication forks has been investigated, the in vivo functions of SMARCAL1, specifically regarding its role in tumorigenesis and the mechanism(s) driving the clinical phenotypes of SIOD, remain unresolved.
Here we report that Smarcal1 is a critical effector of the replication stress response in hematopoietic cells in vivo. In an irradiation (IR)/replication stress-induced model of T-cell lymphomagenesis, a deficiency in Smarcal1 resulted in elevated DNA damage and a significant delay in T-cell lymphoma development. Smarcal1 was required for hematopoietic cell survival during forced proliferation from multiple stimuli and for repopulation of the thymus following IR. Thus, our data establishes Smarcal1 as a critical mediator of hematopoietic cell survival during acute replication stress via its genome-protecting functions. Moreover, these results also offer an explanation behind the immunodeficiency exhibited by SIOD patients.
Results
Smarcal1 knockout mice express a non-functional truncated protein
To investigate the in vivo functions of Smarcal1 during tumor development and acute replication stress, we evaluated Smarcal1 knockout mice (Smarcal1Δ/Δ).18 The targeting construct used to generate the Smarcal1Δ/Δ mice suggests an N-terminal truncated protein lacking amino acids 1-293 could be expressed; this region encodes the RPA-binding domain, the first HARP domain, and a portion of the second HARP domain (Fig. 1A). Western blotting using an N-terminal specific Smarcal1 antibody showed half the levels of the 110 kDa full-length protein in Smarcal1+/Δ MEFs and its expected loss in Smarcal1Δ/Δ MEFs (Fig. 1A). A C-terminal specific antibody detected a ~70 kDa Smarcal1 protein in these same cells (Fig. 1A), verifying the expression of truncated Smarcal1 (Smarcal1Δ).
Figure 1. N-terminal truncated Smarcal1 is non-functional.
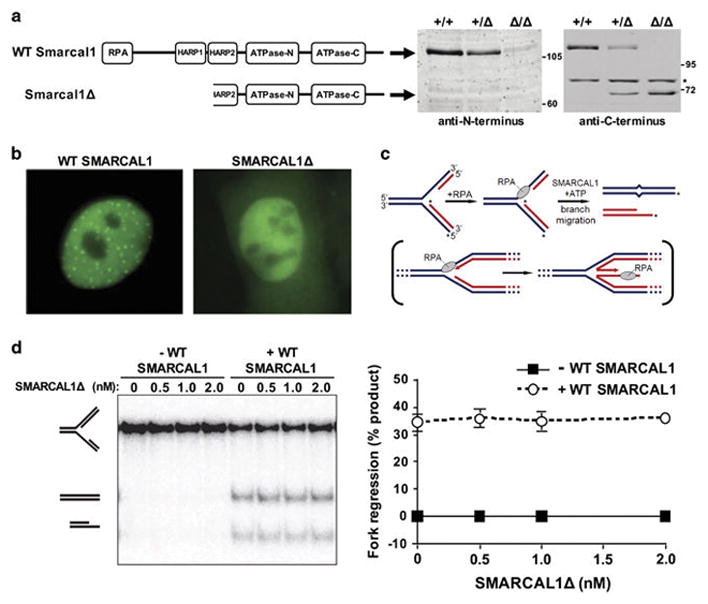
A) Schematic of wild-type (WT) and N-terminal truncated Smarcal1 (Smarcal1Δ) with functional domains indicated. Whole cell lysates from mouse embryonic fibroblasts (MEFs) of the indicated genotypes were Western blotted with antibodies against the N- or C-terminus of Smarcal1. Asterisks denotes location of a non-specific band. B) Representative images of U2OS cells with wild-type (WT) GFP-SMARCAL1 or GFP-SMARCAL1Δ following HU treatment. C) Schematic of the in vitro fork reversal assay with the in vivo physiological reaction shown in brackets. 32P labeled strands are indicated with an asterisk. D) Fork reversal activity of increasing concentrations of SMARCAL1Δ was measured alone and in the presence of wild-type (WT) SMARCAL1. Native gel electrophoresis performed (left). Mean of fork regression quantification using phosphorimaging of three separate experiments is graphed (right); error bars are SEM.
To determine whether Smarcal1Δ retained any functions of the wild-type protein, we performed in vitro analyses. Previous studies demonstrated that wild-type Smarcal1 is localized to stalled replication forks through its interaction with RPA.9,11–14 To assess if Smarcal1Δ, which lacks its RPA binding domain, can localize to stalled replication forks, U2OS cells were transfected with vectors encoding a fusion protein of GFP and either SMARCAL1Δ or wild-type SMARCAL1. GFP foci corresponding to SMARCAL1 localization to stalled replication forks induced by hydroxyurea were observed in cells expressing wild-type GFP-SMARCAL1 (Fig. 1B). In contrast, only diffuse GFP (no GFP foci) was present in cells expressing GFP-SMARCAL1Δ (Fig. 1B). These results indicate Smarcal1Δ is unable to localize to sites of replication stress.
Smarcal1 promotes genome stability through its fork regression and remodeling activities, which require its HARP2 domain.10,15,16 To measure this activity of Smarcal1Δ and to determine whether this truncated protein can exert any dominant negative effects against wild-type Smarcal1, fork reversal assays were performed. Substrates with a leading-strand gap were incubated with increasing concentrations of SMARCAL1Δ alone or in the presence of wild-type SMARCAL1 (Fig. 1C). Fork regression was not observed in reactions containing SMARCAL1Δ alone, whereas wild-type SMARCAL1 induced fork regression regardless of the concentration of SMARCAL1Δ (Fig. 1D). Thus, the N-terminal truncated Smarcal1 protein is functionally dead and appears to exert no dominant negative effects on wild-type Smarcal1.
Loss of Smarcal1 delays gamma irradiation (IR)-induced T-cell lymphomagenesis
Repeated whole body, low-dose IR of young mice induces T-cell lymphoma development, reportedly through the accumulation of DNA mutations in a hematopoietic stem or progenitor cell (HSPC) or an early T-cell progenitor derived from an HSPC.19,20 Following each round of irradiation, HSPCs rapidly proliferate to repopulate the lymphoid compartments depleted by IR-induced apoptosis. The presence of IR-associated DNA damage, coupled with the proliferative burst that occurs after IR exposure, is expected to generate significant levels of replication stress in both cycling HSPCs and the HSPC-derived thymic progenitors repopulating the depleted thymus following each radiation cycle.
