

Abstract
Determination of structure and folding of certain classes of proteins remains intractable by conventional structural characterization strategies and has spurred the development of alternative methodologies. Mass spectrometry-based approaches have a unique capacity to differentiate protein heterogeneity due to the ability to discriminate populations, whether minor or major, featuring modifications or complexation with non-covalent ligands on the basis of m/z. Cleavage of the peptide backbone can be further utilized to obtain residue-specific structural information. Here, hydrogen elimination monitoring (HEM) upon ultraviolet photodissociation (UVPD) of proteins transferred to the gas phase via nativespray ionization is introduced as an innovative approach to deduce backbone hydrogen bonding patterns. Using well-characterized peptides and a series of proteins, prediction of the engagement of the amide carbonyl oxygen of the protein backbone in hydrogen bonding using UVPD-HEM is demonstrated to show significant agreement with the hydrogen-bonding motifs derived from molecular dynamics simulations and X-ray crystal structures.
Graphical Abstract
Hydrogen-bonding motifs of proteins are deciphered by ultraviolet photodissociation mass spectrometry
Introduction
The impact of mass spectrometry in the field of structural biology has increased substantially over the past decade, in large part owing to the complementarity of the information and range of applications offered by mass analysis relative to X-ray crystallography and nuclear magnetic resonance (NMR).1,2 In native mass spectrometry, intact, folded proteins and protein complexes are ionized from native buffers, providing a means to study the native-like species in the gas phase and affording direct stoichiometric and dynamics information. Native mass spectrometry has proven valuable in the development of conditions for crystallization of membrane proteins3,4 and for exploration of the conformational space occupied by intrinsically disordered proteins,5,6 both of which are classes of proteins that are difficult to characterize by conventional structural biology methods. Despite the success of these applications, however, the structural information obtainable from mass spectrometry is inherently low resolution and frequently only sheds light on gross structural features. Ion mobility, hydrogen-deuterium exchange, and a number of other covalent labeling strategies have emerged to help fill this gap,7–11 but these approaches are often restricted to surface exposed regions of the protein, used for comparative analyses, or require modeling or crystal structures to guide interpretation.
The activation and dissociation of native proteins in the gas phase has emerged as a powerful approach for sequencing and structural analysis; consequently, novel activation methods are increasingly being developed to extend the range of applications amenable to mass spectrometry. Collision induced dissociation (CID) is the gold standard for ion fragmentation but suffers from poor sequence coverage (particularly for native proteins in low charge states) and typically promotes protein unfolding prior to fragmentation, thus impeding any type of conformational analysis.12 Electron-based approaches, including electron capture and electron transfer dissociation (ECD and ETD, respectively), have gained popularity for native structure characterization because non-covalent interactions are not disrupted during cleavage of covalent backbone bonds that lead to sequence ions.13–16 ECD studies have been undertaken on several small proteins as a function of unfolding, and it was determined that fragmentation was restricted in the more ordered regions, particularly those stabilized by salt bridges.13–15 ECD to multi-protein systems has also been reported; exclusive observation of fragments from the more flexible, surface exposed regions of the protein provided additional support for the utility of ECD for deciphering unstructured regions of proteins.17,18 This type of structural assessment has some limitations, however, in that it is impossible to deduce whether fragmentation is absent from a particular region due to it being buried in the interior of the protein or due to the protein being highly ordered.
Ultraviolet photon-based activation methods have also emerged as a promising strategy for characterization of native protein structures.19–36 The most popular laser wavelengths are those that coincide with the chromophores of the protein backbone and aromatic sidechains, including 157, 193, 213, and 266 nm.19–36 Virtually all UVPD-based strategies offer fast dissociation dynamics, in theory enabling bond cleavage prior to disruption of peptide or protein structure. This has been well-demonstrated for 193 nm UVPD, for which different fragmentation patterns have been observed for mobility-separated conformers.19,20 Fragmentation efficiency has also been observed to correlate with secondary structural elements, and fragmentation yield is typically higher in loop and turn regions.30 However, because fragmentation efficiency is modulated to some extent by amino acid identity, assignment of structural features based on UVPD fragment abundance alone is difficult. We have recently examined more closely the types and abundances of diagnostic sequence ions created upon UVPD.35,36 We have demonstrated that the ratio of a and a+1 ions is sensitive to amino acid identity and particularly the presence or absence of hydrogen bonds at the amide carbonyl oxygens of the cleaved residues.35 Here, we use UVPD-MS to map hydrogen bonding along the backbone of intact proteins based on the extent of hydrogen elimination from a+1 ions, a process that results in a ions. We term this strategy hydrogen elimination monitoring (HEM). This approach is shown to be useful for diagnosing regions of the backbone engaged in hydrogen bonding in peptides and intact proteins, and we illustrate that the hydrogen bonding motifs of proteins transferred to the gas phase by nativespray ionization are consistent with their solution structures.
Methods
Mass spectrometry
Melittin, bovine ubiquitin, ammonium acetate, methanol, and formic acid were obtained from Sigma Aldrich (St. Louis, MO). Calmodulin (human) was kindly provided by the Dalby group at the University of Texas at Austin, and the human small C-terminal domain phosphatase 1 (Scp1) was provided by the Zhang group at the University of Texas at Austin and was prepared as described previously.37 Guinea pig adrenocorticotropic hormone (ACTH) was purchased from American Peptide Co. (now Bachem, Torrence, CA). Penetratin-Arg and α-synuclein were obtained from Anaspec (Freemont, CA). Lyophilized peptides/protein were suspended in appropriate spray solvent, as noted in the text, without any additional purification.
All UVPD experiments were carried out on a Thermo Scientific Orbitrap Elite mass spectrometer (San Jose, CA) coupled to a 193 nm Coherent Existar XS excimer laser (Santa Clara, CA) as described previously.34 Ions were introduced to the gas phase via a static nanospray source comprised of a pulled uncoated glass capillary tip and a platinum wire. Protein solutions are as indicated in the text. Protein concentrations were generally in the range of 10 uM in 50–100 mM ammonium acetate. Ions were transferred through the source optics using low transfer potentials and were irradiated by a single pulse of 193 nm photons in the HCD cell. The gas pressure in this region was typically 13–15 mTorr (compared to a normal HCD pressure of 5 mTorr) and the pulse energies are as noted in the text, typically between 1.5 and 2 mJ. The m/z of precursor and fragment ions were measured in the Orbitrap mass analyzer using a resolving power of 480 k. It should be noted that data from our previous study was collected on an Orbitrap Fusion mass spectrometer using two pulses at 1.5 mJ delivered per pulse. Because the Orbitrap Elite and Orbitrap Fusion mass spectrometer have different configurations with the lasers aligned in different regions (HCD cell versus linear ion trap), several standard peptides were initially examined by UVPD on the Fusion mass spectrometer and 1.5–2.0 mJ was found to best replicate the results observed previously on the Elite mass spectrometer. Interpretation of mass spectral data was undertaken manually. The αa value ([a]/([a] + [a+1])) of each a ion was computed using a solving algorithm in Microsoft Excel to fit isotope distributions to the isotopic envelope of the a, a+1, and, if present, a+2 ions.35 In instances where a+2 ions were observed, typically entailing cleavages adjacent to Pro residues, the a+2 population was included in the fit but the contributions of the a+2 population were not included in the αa value as production of these ions follows an alternate mechanism unrelated to a ion formation.25,38 Reported values are the average of duplicate data, and fragments having isotopes that overlapped with other fragments are not reported. Criteria for S/N and fragment ion size are discussed in the text. A detailed error analysis for the determination of alpha values is provided in the Supporting Information section with companion Figures S1 and S2. For the present study, all fragment ions having a Δαa=0.1RMSD of less than 0.02 were discarded from the analysis.
Ion mobility (IM) experiments were performed in the Wysocki lab at the Ohio State University using a Waters Synapt G2 mass spectrometer (Milford, MA) for CID-IM experiments and a Synapt G1 mass spectrometer for determination of collisional cross sections (CCS). Ions were generated using a nanospray source similar to that used for UVPD experiments. In the Synapt G1 mass spectrometer, standard wave heights and velocities in the source, trap, IM, and transfer ion guides were used, and typical values in the Synapt G2 mass spectrometer were 300 m/s and 0.1–1 V in the source ion guide, 450–600 m/s and 4–7 V in the trap ion guide, 300–600 m/s and 15–22 V in the ion mobility cell, and 200–300 m/s and 4–7 V in the transfer ion guide. Typical nitrogen gas pressure in the drift cell was 5–10 mbar. External calibration of CCS was performed using ubiquitin, sprayed from 50:50:0.1% H2O/methanol/formic acid, and cross-sections were taken from published values reported using helium.39,40 CID-IM experiments were performed using the quadrupole to isolate the penetratin-Arg precursor (5+) prior to CID activation in the trap travelling wave ion guide (TWIG) using 20–120 eV acceleration energy to activate the precursor and the resulting conformers were separated by ion mobility.
Modeling
Molecular dynamics simulations were carried out using NAMD and the Amber-Cornell forcefield. Trajectories were visualized using VMD. The starting structure of the penetratin-Arg peptide was protonated per experimental results, and starting structures were based on the solution NMR structure (1OMQ) and a fully helical conformer. Two varieties of annealing simulations were used to explore the conformational space of the peptide. Coarse annealing simulations, in which the peptide was held at an elevated temperature (1150 K) for 750 ps in between cooling cycles (300 K), were used to obtain seed conformations for fine annealing simulations in which the peptide was heated to 750 K, held at temperature for 30 ps, and cooled to 300 K for 70 ps. The trajectories were clustered to group similar conformations and potential energies were obtained for ensembles of structures having a backbone alignment with RMSD less than 1 Å. Collisional cross sections were calculated for five representative frames of the 40 lowest energy conformations using MOBCAL, and results from the trajectory method (TJM) are reported.41,42 Hydrogen bonds were determined using Pymol, by application of the distance command to find all C=O---H motifs of the peptide backbone having a donor-acceptor distance of less than 2.7 Å and C=O-H angle of 180° ± 80°.
Ab initio calculations for C-C backbone cleavage and H transfer in model peptides were conducted using density functional theory (DFT) as implemented in the Vienna Ab-Initio Package. Core electrons were described within the projected augmented wave framework; valence electrons were described with a plane wave basis set up to an energy cutoff of 370 eV.43 Convergence tests showed that the energetics of backbone cleavage and H transfer are insensitive to cutoff of the basis set. The generalized gradient approximation in the form of the Perdew, Burke and Ernzehof (PBE) functional was used to model electronic exchange and correlation.44 Spin polarization was used for the calculation of any radical species. Reaction pathways were calculated using the climbing image nudged elastic band method (CI-NEB)45 with double nudging.46
Results and Discussion
From a statistical study of nearly 100 peptides, we have recently shown that the relative abundances of a and a+1 ions produced upon UVPD correlated with both the identity of the C-terminal residue of the a/a+1 ion fragments and the presence or absence of hydrogen bonds involving the amide carbonyl oxygen of the cleaved residue.35 One consequence of this is that the an ion originating from a given cleavage at residue n is lower in abundance (relative to the an+1 ion) when there is active hydrogen bonding to the amide oxygen of residue n. Although our understanding of this phenomenon is incomplete, it is possible that the presence of a hydrogen bond allows delocalization of the radical in the an+1 ion, stabilizing the an+1 ion relative to the an ion. Alternatively, the presence of a hydrogen bond may modulate hydrogen atom migration and thus slow the conversion of a+1 ions to a ions. Other explanations, however, are possible and additional studies are needed to fully understand how hydrogen bonding leads to a reduction in the extent of hydrogen elimination from an+1 ions to generate an ions. Here, efforts are made to exploit this phenomenon and determine the extent to which this phenomenon can be used to decipher local hydrogen bonding interactions in native structures, a series of well-characterized peptides and proteins were investigated by 193 nm UVPD and their structures examined by ion mobility in conjunction with molecular dynamics simulations. In addition, DFT calculations were performed on three Ala8 peptides in an effort to elucidate this phenomenon at a mechanistic level. Following validation of the correlation between αa values ([a]/([a] + [a+1])) derived from UVPD data and the structural results from the other biophysical methods, the method was applied to characterize the hydrogen bonding motifs of both ordered and disordered proteins.
Melittin and Penetratin-Arg
Melittin from honey bee venom is an α-helical amphipathic peptide and has been studied extensively by ion mobility mass spectrometry, making it an ideal model system to examine stable gas-phase structures. Upon ionization, the 3+, 4+, and 5+ charge states are predominantly observed; previous reports suggest that the structure of the lowest of these charge states, 3+, is most helical and has a collisional cross section of 523 Å2.47,48 We have recently used 193 nm UVPD to localize the three charge sites of melittin (3+) and have used molecular dynamics simulations to identify a structure consistent with the charge sites determined by UVPD and with the collisional cross section obtained from ion mobility measurements.49 The resulting structure was the lowest energy one for the protonation scheme derived from UVPD analysis and was within 1.5% of the experimental CCS, thus validating the assignment. UVPD was used to generate fragment ions from the 3+ charge state of melittin. In essence, the distribution of a+1 ions and a ions upon UVPD is termed hydrogen elimination monitoring (HEM) and is mediated by hydrogen bonding motifs of the gas-phase ion.
The αa values, defined as: , where [an] and [an+1] represent the abundances of the a and a+1 ions at each corresponding cleavage site along the backbone, are plotted as a function of peptide sequence in blue and green bars in Figure 1a. Alpha values that approach unity are suggestive of weak or no hydrogen-bond stabilization35 and would be anticipated to reflect more flexible, less ordered regions of the peptide (or protein). Alpha values that approach zero are indicative of strong hydrogen-bond stabilization35 and rigidity and denote more highly ordered regions of the peptide (or protein). The mean αa values associated with the cleaved residue (derived from the compilation reported in ref. 35) are shown to the right of the experimental αa values as solid black or striped bars depending on whether the mean hydrogen bonding αa value (HB, black bars) or mean non-hydrogen bonding αa value (NHB, striped bars) is displayed. The corresponding experimental αa values obtained from the UVPD-HEM analysis are shown as green bars for putative NHB residues or blue bars for putative HB residues, respectively. This color-coded depiction strategy was chosen in order to clearly convey regions engaged in amide carbonyl oxygen hydrogen bonding. From the histogram in Figure 1 for melittin (3+), it is clear that the peptide features extensive hydrogen bonding for residues 6–9 and 11–18 (all displaying low αa values indicative of strong hydrogen bonding of the amide carbonyl oxygens of the cleaved residues). It should be noted that amide NH hydrogen bonding was previously found to contribute to a small reduction of αa values; however, the contribution of this type of hydrogen bond was small (reduction of ≈0.1 on average) and in fact fell well within the natural variation of each αa distribution. Thus, the contributions of amide NH hydrogen bonding are not emphasized here.
Figure 1.

a) The αa values from 193 nm UVPD of melittin (3+) are shown. The αa values in agreement with the mean non-hydrogen bonding (NHB) αa value and mean hydrogen-bonding (HB) αa value of the cleaved residue are shown as green bars and blue bars, respectively, and the corresponding NHB and HB means are shown to the right of the experimental values as solid black bars or striped bars (from ref 35). Selection of black vs. striped bars was made based on whether the experimental αa value was closer to the hydrogen bonded or non-hydrogen bonded mean. Trp is shown with the mean αa bar in grey to denote it having a unimodal distribution. Red crosses shown along the sequence in the x axis denote charge locations. In b) – d), three views of a putative structure of melittin (from ref. 48) are displayed with the backbone carbonyls shown in red and backbone N-H atoms shown in blue. Hydrogen bonds are denoted by purple dashed lines and residues (and for which UVPD was observed) having hydrogen bonds to the backbone amide oxygen are underlined in a).
In Figure 1b – 1d, three views of a putative structure of melittin are shown (from ref. 48) for which the amide carbonyls are colored red and the amide nitrogen and hydrogen atoms are colored blue. Hydrogen bonds are shown by purple dashed lines. Based on agreement with the respective non-hydrogen bonding means, the αa values of residues 5, 10, 19, 21, 23, 24, and 25 are predicted to not engage in hydrogen bonds. Remarkably, the putative structure of melittin (3+) features amide carbonyl oxygen hydrogen bonds at all residues except 5, 10, 21, 22, 23, 24, and 25. Hence, for the residues for which an experimental αa could be determined, the agreement of the αa values (HB and NHB means), is consistent with the proposed structure with two exceptions: Trp19, which is predicted by MD to have the amide carbonyl oxygen hydrogen bonded to the sidechain of Arg24; and Arg22, which is predicted by MD to not be engaged in any hydrogen bonds at the amide oxygen. It is possible that the orientation of the Arg24 sidechain is incorrect in the MD structure, as rotation of the sidechain could easily enable hydrogen bonding to the amide oxygen of Arg22, a factor which would account for both of the observed discrepancies between the UVPD and MD data and would not change the collisional cross section of the peptide substantially. Trp was also found to feature a unimodal αa distribution in our statistical UVPD-MS survey35 and seemed to be insensitive to hydrogen bonding. Studies of the 263 nm UVPD behavior of protonated Trp and Trp-containing dipeptides suggest that fragments arise from cleavage of ions in excited electronic states following hydrogen transfer and by internal conversion and subsequent dissociation via CID-like pathways.50,51 One possibility for the unusual behavior observed here is that internal conversion and vibrational energy re-distribution following 193 nm photoabsorption is particularly prevalent at Trp residues, resulting in unusually abundant b and a ions from more conventional CID-type pathways. It is noteworthy that several of the experimental αa values for melittin, such as those for Lys7, Val8, Leu9, and Leu13, were found to be much lower than the HB means associated with UV photoactivated cleavages at those residues. All of these residues were found in the main body of alpha-helices, and consequently it is possible that the relatively strong hydrogen bonding and inherent rigidity of protein secondary structures such as alpha-helices results in enhanced retention of hydrogen for the a+1 ions upon UVPD. This concept is explored in greater detail using DFT modeling and peptide variants below.