To investigate the role of Smarcal1 in IR/replication stress-mediated T-cell lymphomagenesis, littermate-matched mice of all three Smarcal1 genotypes were subjected to 4 weekly cycles of low-dose IR. Smarcal1 wild-type mice developed T-cell lymphomas at the expected rate with a mean survival of 143 days (Fig. 2A).19 However, Smarcal1+/Δ and Smarcal1Δ/Δ mice had a delay in tumor onset and significantly increased overall survival (Fig. 2A, p=0.0399, log-rank test; mean survival 180 and 237 days respectively). Notably, 500 days after the last dose of IR, 23% of Smarcal1+/Δ mice and 29% of Smarcal1Δ/Δ mice were still alive, whereas by 450 days, all of the Smarcal1+/+ littermates had developed tumors and were sacrificed (Fig. 2A).
Figure 2. Loss of Smarcal1 inhibits radiation-induced T-cell lymphomagenesis.
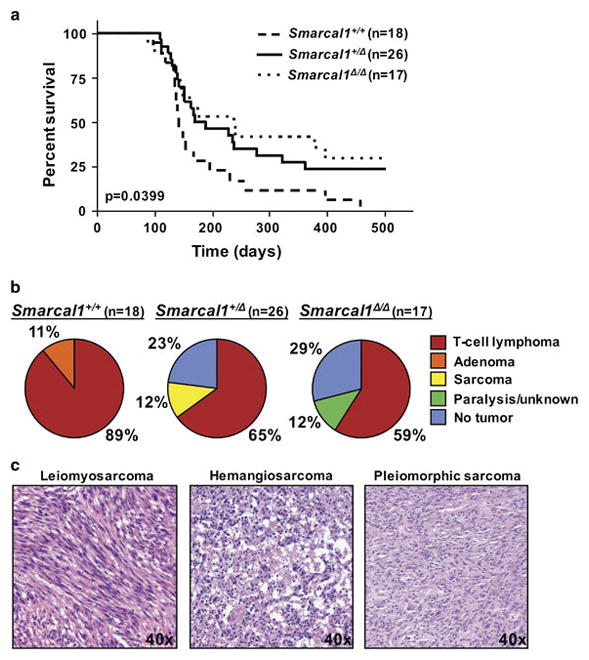
A) Kaplan-Meier survival curves of the indicated genotypes; overall p value denoted on graph; p=0.0307, +/+ vs. +/Δ and p=0.0217, +/+ vs. Δ/Δ; log-rank tests. Number of mice denoted by n. B) Tumor spectrum in the mice in A. C) Representative H&E images of the sarcomas that arose in Smarcal1+/Δ mice.
Genes that influence tumorigenesis can alter the rate of tumor development and/or the types of tumors that develop; therefore we also evaluated the tumor spectrum in the Smarcal1-deficient mice. As expected, most (89%) of the Smarcal1+/+ mice developed T-cell lymphomas, while 11% developed benign adenomas (Fig. 2B). Surprisingly, T-cell lymphomas emerged in only 65% of the Smarcal1+/Δ mice, whereas 12% developed sarcomas (Fig. 2B). Leiomyosarcoma, hemangiosarcoma, and pleiomorphic sarcoma were observed in Smarcal1+/Δ mice (Fig. 2C). Two of these sarcomas occurred in relatively young mice (124 and 230 days old), indicating they did not emerge due to old-age. Only 59% of Smarcal1Δ/Δ mice developed T-cell lymphomas and an additional 12% had undetermined pathology (e.g., hind limb paralysis and death from unknown cause) (Fig. 2B). The reduction in T-cell lymphoma frequency for both Smarcal1+/Δ (24% reduction) and Smarcal1Δ/Δ mice (30% reduction) was significant (p=0.0400 +/+ vs. +/Δ and p=0.0216 +/+ vs. Δ/Δ, t-tests). The T-cell lymphomas that arose in all genotypes were Thy1.2 positive and also typically CD8+ or CD8+/CD4+ positive (Supplemental Fig. 1).
Remarkably, 23% of Smarcal1+/Δ and 29% of Smarcal1Δ/Δ mice never developed a tumor up to 500 days after the last dose of IR, whereas tumors were present in all Smarcal1+/+ mice. This difference in tumor incidence was significant (p=0.0194 +/+ vs. +/Δ and p=0.0089 +/+ vs Δ/Δ, t-tests). Taken together, these data demonstrate that loss of Smarcal1 increased overall survival by inhibiting IR-induced T-cell lymphomagenesis and preventing tumor development altogether in a significant fraction of the mice. Our data also suggest that Smarcal1 haploinsufficiency influences tumor cell of origin, as several Smarcal1+/Δ mice developed sarcomas, which are not typically associated with IR-induced tumorigenesis.
Smarcal1-deficient thymocytes do not have an altered sensitivity to radiation
To gain insight into the biological mechanism behind the delay in tumor development observed in Smarcal1-deficient mice and since studies have disagreed on the requirements of Smarcal1 for the response to IR9,11,13,21, we first evaluated T-cell populations in the thymus in response to IR. We first assessed thymic T cells in unirradiated mice. There were similar percentages of CD4/CD8 double-positive (DP) and CD4 and CD8 single-positive (SP) T cells in Smarcal1-deficient mice compared to wild-type littermates (Fig. 3A and Supplemental Fig. 2A). There appeared to be reduced thymic cellularity in Smarcal1+/Δ and Smarcal1Δ/Δ mice; however, the reductions in total numbers of DP and SP thymocytes were not statistically significant (Fig. 3B and 3C and Supplemental Fig. 2B). Thus, loss of one or both alleles of Smarcal1 does not significantly alter thymic T-cell numbers or the proportion of specific thymocyte populations under normal physiologic conditions.
Figure 3. Deficiency in Smarcal1 does not alter thymocyte sensitivity to radiation.