Penetratin-Arg (PA) was examined by UVPD in the same manner as melittin in order to further explore the correlation of experimental αa value of each amino acid with its engagement in hydrogen bonds. Penetratin is an amphipathic, cell penetrating peptide with antimicrobial activity derived in part from its helical structure in membrane environments. Penetratin-Arg is a sequence analog of penetratin in which four Lys residues (positions 4, 13, 15, and 16) have been substituted for Arg. The peptide was prepared in two different spray solvents, 20 mM ammonium acetate and 50:50 H2O/trifluroethanol, for analysis. The resulting charge state distributions are shown in Figure S3. Trifluroethanol (TFE) is a solvent additive that has been explored extensively in NMR studies and results in the adoption of helical secondary structures; consequently it has been used as a membrane-mimetic solvent.52–54 Addition of TFE to electrospray solvents has also been used to promote helical conformation of intrinsically disordered proteins.6 Spraying from ammonium acetate solution resulted in the observation of charge states 3+ to 6+, and spraying from TFE:H2O solution resulted in the observation of predominantly the 5+ and 6+ charge states of PA. In Figure 2a–d, the experimental αa values of each site cleaved upon UVPD are shown as blue/green bars for the 6+ to 3+ charge states of PA. Black and striped bars are shown to the right of the experimental data to show the NHB and HB means associated with cleavage C-terminal to each amino acid in the sequence, respectively. Selection of the HB or NHB mean αa value displayed was determined based on the value to which the experimental αa showed greatest agreement. The αa values for residues 7 to 12 increase as a function of decreasing charge state, suggesting this region becomes less structured with decreasing charge. In conjunction with the differences in charge state observed upon spraying from ammonium acetate versus TFE, this suggests that the higher 5+ and 6+ charge states may be more helical than the lower 3+ and 4+ charge states.
Figure 2.

The αa values as a function of sequence are plotted for the 6+ to 3+ (a – d) charge states of penetratin-Arg in blue and green bars depending on agreement with the HB and NHB means, respectively. The mean hydrogen bonding and non-hydrogen bonding αa values are shown as striped and solid black bars, respectively. Means for Trp residues are shown in grey to denote unimodal behavior for this amino acid. Red crosses shown along the sequence in each x axis denote charge locations for each of the four charge states examined, as determined from charge site analysis (Figure S4). e) Collisional-cross sections are shown as a function of charge state, and f) the putative structure 288 from MD simulations.
Molecular dynamics simulations, charge site analysis, and ion mobility experiments were undertaken in order to better evaluate whether the more prominent hydrogen bonding features (particularly involving residues 7 – 12) noted for the 5+ and 6+ charge states of PA were consistent with their gas-phase structures. We have previously shown that 193 nm UVPD can be used to localize charge sites in gas-phase proteins.49 In brief, UVPD of native proteins was shown to generate a and x fragment ions with charge states that were consistent with the protonation sites of the intact protein. Based on this strategy, the 5+ charge state of PA was evaluated. The fractional abundance of a and x fragment ion charge states were evaluated as function of peptide sequence, for which the fractional charge state abundance of an a ion is given by, , where p+, q+, and r+ are the observed charge states of a given a ion (an) and the abundance of a given charge state of that a ion is . An analogous equation is used for the x ion analysis. Plots of versus penetratin-Arg sequence are shown for all four charge states of PA in Figure S4. The charging pattern was observed to change somewhat from the 3+ to 6+ charge state of PA, and increasingly lower αa values were observed N-terminal to the site of each additional charge. For example, for PA (3+), charges were found to localize at Arg1, Arg10, and Arg16. The 4+ charge state of PA, however, contained a mixture of two charging schemes, one consistent with localization of two protons at Arg4 and Arg10 and another with the central protons localized at approximately Arg10 and Arg13. Interestingly, by HEM analysis, residues 10–12 feature reduced αa values for the 4+ charge state. Because protonation sites can be modulated by secondary and tertiary structure and protonation at the C-terminus of helices has a stabilizing effect on the helical structure,55 the reduction of αa values N-terminal to the Arg13 protonation site may indicate the formation of a short α-helix in this region.
The 5+ charge state of PA was found to have protonation sites at Arg1, Arg4, Arg10, Arg13, and Arg16, and molecular dynamics simulations were consequently performed on the 5+ charge state with these specific sites protonated. Simulated annealing was performed using a fully helical structure and the solution NMR structure (pdb 1OMQ) as two starting structures. The relative energies of the 40 lowest energy structures are plotted against predicted collisional cross section in Figure S5. Ion mobility measurements were collected for the 3+ to 6+ charge states of PA, sprayed from 20 mM ammonium acetate, and the corresponding collisional cross sections for these charge states are shown in Figure 2e. Consistent with UVPD-HEM data, the 3+ and 4+ charge states feature compact conformations having collisional cross sections of approximately 380 Å2, and the 5+ and 6+ charge states feature more elongated conformations having collisional cross sectional maxima of 496 Å2 and 464 Å2, respectively. It is noteworthy that the 5+ charge state featured three or four distinctive mobility-separable conformations, the most elongated and most abundant of which (496 Å2) was larger than the CCS of the 6+ charge state. This outcome is consistent with the UVPD-HEM data (Figure 2b,c) that predicted the 5+ charge state to feature a wider swath of hydrogen bonding (i.e. low αa values for the FQNRRM section) characteristic of a longer, more stabilized helix. CID-IM experiments were performed on this charge state (5+), as shown in Figure S6, and increasing collisional activation resulted in the formation of fragments that corresponded to depletion of the most elongated conformer, suggesting it is less stable than the compact conformations. Based on this result, we surmised that the structure undergoing preferential fragmentation by UVPD likely corresponded to this more abundant, more labile conformer with a CCS of 496 Å2. Using a ±2% CCS window as a criterion for putative structures, a single structure, structure 288, from the MD simulations was found to best match this CCS (with a predicted CCS of 495 Å2) (see Figure S5). Although not the global minimum, structure 288 is the lowest energy structure having a CCS of greater than 465 Å2, and it is possible that it arises from a kinetically trapped solution structure.
HEM analysis of the 5+ charge state of PA was compared to the hydrogen bonding features of structure 288 (Figure 2f) to evaluate the agreement of HEM with the putative structure. Note that the a6 and a14 ions were not included in this analysis because Trp was not observed to have a bimodal αa distribution previously so could not be used to differentiate hydrogen-bonding and non-hydrogen bonding.35 This Trp effect is also discussed in more detail below. Structure 288 features a helical motif for virtually the entire sequence (Figure 2f); from inspection of the labeled hydrogen bonds, it is clear that hydrogen bonding predicted by UVPD-HEM analysis of residues 2–4 and 7–12 corresponds to the first three turns of the α-helix. Residues 13–16 (omitting Trp14) comprise the last turn of the α-helix and feature amide carbonyl oxygen atoms that lack hydrogen bonding, consistent with the HEM data that predicted no hydrogen bonding for these residues. Cleavage C-terminal to Ile5 generates an unusually abundant a5 ion with three-fold higher abundance than any other identified ion in the UVPD spectrum of penetratin-Arg (5+). The UVPD spectrum of PA (5+) is shown in Figure S7 to demonstrate the unusually large abundance of the a5 fragment. It is possible that unusual fragmentation or an overlapping fragment is distorting the a5/a5+1 distribution, resulting in the unusually high αa value for the Ile5 site and thus making the αa value dubious. Aside from this anomaly, gas-phase structural analysis by UVPD-MS shows the distribution of a versus a+1 ions to be in excellent agreement with the hydrogen bonding motifs of penetratin-Arg.