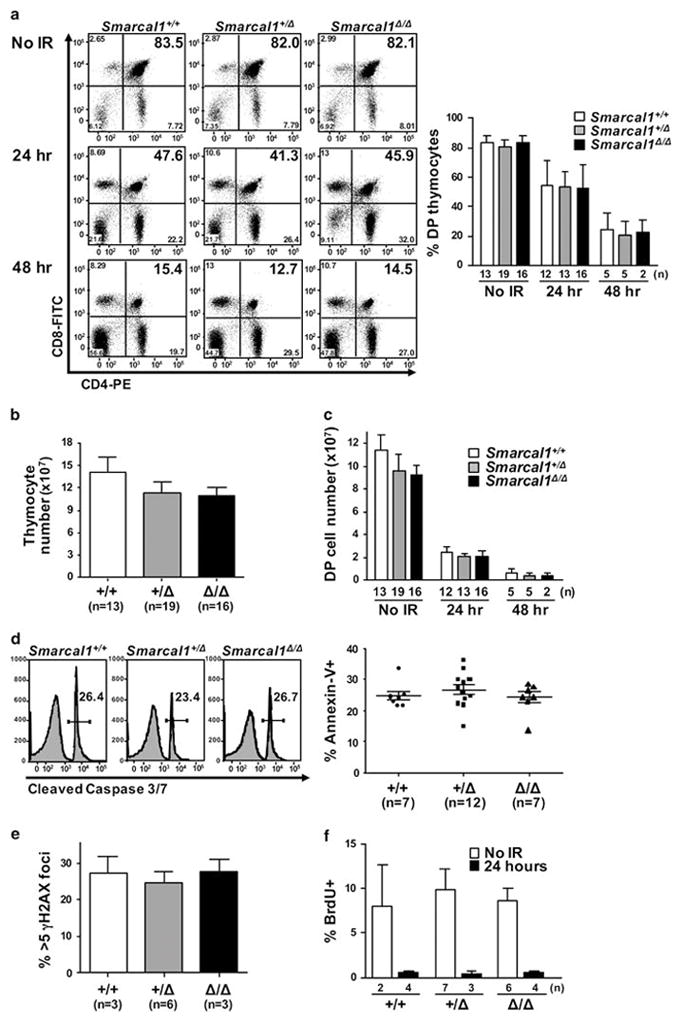
A) Representative dot plots of littermate matched thymic T cells without or at the indicated times following a single dose of 1.75 Gy of IR (left). Mean of data from seven independent experiments (right). B) Total thymic cellularity in unirradiated littermates. C) CD4/CD8 DP thymocytes not subjected to IR or at the indicated times after IR. D-F) Thymi harvested 24 hours after littermates were subjected to a single dose of 1.75 Gy of IR. D) Cleaved caspase 3/7 activity and Annexin V positivity in thymocytes measured by flow cytometry. E) Immunofluorescence for γH2AX was quantified; thymocytes with >5 γH2AX foci graphed. F) Mean BrdU incorporation of three independent litters. Error bars represent SEM; n denotes the number of mice.
Because differences in radiation sensitivity could alter the rate of tumorigenesis in mice, we evaluated thymocytes in littermates during the apoptosis phase induced by IR. Compared to unirradiated mice, all Smarcal1 genotypes showed a reduction in the percentage of DP thymocytes of ~30% at 24 hours and ~60% at 48 hours after a single 1.75 Gy dose of IR (Fig. 3A). The numbers of DP and SP thymocytes fell precipitously at each interval evaluated after IR (Fig. 3C and Supplemental Fig. 2A and 2B). Total DP numbers were reduced by >95% in all genotypes 48 hours post-IR, indicating a similar ablation of the thymic compartment for all mice (Fig. 3C). Thymocyte apoptosis, as measured by cleaved caspase 3/7 and Annexin V, was analogous between all Smarcal1 genotypes 24 hours after IR (Fig. 3D). Similarly, thymocytes from littermates of all genotypes showed comparable amounts of phosphorylated histone H2AX (γH2AX), a marker of DNA breaks 24 hours post-IR (Fig. 3E). Therefore, a deficiency in Smarcal1 does not appear to alter sensitivity to IR.
To determine if a loss of Smarcal1 would impact the cell cycle arrest that occurs upon radiation exposure, BrdU incorporation was measured in DP thymocytes. All Smarcal1 genotypes showed a similar percentage of BrdU positive DP thymocytes in the absence of IR (Fig. 3F). Twenty-four hours after IR, DP thymocytes in littermates of all genotypes had <1% BrdU incorporation (Fig. 3F and Supplemental Fig. 2C), demonstrating that Smarcal1-deficient cells have an intact and functioning DNA-damage induced cell cycle arrest response. Therefore, the early response to IR-induced DNA damage that results in cell cycle arrest and apoptosis is not altered with loss of Smarcal1 and likely does not contribute to the observed delay in T-cell lymphoma development in the Smarcal1+/Δ and Smarcal1Δ/Δ mice.
Smarcal1-deficient mice have reduced numbers of T cells during the proliferative response to IR
Following whole-body IR, thymocytes undergo apoptosis within 48 hours (Fig. 3), requiring a replicative burst of HSPCs to generate precursor T cells that then proliferate and differentiate to repopulate the thymus, which is observed 72 hours after IR.22 To evaluate whether Smarcal1-deficient thymocytes are impaired during this proliferative burst, we evaluated thymocyte populations 72 hours following IR. Compared to wild-type littermates, there was a significant decrease in both the percentage and total number of DP T cells and a reduction in the number of SP thymocytes in Smarcal1+/Δ and Smarcal1Δ/Δ mice (Fig. 4A, Supplemental Fig. 3). These data indicate Smarcal1-deficient mice have an impaired ability to repopulate the thymus after IR.
Figure 4. Loss of Smarcal1 increases thymocyte sensitivity to replication stress.