From inspection of the residues flanking Trp in Figure 2, it appears that higher αa values are typically observed for backbone cleavages adjacent to tryptophan. The side-chain of Trp may act as a UV chromophore, and we have observed enhanced fragmentation C-terminal to this residue. Photoabsorption by the indole group of tryptophan (e.g., π to π* transition) may allow access to different excited electronic states which may favor alternative fragmentation pathways or couple to other dissociative excited states that lead to electron and/or hydrogen migration.56 The latter process is modulated by spatial constraints related to the indole ring and the peptide secondary structure,56 and may explain some of the variations of the αa values that appear to arise from the presence of tryptophan in the present study. It is also possible that the different electronic states accessed by tryptophan alter the subsequent rate of fragmentation from the excited state relative to internal conversion to a lower electronic state, thus shifting the ratio of a to a+1 ions. To better evaluate the influence of Trp on αa values at residues adjacent to Trp, three penetratin-Arg variants were examined in which one or both Trp residues were replaced by Ala. The 3+ to 6+ charge states of each penetratin-Arg variant were studied by UVPD-MS, and the αa values from HEM analysis (Figure S8–S11) were compared to those of the wildtype (WT) penetratin-Arg peptide in Figure S12 as a function of Δαa, defined as: αa[variant] - αa[WT].
The 3+ and 4+ charge states of the penetratin-Arg variants, which are expected to adopt more globular conformations, exhibited similar behaviors to one another. The 5+ and 6+ charge states, which are expected to be more helical, also displayed similar behaviors to one another for all three variants. For the helical charge states, the W6A and W14A substitutions resulted in notably lower αa values at the substituted residue as well as the preceding residue, suggesting tryptophan may indeed result in distortion of the αa value of the preceding residue. Interestingly, the αa values of residues 7–12 were also found to be slightly (0.05–0.1) lower than in the WT peptide. As Ala residues are known to have a high propensity to form helices, it is possible that this substitution results in some degree of stabilization of the helix. In contrast to the 5+ and 6+ charge states, the 4+ charge state of the penetratin-Arg variants featured inconsistent changes in the αa values across the peptide, with reduction occurring for some residues and elevation for others. The 3+ charge state, on the other hand, exhibited relatively large reductions in αa across the length of the penetratin-Arg variants relative to the WT peptide. The inconsistent effects of Trp→Ala substitution across the four charge states of penetratin-Arg suggests that photon absorption by the tryptophan side-chain alone does not account for the unimodal behavior of the residue and that the observed differences in αa values are the result of conformational changes. Moreover, the inconsistent impact of Trp→Ala substitution on the αa values of adjacent residues suggests that Trp does not exert a universal influence. However, owing to the similar behavior observed for the charge states expected to be more helical (5+, 6+), it remains possible that Trp can influence the observed αa of the preceding residue, and a more extensive study across a larger set of peptides is needed to fully evaluate this possibility.
Computational Modeling
In order to better understand the underlying processes that govern the observed reduction in αa values in ordered, helical regions of peptides, DFT computations were undertaken. The minimum (ground state) energy pathways for C-C backbone cleavage and hydrogen transfer were calculated in order to provide information about the likely mechanism of dissociation and the expected products. While this approach does not capture the excited state dynamics that is responsible for backbone cleavage, as other models do,56 we show that the calculated ground state reaction mechanisms provide a simple and intuitive explanation for the observed fragmentation products. A simple Ala8 sequence was selected as a model peptide for these calculations to minimize computational cost. Beta hydrogen and amide hydrogen transfer have been shown previously35,57 to comprise the bulk of the hydrogen transfer associated with formation of a- and x-type ions upon 157 nm and 193 nm UVPD, and these pathways, depicted in Ref. 35, were explored in detail here. Owing to the proximity of the alpha hydrogen to the cleavage site, a pathway in which this atom was transferred to the x-type product ion was also explored but was found to be unfavorable for all peptide structures. Three structures of an Ala8 peptide were considered to assess possible correlations between secondary structure and the dissociation products. The structures, shown in Figure 3, include an unstructured linear peptide, a hairpin turn, and a helix. The unstructured peptide lacks structural hydrogen bonds, whereas the hairpin and helix contain an array of intramolecular hydrogen bonds that contribute to the secondary level of organization. The fifth residue of each Ala8 peptide structure was selected for the site of cleavage, producing complementary a5/x3 fragment ions, and corresponding to cleavage in the center of the helix or in the middle of the hairpin turn. Notably, the amide NH and C=O of the fragmenting residue were strongly hydrogen bonded in the helical structure, whereas only the amide NH of the fragmenting residue was weakly hydrogen bonded (bond angle of ≈90°) in the hairpin turn.
Figure 3.

Three Ala8 peptide structures used as starting structures for DFT modeling: (a) unstructured linear, (b) hairpin turn, and (c) alpha helix.
Prior to investigating the mechanisms of dissociation, the relative energies of the reactant and possible product structures of the three peptide conformers were calculated and are shown in Table 1. All energies are reported relative to the linear unstructured peptide. The hairpin structure is stabilized by hydrogen bonding, but this energy is largely negated by the strain of the hairpin turn. The helix features the most extensive hydrogen bonding and in fact is predicted to be the most stable structure. The energies of the a+1/x+1 products were found to be significantly higher than those of the peptide precursors and the alternative a/x+2 products, consistent with the a+1/x+1 structures being unstable radicals. The beta hydrogen transfer pathway in which an alkene-type a ion is produced was found to be most thermodynamically favorable for each Ala8 conformer. It is clear from these results that the energy differences between the radical a+1/x+1 and a/x+2 product structures are significantly larger than the energy differences between the different peptide geometries. Given that a significant fraction of radical a+1 and x+1 ions are observed experimentally, we can conclude that the ratio of a+1 ions to a ions are not determined primarily by these thermodynamic energies.
Table 1.
Relative energies of the Ala8 peptide structures and products (kJ/mol) upon UVPD.
| Geometry | Reactant | a+1/x+1 radicals | a/x+2 (amide hydrogen transfer: imine a product) | a/x+2 (beta hydrogen transfer: alkene a product) |
|---|---|---|---|---|
| Linear (unstructured) | 0 (reference) | 412 | 152 | 90 |
| Hairpin | −2 | 488 | 192 | 57 |
| Helix | −17 | 394 | 157 | 64 |
An alternate explanation for the generation of the observed fragment ion product ratios (i.e. a versus a+1) is that hydrogen atom transfer is kinetically controlled. To investigate this possibility, the minimum energy pathway (MEP) for the dissociation of each peptide structure and the subsequent formation of the a/x+2 products was calculated. Three general pathways were considered for each conformer, resulting in cleavage of the C-Cα bond of the peptide backbone in conjunction with hydrogen transfer from amide, alpha or beta positions. Figure 4 shows the lowest energy pathway for each structure (with fragment ion structures shown in Figure S13); the other pathways are included in Figure S14. Multiple barriers were found for each MEP, and the kinetics of the reaction were considered in terms of the highest energy barrier prior to separation of the nascent product ions; these values are reported in Table S1. For example, two barriers were observed for the amide hydrogen pathway of the unstructured peptide (Figure 4a), one for simultaneous cleavage of the peptide backbone and transfer of the amide hydrogen, and a second, higher barrier corresponding to hydrogen transfer within the x+2 product ion. With the exception of the amide hydrogen transfer pathway of the helix conformer, for which a barrier to C-C cleavage of approximately 400 kJ/mol was found, cleavage of Cα-C bond was estimated to have a barrier of roughly 300 kJ/mol for every pathway for all three conformers.
Figure 4.
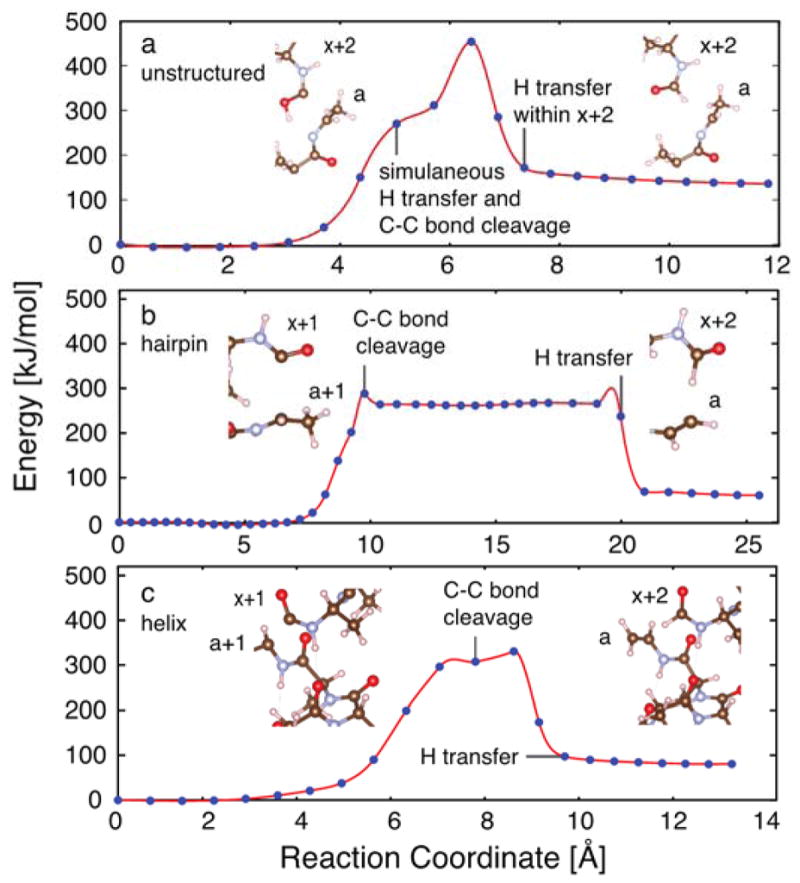
Minimum energy pathway of C-C bond cleavage for (a) an unstructured linear peptide via amide hydrogen transfer, (b) a hairpin turn via beta hydrogen transfer, and (c) an alpha helix via beta hydrogen transfer. The reaction coordinate is the collective distance of atomic motion along the minimum energy pathway. Inset pictures of truncated fragment ion structures were prepared using VESTA.80 The full fragment ion structures are shown in Figure S13.