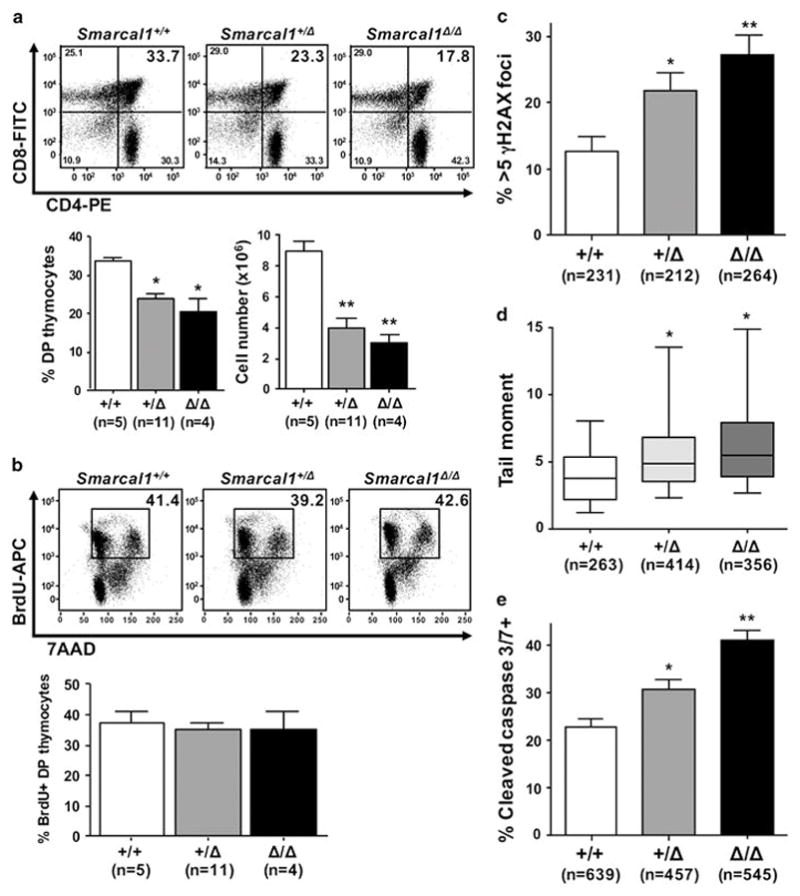
Thymi from littermates were harvested 72 hours after a single dose of 1.75 Gy IR. A) Representative dot plots of thymocytes (top), and mean percentage of DP thymocytes from three independent experiments (bottom left). Mean total DP thymocytes from each genotype from three independent experiments (bottom right). B) Representative dot plots of BrdU incorporation and DNA content (7AAD) of thymocytes (top). Mean of three independent experiments (bottom). C) Quantification of thymocytes with >5 γH2AX foci 72 hours after IR from 4 +/+, 3 +/Δ, and 4 Δ/Δ mice from two litters; n denotes the number of cells analyzed. D) Box-and-whisker plots of tail moments from neutral comet assays of thymocytes isolated from mice of the indicated genotypes. 3 +/+, 5 +/Δ and 4 Δ/Δ mice from two separate litters were evaluated. Boxes are the 25th and 75th percentiles, whiskers are 10th and 90th percentiles, and the lines are the medians. The number of cells evaluated is indicated by n. E) Mean percentage of cleaved caspase 3/7-positive thymocytes from a representative litter (3 +/+, 2 +/Δ, and 2 Δ/Δ) of 2 independent experiments; the number of cells analyzed is indicated by n. Error bars are SEM; A, *p<0.01, **p<0.001; C, *p<0.05, **p<0.0001; D, *p<0.001; E, *p<0.01, **p<0.0001; one-way ANOVA.
To investigate the possible explanations for the decrease in thymocytes detected in mice lacking Smarcal1 during the proliferative burst, we first measured BrdU incorporation. At 72 hours post-IR, all Smarcal1 genotypes showed an analogous percentage (~40%) of DP thymocytes had incorporated BrdU (Fig. 4B). This suggested there were similar numbers of thymocytes cycling in Smarcal1-deficient mice as in wild-type littermates. We then evaluated DNA damage and apoptosis, both of which are consequences of unresolved replications stress. As compared to wild-type thymocytes, there were significantly increased numbers of γH2AX foci (Fig. 4C) and DNA breaks (Fig. 4D) in Smarcal1+/Δ and Smarcal1Δ/Δ littermates, 72 hours after IR. Additionally, Smarcal1-deficient thymocytes had elevated levels of apoptosis, as measured by cleaved caspase 3/7 activity (Fig. 4E). Thus, thymocytes lacking one or both alleles of Smarcal1 have increased DNA damage and apoptosis during a time of rapid proliferation, indicating increased sensitivity to replication stress. These data also suggest the decreased ability to respond to replication stress caused from the proliferative burst following IR likely contributes to the delay in T-cell lymphomagenesis in Smarcal1-deficient mice.
Loss of Smarcal1 results in decreased HSPCs following IR
Because Smarcal1-deficient mice showed defects in repopulating the thymus after IR and thymic progenitor cells are derived from HSPCs that have been driven out of quiescence22,23, we assessed HSPC populations. We identified the HSPC-enriched LSK population (lineage-, cKit+, Sca1+), which we further refined into multi-potent progenitors (MPPs; lineage-, cKit+, Sca1+, CD48+, CD150-) and long-term hematopoietic stem cells (LT-HSCs; lineage-, cKit+, Sca1+, CD48-, CD150+) (Supplemental Fig. 4A).24,25 To determine whether there were differences in HSPCs in unstressed Smarcal1-deficient mice, we evaluated unirradiated mice. There were similar numbers of LSKs, MPPs, and LT-HSCs in Smarcal1-deficient mice compared to wild-type littermates (Fig. 5A and 5B). The percentages of these HSPC populations were also analogous between genotypes (Supplemental Fig. 4B).
Figure 5. Reduced HSPCs in mice lacking one or both alleles of Smarcal1 following forced proliferation.
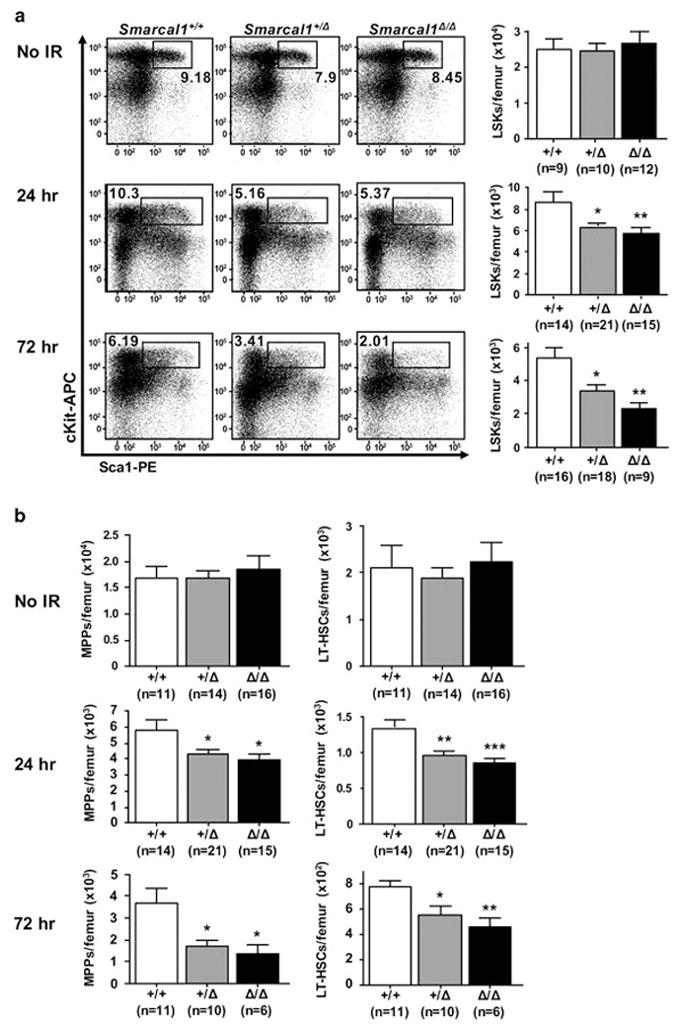
Bone marrow harvested from littermates unirradiated or at the indicated interval after IR. A) Representative dot plots of LSKs (left). Mean total LSKs per femur (right). B) Mean total MPPs (left) and LT-HSCs (right) at the indicated intervals following IR. Data are from six independent experiments. Error bars are SEM; A, *p<0.05, **p<0.01; B, *p<0.05, **p<0.01, ***p<0.001; one-way ANOVA.
To determine whether loss of Smarcal1 affected rapidly cycling HSPCs, we evaluated HSPC populations at intervals following IR when HSPCs are induced to proliferate. Compared to wild-type littermates, there was a significant decrease (~30%) in each of the LSK, MPP, and LT-HSC populations in Smarcal1+/Δ and Smarcal1Δ/Δ mice 24 hours after IR (Fig. 5A, 5B and Supplemental Fig. 4C). At 72 hours after IR, there was a ~40%, ~55%, and ~30% reduction in the LSK, MPP, and LT-HSC populations, respectively, in the Smarcal1+/Δ mice compared to wild-type littermates (Fig. 5A and 5B). Similarly, the Smarcal1Δ/Δ mice showed a ~60%, ~65% and ~40% decrease, respectively, in these same populations compared to wild-type mice (Fig. 5A and 5B). Therefore, the decreased numbers of HSPCs likely contributes to the delay in repopulation of the thymus and the inhibition of lymphomagenesis observed in Smarcal1-deficient mice as there are fewer HSPCs with the potential to undergo transformation.
To determine if the HSPC reduction observed in Smarcal1-deficient mice was due to differences in proliferation rates and/or increased sensitivity to replication stress, we measured BrdU incorporation in the LSK compartment at intervals following whole-body IR. At 24 hours post-IR, we observed an analogous significant increase in BrdU positive LSKs, MPPs and LT-HSCs in all Smarcal1 genotypes compared to unirradiated mice, and the percentage of BrdU positive cells remained elevated above steady-state in all genotypes 72 hours post IR (Fig. 6A). Since Smarcal1+/Δ and Smarcal1Δ/Δ LSKs appeared to be proliferating at equal rates to wild-type Smarcal1 LSKs in response to IR, we measured DNA damage and apoptosis in bone marrow cells during this forced proliferative stress. Both Smarcal1+/Δ and Smarcal1Δ/Δ bone marrow cells had significantly increased numbers of cells with γH2AX foci (Fig 6B) and elevated amounts of broken DNA (Fig. 6C) 24 hours after IR. Subsequently, Smarcal1-deficient bone marrow showed increased numbers of apoptotic cells as detected by cleaved caspase 3/7 activity 72 hours following IR (Fig. 6D). These data indicate Smarcal1 is required to respond to proliferative stress in HSPCs, and loss of one or both alleles of Smarcal1 increases HSPC susceptibility to DNA breakage caused by replication stress, which results in HSPC apoptosis.
Figure 6. Smarcal1-deficient HSPCs have increased sensitivity to replication stress.
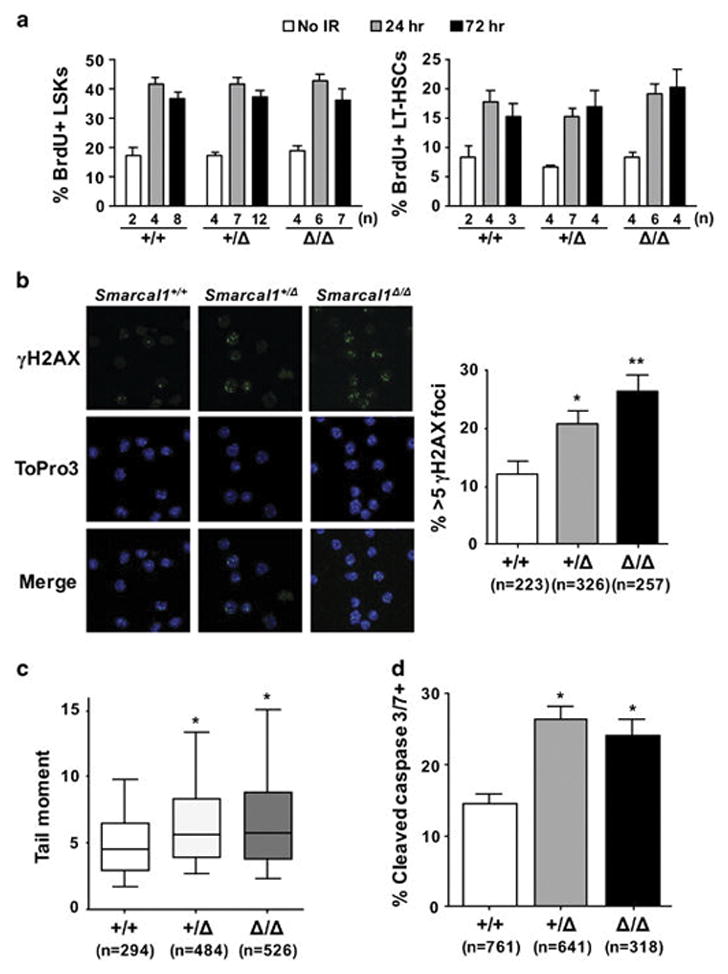
A) Bone marrow harvested from littermates unirradiated or at the indicated interval after IR. Mean percentage of BrdU positive LSKs (left) or LT-HSCs (right). B) Representative images of γH2AX immunofluorescence of bone marrow cells from littermates of the indicated genotypes (left) Quantification of bone marrow cells with >5 γH2AX foci 24 hours after IR from 3 +/+, 4 +/Δ, and 3 Δ/Δ littermates from two litters; n denotes the number of individual cells analyzed. C) Box-and-whisker plots of tail moments from neutral comet assays of bone marrow cells isolated from littermates of the indicated genotypes 24 hours after IR. Boxes are the 25th and 75th percentiles, whiskers are 10th and 90th percentiles, and the lines are the medians. The number of individual cells analyzed is indicated by n from 3 +/+, 5 +/Δ, and 3 Δ/Δ littermates from two litters. D) Mean percentage of cleaved caspase 3/7-positive bone marrow cells from a representative litter (3 +/+, 2 +/Δ, and 2 Δ/Δ); 2 independent experiments; the number of cells analyzed is indicated by n.. Error bars are SEM; B, *p<0.05, **p<0.001; C, *p<0.001; D, *p<0.0001, one-way ANOVA.