For the hairpin turn and helix conformers, the beta hydrogen atom transfer pathway was lowest in energy, making the resulting alkene product both kinetically and thermodynamically favored (Figure 4b,c). In contrast, amide hydrogen transfer was found to be the lowest energy pathway for the unstructured conformer. Two features of these pathways were striking. First, temporal constraints limit hydrogen transfer in the beta hydrogen atom transfer pathways such that transfer of the hydrogen to generate the alkene-type a product occurs as a separate event following cleavage of the Cα-C backbone bond. This was also true to some extent for the alpha hydrogen transfer pathways. In these cases, spatial separation of the incipient radical a+1/x+1 product ions upon cleavage of the Cα-C backbone bond may impede transfer of a hydrogen atom to form a/x+2 products, despite these products being energetically favorable. In contrast, transfer of the amide hydrogen atom was found to occur simultaneously with cleavage of the Cα-C backbone bond for all three conformers, therefore facilitating generation of the a/x+2 products. Thus, the yield of a/x+2 product ions is expected to be enhanced for peptides in which cleavage by the amide hydrogen transfer pathway is preferred. Second, it is noteworthy that the differences in the barriers found for each pathway of each conformer, varying from 300 to 400 kJ/mol, are smaller than the amount of energy deposited by a single or multiple 193 nm photons (>600 kJ/mol). Consequently, it is feasible that all pathways are viable; however, the branching ratios are expected to vary with the relative heights of the Cα-C bond cleavage barriers. For the unstructured peptide, the amide hydrogen transfer pathway was energetically more favorable compared to the alpha and beta hydrogen transfer pathways. In contrast, the beta hydrogen transfer pathway was more favorable for both the hairpin and alpha helix conformers relative to the alpha hydrogen transfer pathway and the amide hydrogen pathway. In sum, the preferential formation of product ions by the amide pathway is expected to follow the trend: unstructured ≫ hairpin > helix. This is consistent with the experimental findings of Zhang and co-workers for small unstructured peptides,57 in which mixtures of beta hydrogen and amide hydrogen transfer pathways were observed.
Qualitative inspection of the amide hydrogen transfer reaction pathways suggests that a key difference between the unstructured peptide and the more ordered hairpin and helix structures is flexibility along the backbone and whether or not the more ordered arrangement of the atoms in these latter two cases is optimal for amide transfer. The unstructured peptide is able to rotate the donor amide hydrogen atom more readily into a favorable geometry for transfer as the Cα-C bond breaks without incurring the penalty associated with breaking an intramolecular hydrogen bond, thus avoiding the formation of radical intermediates. In the more structured hairpin and helix structures, this rotation incurs an energetic penalty associated with breaking the intramolecular hydrogen bond in which the amide NH is engaged. For the unstructured peptide, the geometry for transfer of a hydrogen atom to the amide oxygen and subsequent rearrangement was found to be more favorable. In contrast, in the hairpin turn and helix structures, amide hydrogen atom transfer was observed to proceed optimally via approach from opposite the carbonyl group of the amide bond. However, in the hairpin turn, the extent to which the peptide backbone must reorient was significantly less than that of the helix because the inherent geometry of a hairpin makes it more optimal for amide hydrogen transfer. Thus, owing to geometry, the amide hydrogen transfer pathway is not as energetically disfavored in tight hairpin turns as it is in helices because the peptide exists in a conformation suitable to transfer the hydrogen atom from opposite the carbonyl. This difference provides one qualitative explanation for the greater abundance of radical fragments (a+1, x+1) that are formed from highly structured, helical regions of proteins, for which amide hydrogen atom transfer is not possible without significant structural reorganization.
Structured Proteins
Based on the UVPD-HEM behavior of peptides possessing structures and secondary structural motifs supported by DFT modelling, a correlation between the extent of structural rigidity/hydrogen bonding and the αa values derived from HEM analysis of the UVPD fragmentation patterns was observed. These results suggest that disordered/unstructured regions of proteins feature αa values consistent with unstructured peptides and that αa values of more ordered regions such as helices, turns, and beta strands exhibit significant reduction in the αa values. To investigate the extent to which the HEM strategy can be applied to intact proteins in the gas phase, two ordered and two disordered proteins were studied by UVPD-MS.
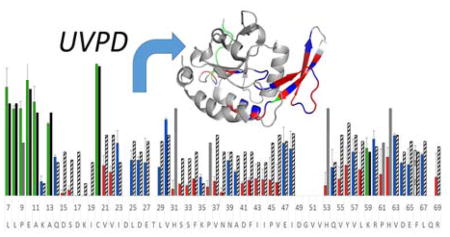
A structurally rigid protein was studied by UVPD-HEM to examine how well hydrogen bonding interactions can be studied in intact proteins using the approach described for melittin and penetratin-Arg. The small C-terminal domain phosphatase 1 (Scp1) regulates the dephosphorylation of C-terminal domain (CTD) of eukaryotic RNA polymerase II and is comprised of a central β-sheet that is flanked by two multi-turn α-helices, a small β-sheet that serves as the recognition and catalytic region, and a 3–10 helix.58 Several small 3–10 helices and α-helices decorate the periphery of the protein, and a number of multi-residue loops connect these different structural features.58 In addition, the ten residues at the N-terminus constitute an unstructured region; hence, Scp1 contains a diverse array of secondary structural elements and is therefore ideal for assessing the degree to which structural information is reflected by HEM analysis. These residues encompass the functional sequence of Scp1, including the substrate recognition and catalytic site (residue 102–133). The Scp variant used here is human Scp isoform 1 and includes residue 78–261, based on the residue numbering used in the NCBI database. The 7+ to 10+ charge states of monomeric Scp1 were observed following electrospray ionization of Scp1 from 50 mM ammonium acetate, and the most abundant charge state (9+) was selected for further examination by UVPD and HEM analysis. UV photoactivation resulted in an abundant a ion series covering the first 75 residues (residue 78–153 in human Scp1 which includes the active site, phosphoryl-transfer reaction site, and substrate recognition groove), and HEM analysis was performed on the first 63 residues as summarized in Figure 5 (residue Tyr78 to Arg141). Although a ion fragments were observed beyond Arg141, these ions were of low abundance and the isotope distributions could not be reliably deconvolved. Description of the metrics for inclusion of a ions in the HEM analysis are provided as Supporting Information.
Figure 5.

HEM analysis of the first 63 residues of Scp1 (9+) following 193 nm UVPD (one pulse, 2 mJ). Experimental αa values are shown in a) in green/blue/red for Scp1, and the HB and NHB means associated with cleavage at a given residue are shown as striped and solid black bars, respectively. Experimental bars are colored green for instances of αa consistent with the NHB mean, blue for instances of αa consistent with the HB mean, and red for instances of αa consistent with the hyper-HB mean (extremely strong hydrogen-bonding). Residues engaged in amide oxygen hydrogen bonds in the crystal structure are underlined. In b) the crystal structure of Scp1 (pdb 3PGL) is colored according to the data in a) such that regions predicted by HEM to lack hydrogen bonding are green, regions with “typical” hydrogen bonding are blue, and regions with unusually strong hydrogen bonding are red. (Note that the first five residues of the isoform described herein are engineered from a protease cutting site and are not part of the native Scp1 sequence and are designated residues i–v in a.)