Smarcal1 deficient HSPCs are more sensitive to replication stress
To further examine HSPC function and sensitivity to replication stress in Smarcal1-deficient mice, we utilized stimuli other than IR to induce in vivo replication stress. We subjected a cohort of Smarcal1+/+, Smarcal1+/Δ and Smarcal1Δ/Δ littermates to repeated injections of 5-fluorouracil (5-FU), a pyrimidine analogue that kills cycling hematopoietic cells, and thereby drives a burst of HSPC proliferation.26 Compared to wild-type littermates, Smarcal1-deficient mice showed significantly reduced survival when challenged with repeated replication stress from 5-FU (Fig. 7A; p=0.0194, log-rank test). Thirty-five days after the first 5-FU injection, 52% of wild-type mice were still alive, whereas only 5% of Smarcal1+/Δ and 16% of Smarcal1Δ/Δ mice were alive. These data show bone marrow cells lacking one or both alleles of Smarcal1 have increased sensitivity to a form of repeated, acute replication stress distinct from IR, demonstrating a requirement of Smarcal1 to respond to multiple forms of replication stress.
Figure 7. Loss of Smarcal1 impairs HSPC function.

A) Kaplan-Meier survival curves of the indicated genotypes following 5-FU injections every week for 5 weeks beginning at time 0; number of mice denoted by n; overall p value denoted on graph; p=0.0031 +/+ vs. +/Δ and p=0.0203 +/+ vs. Δ/Δ, log-rank tests. B) Mean percentage of CD45.2+ peripheral leukocytes in mice of the indicated genotype were determined at intervals following competitive bone marrow transplantation; n denotes the number of mice; error bars are SEM; p value determined by a two way ANOVA. C) Quantification of the percentage of CD45.2+ DP and SP thymocytes 16 weeks post-transplant. D) Quantification of the total numbers of CD45.2+ BMCs, LSKs, MPPs and LT-HSCs 16 weeks post-transplant. Error bars are SEM from n=7 Smarcal1+/+, n=8 Smarcal1+/Δ, and n=7 Smarcal1Δ/Δ mice for C and D; p values for C and D indicated.
To directly compare the functionality of Smarcal1-deficient HSPCs in response to forced proliferation, we performed competitive bone marrow transplants. Littermate donor CD45.2 Smarcal1+/+, Smarcal1+/Δ, and Smarcal1Δ/Δ bone marrow cells were injected with CD45.1 wild-type bone marrow cells (1:1 ratio) into lethally irradiated CD45.1 recipients. Four weeks post-transplant, and at each of the subsequent analyses, we detected a significant decrease in the percentage of CD45.2 peripheral leukocytes in recipient mice that received Smarcal1+/Δ or Smarcal1Δ/Δ bone marrow compared to mice that received wild-type bone marrow (Fig. 7B). By week 16, mice that received wild-type bone marrow had ~43% of circulating CD45.2 expressing leukocytes, whereas only ~32% of circulating leukocytes expressed CD45.2 in mice that received Smarcal1+/Δ or Smarcal1Δ/Δ bone marrow (Fig. 7B).
We also evaluated CD45.2 expression in thymocytes and bone marrow cells in recipient mice at sacrifice (16 weeks post-transplant). Compared to mice that received wild-type bone marrow, we observed a decrease in the percentage of CD45.2 positive cells in the DP and SP T-cell compartments within the thymus of mice that received Smarcal1+/Δ or Smarcal1Δ/Δ bone marrow (Fig. 7C). Moreover, analysis of bone marrow revealed a significant reduction in the number of CD45.2 positive total bone marrow cells in mice that received Smarcal1-deficient cells with decreases in the numbers of LSKs and MPPs (Fig. 7D). There was not a significant reduction in the less proliferative LT-HSCs in mice that received Smarcal1-deficient bone marrow (Fig. 7D). Therefore, these data demonstrate Smarcal1-deficient HSPCs are less functionally fit relative to wild-type HSPCs when challenged to repopulate the hematopoietic system, providing further evidence that Smarcal1 is required by HSPCs to mediate a normal response to replication stress.
Discussion
Biochemical and cellular analysis of Smarcal1 has shown it is activated by DNA replication stress and recruited to stalled replication forks. There Smarcal1 facilitates the completion of DNA synthesis by catalyzing fork remodeling, which is thought to promote genome stability.9–16 However, the in vivo significance of these findings, particularly in relationship to the development of malignancies and SIOD, the disease associated with mutant Smarcal1, remained unresolved. Our data show that functional Smarcal1 is necessary for cellular viability during acute replication stress in hematopoietic cells. A lack of even one allele of Smarcal1 was sufficient to confer sensitivity to multiple forms of replication stress in hematopoietic cells, leading to increased DNA damage and apoptosis. However, our data also unexpectedly revealed that being able to respond properly to replication stress contributes to tumorigenesis. Loss of one or both alleles of Smarcal1 significantly delayed DNA-damage induced T-cell lymphomagenesis and prevented lymphoma development altogether in a quarter of the mice. Thus, a Smarcal1 deficiency protected mice from IR-induced lymphomagenesis, indicating that a disabled replication stress response could shield against DNA replication stress-induced tumorigenesis.
IR-induced T-cell lymphomagenesis is reportedly due to the combined effects of acquired mutations in an HSPC, resulting in its cellular transformation, and the induced proliferation of this cell from signals that indicate lymphoid compartments need to be repopulated.20 Reducing HSPC proliferation by blocking lymphocyte apoptosis after IR inhibited lymphoma development, demonstrating the critical role of the apoptotic response during IR-mediated lymphomagenesis.22,23 Our data show that more DNA breaks and apoptosis occur in lymphocytes and bone marrow cells that have lost Smarcal1 during the proliferative, repopulation phase of the IR response, but that Smarcal1-deficient hematopoietic cells are as equally sensitive to the immediate apoptotic effects of IR as wild-type hematopoietic cells. These results are in contrast to data with other cell systems (shRNA and knockout chicken cell lines) that indicate a reduction or loss of Smarcal1 increases radiosensitivity.11,21 Our results indicate Smarcal1 does not contribute to gamma radiation sensitivity, but it is necessary for the proliferation that ensues as a consequence of the IR. Our data show the replication stress response that occurs due to HSPCs being forced out of quiescence necessitates Smarcal1 be functional to aid in the repair and restart of replication forks. Similarly, as they attempt to repopulate the thymus, HSPC-derived precursor T cells also experience proliferative stress that needs functional Smarcal1 to survive. With a haploinsufficiency or loss of both alleles of Smarcal1, both mature and precursor hematopoietic cells default to apoptosis from the replication stress due to the increased amount of unrepaired DNA damage. This leads to reduced pools of hematopoietic cells in the thymus and the bone marrow with mutated DNA. In support of this concept, we observed a significant reduction in the number of Smarcal1-deficient DP thymocytes and HSPCs 72 hours after IR compared to wild-type littermates. Additionally, Smarcal1 binding to RPA is reported necessary to facilitate the repair of double-strand DNA breaks.21 Therefore, although the initial apoptotic response of lymphocytes is required for IR-induced T-cell lymphomagenesis22,23, the clearance of proliferating progenitors with damaged DNA also conferred protection against lymphoma development.
Although sarcomas arise from mesenchymal cells, the actual cell of origin of sarcomas is controversial. It is hypothesized that a sarcoma arises from a mesenchymal stem or progenitor cell that has the potential to differentiate into osteoblasts, chondroblasts, and adipocytes.27 Sarcoma development is extremely rare in the IR model we used; yet, 12% of the Smarcal1 heterozygous mice developed sarcomas rather than T-cell lymphomas in response to IR. This was not because these 12% lived longer than the rest of the cohort and developed sarcoma due to age. Instead, these data suggest that a Smarcal1 haploinsufficiency conferred an increased susceptibility to the development of sarcoma from IR. If a mesenchymal stem or progenitor cell is the cell of origin of sarcomas, a reduced ability of these cells to respond to replication stress during a time of increased mesenchymal cell development (weeks 4–8 of mouse life) occurred preferentially in the heterozygous mice and resulted in the acquisition of mutations that allowed it to transform and not die. It is unclear why the Smarcal1Δ/Δ mice also did not develop sarcomas, but suggests that complete loss of functional Smarcal1 conferred protection against sarcoma development. Future investigations are needed to determine the reasons for the development of sarcomas in Smarcal1 heterozygous mice.
While our results have demonstrated a critical function for Smarcal1 during tumor development, our findings also have significant implications for other fields of research and particularly, SIOD patients. For example, hematopoietic cell proliferation and replication stress occurs in response to multiple stimuli, including infection, injury, and aging.28–30 Physiological stimuli that drive hematopoietic stem cells out of quiescence lead to the accrual of DNA damage, apoptosis, and stem cell attrition.28,31–33 Over time, proliferative stress results in DNA damage and stem cell loss or dysfunction. Our data reveal that Smarcal1 is critical for normal HSPC function in response to multiple forms of proliferative stimuli (IR, 5-FU, and competitive transplantation). With each of these stimuli, Smarcal1-deficient HSPCs were unable to respond as well as wild-type HSPCs and this resulted in reduced numbers of HSPCs, leading to reduced tumorigenesis, diminished cell expansion, and cell death.
Our data also provide a significant increase in understanding of the pathophysiology of SIOD. SIOD patients with homozygous mutations in SMARCAL1 are characterized by a severe, progressive immunodeficiency and increased rates of infection.17,34 Our data demonstrate a lack of Smarcal1 in rapidly cycling hematopoietic cells results in elevated DNA damage and loss of hematopoietic cells. When responding to infection, lymphocytes rapidly proliferate, which likely leads to increased replication stress and elevated lymphocyte apoptosis in SIOD patients. HSPCs would then need to proliferate to repopulate the lymphocyte compartments, resulting in elevated HSPC replication stress and apoptosis, which leads to a further decrease in lymphocytes and increased susceptibility to infection. Therefore, a reduced ability to repopulate lymphocyte compartments following normal childhood infections may explain the progressive lymphopenia observed in SIOD patients.17,34 Additionally, a recent report suggests that T cells in SIOD patients may have decreased ability to proliferate due to reduced expression of the IL-7α receptor.35 We determined that neither circulating nor pre-cursor thymic T cells in Smarcal1-deficient mice had a decrease in IL-7α receptor (data not shown). However, they did have defects in their ability to respond to replication stress and repopulate the thymus.
Therefore, our data significantly increase our understanding of the function of Smarcal1 in replication stress in vivo by revealing its requirement for mediating replication stress to protect from hematopoietic cell loss. Our results also indicate that inhibiting DNA replication can provide a protective function against tumorigenesis caused from replication stress. Finally, our data likely reveal the biological mechanism behind the lymphoid deficiencies of SIOD patients as being an HSPC defect.
Materials and Methods
Mice
C57Bl/6 Smarcal1+/Δ mice were from Dr. Cornelius Boerkoel (University of British Columbia). Littermates (male and female) were used for all experiments. For IR-induced T-cell lymphomagenesis, littermates were irradiated (1.75 gray, 137Cs) once weekly for four weeks at 28 days of age (+/−2 days). Mice were monitored and sacrificed upon tumor development and/or signs of illness. At 500 days, any mouse that was still alive was sacrificed. Tissues from all mice in the study were harvested for histological/pathological analysis (see Supplemental Material). For 5-fluorouracil (5-FU) experiments, 6–8 week-old littermates were intraperitoneally injected with 5-FU (150 mg) once weekly for 5 weeks and sacrificed at humane endpoints. Competitive (1:1 ratio) bone marrow transplants were performed following standard procedures (details in Supplemental Material). Mouse studies were approved by the Vanderbilt University and Thomas Jefferson University Institutional Animal Care and Use Committees and adhered to all state and federal guidelines.
Cell culture and vectors
Mouse embryonic fibroblasts (MEFs) were isolated and cultured as previously described.36 U2OS cells (ATCC) were cultured in DMEM plus 7.5% FBS. To induce fork stalling, U2OS cells were transfected with vectors encoding wild-type GFP-SMARCAL1 or GFP-SMARCAL1Δ and treated with 2 mM hydroxyurea (HU) for 4 hours as previously described.9
Flow cytometry
HSPCs were harvested from femurs and identified with a biotinylated hematopoietic lineage kit (B220, CD3, Gr-1, CD11b and Ter119; eBioscience) and a panel of antibodies against specific HSPC surface markers (details in Supplemental Material). BrdU incorporation in thymi and bone marrow from 6–8 week old mice 1–3 days after a single 1.75 Gy dose of IR was performed according to manufacturer’s instructions (BD Biosciences) four hours after intraperitoneal injection (1mg). Cells were incubated with FITC-Annexin-V and 7-AAD or Caspase 3/7 detection reagent according to the manufacturer (BD Biosciences or ThermoFisher, respectively). All samples were evaluated (blinded) on a FACScalibur (BD Biosciences) and analyzed using FlowJo.