Inspection of Figure 5 suggest αa values occupy a wide range and were observed to fall into three categories: approximately the NHB mean (high αa values), approximately the HB mean (low αa values), and significantly below the HB mean (very low αa values signifying extremely enhanced hydrogen bonding). To ease interpretation, the term hyper-HB mean is defined as 50% of the HB mean for all residues excluding Gly and Lys, which feature HB means near zero; hyper-HB denotes extremely enhanced hydrogen bonding. The experimental αa values are consequently shown as green, blue, and red bars in Figure 5 to indicate αa values consistent with the NHB mean, the HB mean, and hyper-HB mean, respectively. Hydrogen bonding and non-hydrogen bonding means for each cleaved residue are shown in striped and solid bars to the right of the experimental data, and unimodal means are shown in grey. Residues for which the backbone amide oxygen is hydrogen bonded in the crystal structure are underlined in Figure 5a. The experimentally determined αa values of the first 16 residues of the protein were found to be largely consistent with no hydrogen bonding, which is in good agreement with the crystal structure (pdb 3PGL) in which the N-terminus of the protein is devoid of ordered secondary structure. The crystal structure is highlighted in green in Figure 5b (front and back views of the protein) to denote regions predicted by HEM to lack hydrogen bonding. The bulk of the protein from Asp98 to Arg141 is predicted by UVPD-HEM to be either moderately or strongly hydrogen bonded along the protein backbone (blue and red bars). Regions in which the observed αa values were consistent with the hyper-HB mean are highlighted in red on the crystal structure, and regions with the observed αa values consistent with HB means are shown in blue.
From inspection of the two views of the crystal structure in Figure 5b, it is clear that most of the strongest hydrogen bonding regions predicted by HEM analysis to be the most rigid occur along the β-strands of Scp1. Interestingly, alternating (oxygen) hydrogen bonds are found along a β-strand spanning residues 113–118 while low αa values were found for the entire span. As indicated in a previous study that examined αa values for peptides,35 hydrogen bonding to both the amide oxygen and amide nitrogen of the peptide backbone result in a reduced αa value, but amide oxygen hydrogen bonding caused a more pronounced effect. In light of the consistent reduction of αa values in the 113–118 stretch of Scp1, however, it is possible that stable NH hydrogen bonds may contribute more substantially to the reduction in αa values. This concept, however, is not explored in further detail here and remains speculative. In contrast to the β-strands, along which αa values are found to be low, HEM suggests the loop regions to be less strongly hydrogen bonded. Although substantial hydrogen bonding along the loop regions was a bit unexpected, this trend can be rationalized by considering that flexible regions occupy a medley of conformation and exhibit greater heterogeneity among hydrogen bonding motifs. Some fraction of the protein population may be transiently hydrogen bonding in these regions while another population is not. Given that a loop region may occupy several different conformational states, it is not unreasonable that every residue would be hydrogen bonded to some extent in one or more of these different forms. This can be particularly true for hairpin turns and other loop/turn motifs that feature strong hydrogen bonding. It is interesting to note that three residues (Val102, Glu119, and Gln126) found in the exterior β-sheet (which spans residues Val102 to Leu130) were predicted by HEM to be hydrogen bonded, but not as strongly as the adjacent residues, such as Phe114 and Asp113-Pro117. In all cases these residues (Val102, Glu119, and Gln126) were weakly hydrogen bonded in the crystal structure to remote secondary structures and were not a part of the hydrogen bonding motif of the β-sheet itself. Val102 in particular is part of the hinge region of the beta-sheet and plays a role in substrate recognition, necessitating a degree of flexibility that appears to be reflected well in the HEM analysis. Two loop regions, including Pro108-Asn110 and Asp87-Ile91, were predicted by UVPD-HEM to be strongly hydrogen bonded. Deeper inspection, however, shows a hydrogen bond between the sidechain of Lys107 and Pro108, and a turn along residues Val109 to Asn111 causes possible backbone hydrogen bonding that could be enhanced in the gas phase. The region between residue Gln86 and Ile91 forms a coil in the crystal structure with backbone hydrogen bonding intermittingly present. Hence, UVPD-HEM of the N-terminal 63 residues of Scp1 shows relatively good agreement with the secondary structure of the crystallized protein. Dissecting the UVPD-HEM results into three general categories, no hydrogen bonding, average or transient hydrogen bonding, and strong hydrogen bonding, provides insight into the three-dimensional arrangement of the protein and suggests HEM is a compelling strategy for making predictions as to which protein regions are engaged in stable secondary structures such as β-sheets.
To further explore the utility of HEM for characterization of hydrogen bonding motifs and secondary structures in ordered proteins, a primarily helical protein, calmodulin, was studied by UVPD. HEM analysis undertaken for the 7+ charge state, for which sufficient abundances of a ions were detected to cover the first 57 residues. The resulting HEM plot is shown in Figure 6a, and the crystal structure of apo calmodulin (pbd 1CFC) is shown in Figure 6b. The color coding for the HEM plot and crystal structure are the same as was used for Scp1. Hydrogen bonding is predicted for residues 13–16, which are engaged in an alpha helix in the crystal structure. Residues 17, 19, and 20 are predicted to be free of hydrogen bonding based on the αa values from HEM analysis. These particular residues are located at the C-terminal turn of the first alpha helix in the crystal structure and consequently the associated amide carbonyl oxygen atoms are oriented in a manner that prevents hydrogen bonding. HEM analysis predicts residues 21–38 to be hydrogen bonded; as this region corresponds to a tightly hydrogen bonded hairpin turn, β-strand, and α-helix, this result is also consistent with the crystal structure. Residues 39, 41, and 42 are predicted by HEM analysis to lack hydrogen bonding or feature weak hydrogen bonding (all three are green bars), and in fact correspond to an unstructured region of the calmodulin crystal structure. Based on the αa values, residues 48–52 are predicted to be hydrogen bonded (all are blue bars) and correspond to helix 3 of the crystal structure. In contrast, the αa values for residues 53–57 reflect little or no engagement in hydrogen bonding and correspond to an unstructured loop in the crystal structure. Across the board, the HEM results are in good agreement with the reported crystal structure of apo calmodulin, featuring hydrogen bonding along the secondary structures where hydrogen bonding would be expected for the regions of the protein sequence for which sufficiently abundant a ions were generated to apply HEM analysis. Interestingly, however, the N-terminus of helix 3 (residues 45–47) is predicted to be unstructured or relatively flexible by HEM analysis of the αa values, and this is the only portion of the HEM data that contradicts the crystal structure, which predicts this region to be helical. In order to assess this discrepancy, the B-factors of apo calmodulin were examined and are plotted in Figure S15 (higher B-factors indicate regions of greater flexibility). In general, apo calmodulin is relatively rigid with low B-factors for much of the sequence, with the exception of the two termini and a loop region from residue 76–80. Interestingly, however, the region from residue 40 to 50 is one of the most flexible regions of the protein, and consequently is not unreasonable for this region to either fully unravel in the gas phase or exist in multiple conformations, only some of which are helical. HEM analysis of the N-terminus of apo calmodulin is thus in good agreement with the crystal structure of the protein as flexible regions are found to be associated with no hydrogen bonding in many areas and areas predicted by HEM to be moderately hydrogen bonded generally correspond to tight loops and helices.
Figure 6.

HEM analysis of the first 57 residues of apo calmodulin (7+) following 193 nm UVPD (one pulse, 2 mJ). Experimental αa values are shown in a) in green/blue/red for calmodulin and the HB and NHB means associated with cleavage at a given residue are shown as striped and solid black bars, respectively. Experimental bars are colored green for those that agree with the NHB mean, blue for those that agree with the HB mean, and red for those that agree with the hyper-HB mean (extremely strong hydrogen-bonding). Residues in which the amide oxygen is hydrogen bonded in the crystal structure are underlined. In b) the crystal structure of apo calmodulin (pdb 1CFC) is colored according to the data in a) such that regions predicted by HEM to lack hydrogen bonding are green, regions with “typical” hydrogen bonding are blue, and regions with unusually strong hydrogen bonding are red.
Disordered Systems
Intrinsically disordered proteins (IDPs) have recently emerged as an integral component to a number of biological disorders, such as Parkinson’s disease and Alzheimer’s disease,59 and the precepts emerging from the canonical structure-function relationship of proteins have been modified to account for these unique systems that have been generally regarded as lacking discrete structure. Recent studies indicate that many of these proteins are likely composed of a composite of interconverting structures and that the diversity of structures is critical to cell function and regulation.60–63 IDPs feature a diverse array of binding partners and frequent transitions between ordered and disordered states. Perhaps as a consequence of the rapidly changing structures of IDPs, self-aggregation is also common. As a result of these characteristics, characterization of IDPs by conventional methods is problematic and little is typically known about the three-dimensional structures of these proteins. As these proteins occupy the opposite end of the structural spectrum compared to Scp1 and calmodulin, two unstructured/disordered proteins were studied by UVPD-HEM analysis in order to evaluate the applicability of the technique to less ordered structures.
α-synuclein (α-syn), an amyloid-forming protein responsible for plaque formation in Parkinson’s Disease, is intrinsically disordered in aqueous solvents and has been studied by mass spectrometry in both the negative and positive polarities.5,6,64 Multimodal charge state distributions of protonated α-syn have been observed previously by nativespray mass spectrometry, and the different charge state populations were found to correspond to solution states.6 α-Synuclein contains seven imperfect sequence repeats (KTKE(Q)GV).65,66 The mass spectrum of α-synuclein in ammonium acetate (pH 7) is shown in Figure S16. Two charge states, 7+ and 10+, were selected for UVPD-HEM analysis. In Figure 7a and 7b, αa values from HEM analysis are shown for the first 60 residues of α-synuclein (7+ and 10+ charge states). HEM of the 7+ charge state suggests a mixture of significant and moderate hydrogen bonding from residues 6 to 14 and 21 to 51, which encompass several of the sequence repeats. The N-terminus, residues 15 to 20, and residues 54 to 58 were found to feature αa values consistent with the non-hydrogen bonding means, and suggests that these areas are more flexible or disordered. Interestingly, a different HEM pattern is observed for the 10+ charge state of α-synuclein, with αa values that are consistent with hydrogen bonding for residues 6 to 28 and 32 to 41. The remainder of the sequence, particularly residues 43–60, features high αa values consistent with little hydrogen bonding. From ion mobility and variable pH measurements, the Kaltashov group proposed that the compact conformation spans charge states 5+ to 9+ and exhibited extensive helical character.6,64 It is possible that the hydrogen bonded region predicted by HEM that spans residues 21 to 53 is largely helical and exhibits kinks or turns at residues 28, 41, and 45, which were predicted to lack hydrogen bonding. Based on solvent modulation studies, a population featuring intermediate elongation, termed I2, was found to be centered at the 10+ charge state and was proposed to contain significant β-sheet character.6 The difference in HEM patterns for the 7+ and 10+ populations is in agreement with this charge state featuring significantly different structural motifs. In Scp1, extremely low αa values were found for the portions of the backbone that formed a β-sheet in the crystal structure; similarly low αa values are observed from residue 5 to 16 and 34 to 41 of the 10+ charge state of α-synuclein and may suggest these regions comprise β-strands. Thus, the HEM results agree with solution and ion mobility studies of α-synuclein, and these results demonstrate the utility of HEM as a novel gas-phase characterization approach, even for the relatively small regions of protein sequence that can be currently accessed.
Figure 7.

HEM analysis of the first 60 residues of a) α-synuclein (7+), b) α-synuclein (10+) and c) guinea pig ACTH (4+) using UVPD (one pulse, 1.8 mJ). Green/blue/red bars indicate the experimental data. The HB and NHB means associated with cleavage at a given residue are shown as striped and solid black bars, respectively. Experimental bars are colored green for those αa values in agreement with the NHB mean, blue for those αa values in agreement with the HB mean, and red for those αa values in agreement with the hyper-HB mean (extremely strong hydrogen-bonding). The predicted (mean) αa value for His, Pro, and Trp residues are shown in grey because these amino acids feature unimodal distributions.
Adrenocorticotropic hormone (ACTH) is a 39 amino acid pituitary hormone responsible for stimulating the secretion of glucocorticoid hormones by binding a G protein-coupled receptor at the surface of adrenal cortex cells.67 Although the overall structure of this peptide hormone has been simply described as linear, the N-terminal 24 residues have been linked to receptor recognition and activation, and initial binding is thought to induce a conformational change connected to the function of the hormone.67 The solution structure of the free hormone is thought to be largely unstructured and thus provides an ideal example for UVPD-HEM analysis. Guinea pig ACTH was sprayed from 100% H2O, and the 4+ charge state was fragmented by 193 nm UVPD using a single 1.8 mJ pulse. The resulting HEM analysis is shown in Figure 7c. HEM of ACTH is predominantly comprised of αa values consistent with no hydrogen bonding of the backbone amide carbonyl oxygen atoms, in good agreement with solution-based studies that have suggested the absence of a single specific conformation. Sporadic hydrogen bonding is observed throughout the peptide, at Arg8, Lys11, Lys16, Arg17, Glu30, Phe35, and Leu37, suggestive of random coils/turns or hydrogen bonds between the peptide backbone and adjacent sidechains. Because NMR and x-ray crystallography are often unsuitable for structural analysis of disordered species, the UVPD-HEM approach offers a potential strategy for developing models with atomic level detail for IDP structure(s). However, this level of computational modeling is beyond the scope of the present study. Nonetheless, HEM analysis of α-synuclein and ACTH exhibits excellent agreement with available solution structural data, suggesting that ACTH is completely disordered and that different charge states of α-synuclein represent different conformational families.
Ubiquitin (12+): Discriminating structural differences of gas phase conformers
Although crystal structures can be obtained for the majority of globular proteins, limited sample quantity and environment challenges for some proteins, such as membrane proteins, can impede the applicability of x-ray crystallography. In order to assess whether useful information could be obtained using UVPD-HEM for a protein lacking a known structure, a protein having a wide variety of gas-phase conformations was studied. Virtually all charge states of gas-phase ubiquitin have been studied extensively by mass spectrometry, and numerous studies have shown that different gas-phase structures can be obtained by modulating solution conditions and ion energies.68–70 Ubiquitin has been shown by NMR to adopt two solution conformations: a globular N state comprised of a five strand beta sheet, α-helix, and 3–10 helix, and an extended A state in which the C-terminal half of the protein is α-helical.71 Gas-phase studies indicate that addition of methanol and acidification of the spray solution results in preferential formation of conformational families associated with the helical A state, which predominantly occupies the 7+ and 8+ charge states; a variation of the A state termed the B state; and an unfolded U state, which occupies the 8+ to 10+ and 12+ charge states at the highest methanol concentrations.72 The 12+ charge state has additionally been studied by two-dimensional ion mobility spectrometry, trapping ion mobility spectrometry (TIMS), field asymmetric ion mobility spectrometry (FAIMS), and hydrogen-deuterium exchange (HDX).73–76 Ion mobility measurements suggest the presence of up to four conformers, two of which are dominant and feature similar cross-sections and two of which are much lower in abundance.73,76 Two forms of ubiquitin (12+) were also detected by HDX, and it was proposed that the two conformations differed as a function of the locations of protonation sites.75 193 nm UVPD was thus performed on the 12+ charge state of ubiquitin (sprayed from 50:50:0.1% methanol/H2O/formic acid), and the charge sites were localized by considering the charge states of the a and x ion fragments. In Figure 8, the fractional abundances of a and x fragment ion charge states are shown as function of sequence and used to infer the charge sites in a manner described previously.49 Two charging schemes are evident from the splitting in observed charge states between Lys11 and His68 and are denoted A and B, with A referring to the more abundant population. For example, the bar at Gln49 in Figure 8 reflects the a49 ion population for which ~60% is detected in the 7+ charge state (thus representative of A) and 40% is detected in the 8+ charge state (representing B). The relative abundance of these two charge site isoforms was assessed by averaging the relative abundance of the a ion charge states for which two charge states were present. The sequence of ubiquitin is shown below the segmented histograms with the localized charge sites denoted in bold red font. Conformer B features two charge sites between residues 11 and 27 (at Ser20 and Lys27), whereas conformer A features a single charge site at Lys11. The difference in charging schemes in this region could indicate that structural differences between the two populations are particularly prominent in this region. A beneficial consequence of these charging schemes is that the two populations can be crudely separated by individually considering the two charge states of a ions for which the charge states are split. For example, the a35 ion arising from cleavage at Gly35 is observed in two charge states: 5+ and 6+. The a356+ can be used to represent isomer B, and the a355+ ion can be used to represent isomer A.
Figure 8.

Relative abundances of the charge states of the a) a and b) x fragments of ubiquitin (12+). In c) the locations of the charges of the two isomers are shown in red font on the sequence of ubiquitin. Charge site isomer A refers to the more abundant population (≈60%) and charge site isomer B refers to the less abundant population (≈40%).