Western blotting
Western blotting of whole cell lysates from MEFs isolated from Smarcal1+/+, Smarcal1+/Δ, and Smarcal1Δ/Δ embryos was performed as previously described.37 Antibodies directed against the N- and C-terminus of Smarcal1 were used as previously described.9
Fork reversal assay
Fork reversal assays were completed using gel-purified, radio-labeled DNA substrates containing a leading strand gap and 0.5–2nM SMARCAL1Δ with or without 2nM wild-type SMARCAL1 in fork reversal reaction buffer as previously described.16
Immunofluorescence
Immunofluorescence for γH2AX was performed using a standard protocol (details in Supplemental Material); a minimum of 40 cells per mouse were evaluated (blinded) for each experiment. Activated Caspase 3/7 was measured with the CellEvent Caspase 3/7 detection reagent (ThermoFisher) according to the manufacturer’s instructions. Nuclei were counterstained with DAPI (Sigma-Aldrich) or ToPro3-iodide (Invitrogen) and images captured using microscopy (Zeiss LSM 510, Nikon A1R, or Nikon Eclipse 80i); a minimum of 110 cells per mouse were evaluated (blinded) for each experiment.
Neutral Comet Assays
Neutral comet assays with thymocytes and total bone marrow cells were performed as previously described.38,39 A minimum of 50 cells per mouse were evaluated (blinded) per experiment.
Statistics
Log-rank tests (Figures 2A and 7A), t-tests (Figures 2B, 7C, and 7D), one-way ANOVA followed by a Bonferroni correction (Figures 4A, 4C, 4D, 4E, 5A, 5B, 6B, 6C, and 6D), and two-way ANOVA (Figure 7B) were used to determine statistical significance.
Supplementary Material
Acknowledgments
Financial support:
F30CA189433 (MVP), T32GM007347 (MVP), Ann Melly Scholarship (MVP), R01CA160432 (CME and DC), and NCI Cancer Center Support Grants P30CA068485 and P30CA056036.
The authors would like to thank Dr. Cornelius Boerkoel for providing the Smarcal1+/Δ mice, Carol Bansbach for technical assistance with figure 1B, Catherine Alford for technical assistance with flow cytometry, the Bioimaging Shared Resource of the Sidney Kimmel Cancer Center, and Dr. Sandy Zinkel, Dr. Scott Hiebert, and members of the Eischen lab for helpful discussions.
Footnotes
Conflict of Interest:
The authors disclose no conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 5.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 6.Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- 7.Nam EA, Cortez D. ATR signalling: more than meeting at the fork. Biochem J. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yusufzai T, Kadonaga JT. HARP is an ATP-driven annealing helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, et al. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Betous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3:1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30:215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 18.Baradaran-Heravi A, Cho KS, Tolhuis B, Sanyal M, Morozova O, Morimoto M, et al. Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Hum Mol Genet. 2012;21:2572–2587. doi: 10.1093/hmg/dds083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan HS, Brown MB. A quantitative dose-response study of lymphoid-tumor development in irradiated C 57 black mice. J Natl Cancer Inst. 1952;13:185–208. [PubMed] [Google Scholar]
- 20.Kominami R, Niwa O. Radiation carcinogenesis in mouse thymic lymphomas. Cancer Sci. 2006;97:575–581. doi: 10.1111/j.1349-7006.2006.00218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keka IS, Mohiuddin, Maede Y, Rahman MM, Sakuma T, Honma M, et al. Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Res. 2015;43:6359–6372. doi: 10.1093/nar/gkv621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labi V, Erlacher M, Krumschnabel G, Manzl C, Tzankov A, Pinon J, et al. Apoptosis of leukocytes triggered by acute DNA damage promotes lymphoma formation. Genes Dev. 2010;24:1602–1607. doi: 10.1101/gad.1940210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michalak EM, Vandenberg CJ, Delbridge AR, Wu L, Scott CL, Adams JM, et al. Apoptosis-promoted tumorigenesis: gamma-irradiation-induced thymic lymphomagenesis requires Puma-driven leukocyte death. Genes Dev. 2010;24:1608–1613. doi: 10.1101/gad.1940110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 25.Yilmaz OH, Kiel MJ, Morrison SJ. SLAM family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood. 2006;107:924–930. doi: 10.1182/blood-2005-05-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 27.Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–131. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520:549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- 29.Cheshier SH, Prohaska SS, Weissman IL. The effect of bleeding on hematopoietic stem cell cycling and self-renewal. Stem Cells Dev. 2007;16:707–717. doi: 10.1089/scd.2007.0017. [DOI] [PubMed] [Google Scholar]
- 30.Adams PD, Jasper H, Rudolph KL. Aging-Induced Stem Cell Mutations as Drivers for Disease and Cancer. Cell Stem Cell. 2015;16:601–612. doi: 10.1016/j.stem.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512:198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alvarez S, Diaz M, Flach J, Rodriguez-Acebes S, Lopez-Contreras AJ, Martinez D, et al. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun. 2015;6:8548. doi: 10.1038/ncomms9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spranger J, Hinkel GK, Stoss H, Thoenes W, Wargowski D, Zepp F. Schimke immuno-osseous dysplasia: a newly recognized multisystem disease. J Pediatr. 1991;119:64–72. doi: 10.1016/s0022-3476(05)81040-6. [DOI] [PubMed] [Google Scholar]
- 35.Sanyal M, Morimoto M, Baradaran-Heravi A, Choi K, Kambham N, Jensen K, et al. Lack of IL7Ralpha expression in T cells is a hallmark of T-cell immunodeficiency in Schimke immuno-osseous dysplasia (SIOD) Clin Immunol. 2015;161:355–365. doi: 10.1016/j.clim.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 36.Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alt JR, Bouska A, Fernandez MR, Cerny RL, Xiao H, Eischen CM. Mdm2 binds to Nbs1 at sites of DNA damage and regulates double strand break repair. J Biol Chem. 2005;280:18771–18781. doi: 10.1074/jbc.M413387200. [DOI] [PubMed] [Google Scholar]
- 39.Bouska A, Lushnikova T, Plaza S, Eischen CM. Mdm2 promotes genetic instability and transformation independent of p53. Mol Cell Biol. 2008;28:4862–4874. doi: 10.1128/MCB.01584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.