Hydrogen elimination monitoring was used to examine the differential behavior of conformer A and B of the 12+ charge state of ubiquitin using the split charge states of the a ion series to separate the A and B populations. Histograms showing the experimental αa values and hydrogen bonding/non-hydrogen bonding means are shown in Figure 9. For a ions for which split charge states were not present, the αa value is delegated to conformer A, which was the more abundant of the two conformations. In general the experimental αa values were found to be extraordinarily low (“hyper HB”), suggestive of extremely strong hydrogen bonding. Gas-phase hydrogen/deuterium exchange has previously been used to probe the two populations of ubiquitin (12+), and two populations were differentiated: a more dominant one feature only 2–3 exchanges and a less dominant one featuring approximately 20 exchanges.77 The relatively low number of exchangeable protons observed for these two populations is consistent with the low αa values observed for both charge site isomers in the present study, suggesting that the conformations of the 12+ charge state of ubiquitin are strongly hydrogen bonded. Together, these data suggest that neither conformation of ubiquitin (12+) is an unraveled and disordered structure. Comparison of the αa values as a function of sequence in Figure 9 suggests that isomer A features significantly stronger hydrogen bonding, as evidenced by the near zero αa values observed for the majority of cleavages. Based on this information, it is possible that conformer A adopts a more rigid structure, one specially stabilized by hydrogen bonds, than does conformer B. This is particularly true for the stretch spanning residues 15 to 19, for which conformer B features αa values consistent with “moderate” hydrogen bonding, whereas conformer A features αa values consistent with very strong hydrogen bonding. This region also exhibits the greatest differences in charge site locations (see Figure 8), which further implies that structural differences are present for residues 6–25. Upon examination of Scp1 and apo calmodulin, moderate hydrogen bonding was associated with rigid turns and α-helices, whereas strong hydrogen bonding was associated with β-sheets. Considering what is known about ubiquitin, it seems unlikely that the 12+ conformations of ubiquitin form β-sheets; we therefore propose that conformer A is almost fully helical, perhaps with kinks at residues 22 and 35, both of which were found to lack hydrogen bonding. In contrast, conformer B, while also likely helical for the bulk of the protein, may have a turn region from residue 16 to 20. Additional studies using IM-UVPD-HEM could provide experimental separation of the conformers and allow deeper investigation of conformer B. Nonetheless, UVPD-HEM provides some regional specificity for the conformational differences between protonation isomers A and B of ubiquitin (12+).
Figure 9.

UVPD-HEM of a) conformer A and b) conformer B of the 12+ charge state of ubiquitin, using the split charge states of the a ions to differentiate isomers A and B. Experimental αa values are shown in a) in green/blue/red for conformers A and B, and the HB and NHB means associated with cleavage at a given residue are shown in striped and solid black bars, respectively. Experimental bars are colored green for those that agree with the NHB mean, blue for those that agree with the HB mean, and red for those that agree with the hyper-HB mean (extremely strong hydrogen-bonding). Grey bars denote mean αa values for which only unimodal trends were previously observed.
Limitations and outlook for HEM analysis
The seven systems on which HEM analysis was performed herein offer compelling evidence to suggest that UVPD and subsequent analysis of αa values can be used to probe the hydrogen bonding interactions of the amide carbonyl groups of the protein backbone. There are, however, several limitations to this approach. As noted above and in the Supporting Information, accurate fitting and de-isotoping of the a and a+1 ion isotope distributions is contingent upon the quality of the ion signal and the complexity of the isotope pattern (the latter directly related to the size of the fragment, with the approximate upper limit being around 8 kDa). At present, this restricts the approach to the N-terminus of proteins or to very small proteins. In addition, cleavages for which the abundances of the a ions are very low, which we have observed to occur with some frequency C-terminal to Thr and Ser residues, must also be excluded from the analysis, which restricts the extent of information obtainable in some cases. Finally, Trp causes somewhat aberrant behavior, and although this residue is not terribly common, this too limits HEM coverage. However, in general hydrogen elimination from a given a+1 ion is expected to entail hydrogen transfer to the corresponding x+1 ion, and it is possible that HEM coverage may be significantly extended by inclusion of the corresponding information buried in the x ion series.
Despite the limitations, the results shown for ordered systems and disordered systems provide compelling evidence that αa values correlate well with the presence or absence of hydrogen bonding to the amide carbonyl oxygen of the protein backbone and imply that HEM is sensitive to local order in proteins. In particular, comparison of the secondary and tertiary structure of Scp1 with the experimental αa values suggested that extreme reduction of the αa values occurs for residues engaged in β-sheets in the protein crystal structure, even for residues having exclusively NH hydrogen bonds. Hence, additional study of β-sheet-containing proteins may permit refinement of the influence of hydrogen bonding and secondary structure on the observed reduction in the αa value for a given residue. In this way, UVPD-HEM has the potential to complement conventional and emerging structural biology technologies, particularly for those classes of protein that are challenging to characterize by traditional methodologies. For example, intrinsically disordered proteins are thought to interconvert between structural families on a timescale incompatible with NMR and are difficult to crystallize for X-ray analysis. Circular dichroism, FRET, hydrogen-deuterium exchange mass spectrometry, ion mobility, and ECD have been used in various combinations to assess global and local flexibility for disordered systems,5,6,63,78,79 and UVPD-HEM offers the potential to provide an orthogonal structural validation approach for the results obtained by these methods.
Conclusions
UVPD-HEM was performed on a pair of peptides having well-defined gas-phase structures and a series of proteins known to adopt different structures. In general, the hydrogen bonding motifs predicted based on the observed αa values at each backbone cleavage were found to be in excellent agreement with structures predicted by MD simulations and X-ray crystal analysis. Application of HEM to the structured proteins Scp1 and calmodulin showed that the regions with the strongest hydrogen bonding in the crystal structures corresponded to the largest reduction in αa values. Comparison to a disordered peptide further supports this correlation, as large regions lacking hydrogen bonding were observed in accord with solution structures. DFT modeling of the minimum energy pathways was performed on three Ala8 conformers and showed hydrogen transfer occurs simultaneously with C-C backbone cleavage for the amide hydrogen transfer pathway rather than as a separate event as was the case for the beta hydrogen transfer pathway. The amide hydrogen transfer pathway was only found to be the lowest energy pathway for the unstructured peptide.
Application of UVPD-HEM to protonated ubiquitin (12+), which features two protonation site isomers with similar collisional cross sections, suggests that the impact of hydrogen bonding of the N-terminus, particularly residues 16–21, accounts for at least some of the conformational differences. Hence, hydrogen elimination monitoring is introduced as an innovative characterization approach for native-like proteins in the gas phase, complementary to the array of solution and gas-phase structural characterization techniques already available. Utilization of HEM in conjunction with HDX or IM methods may allow determination of even more refined structural assignments. Additional comparisons between HEM and crystal structures of ordered systems, including protein-ligand and multi-protein complexes, may permit more comprehensive interpretation of some of the nuances of αa values. The development of hydrogen elimination monitoring using the complementary x ion series may also enable HEM to be applied to the C-terminal sections of proteins.
Supplementary Material
Acknowledgments
Funding from the NSF (CHE-1402753), the Welch Foundation (F-1155 to JSB and F-1841 to GH) and NIH 1K12GM102745 (fellowship to LM) is acknowledged. We would like to thank Professor Yan Zhang for kindly supplying the Scp1 protein and providing the biological information relevant to the protein. We also thank Professor Dalby for providing the calmodulin protein and acknowledge the Texas Advanced Computing Center, on which the MOBCAL simulations were performed. Special thanks are given to Drs. Vicki Wysocki and Sophie Harvey for providing assistance and access to the Synapt G2 mass spectrometer and collecting the data on the Synapt G1 mass spectrometer at Ohio State University. Computational resources were provided by the Texas Advanced Computing Center.
Footnotes
Supplemental Information: Supplemental information includes ESI-mass spectra of penetratin-arg, histograms created for charge site analysis of penetratin-Arg, collision cross sections and relative energies of penetratin-Arg from MD-modelling, CID-IM drift time distributions of penetratin-Arg, the UVPD mass spectrum of penetratin-Arg (5+), a description of the error analysis strategy for determination of αa values with accompanying isotope fitting and RMSD figures, and a plot of B factors for apo calmodulin.
References
- 1.Heck AJR. Nat Meth. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 2.Chait BT, Cadene M, Olinares PD, Rout MP, Shi Y. J Am Soc Mass Spectrom. 2016;27:952–965. doi: 10.1007/s13361-016-1385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laganowsky A, Reading E, Hopper JTS, Robinson CV. Nat Protoc. 2013;8:639–651. doi: 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Konijnenberg A, van Dyck JF, Kailing LL, Sobott F. Biol Chem. 2015;396:991–1002. doi: 10.1515/hsz-2015-0136. [DOI] [PubMed] [Google Scholar]
- 5.Dickinson ER, Barran PE, Jurneczko E, Nicholson J, Hupp TR, Zawacka-Pankau J, Selivanova G. Front Mol Biosci. 2015;2:39. doi: 10.3389/fmolb.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frimpong AK, Abzalimov RR, Uversky VN, Kaltashov IA. Proteins: Struct, Funct, and Bioinf. 2010;78:714–722. doi: 10.1002/prot.22604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konermann L, Pan J, Liu YH. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 8.Pirrone GF, Iacob RE, Engen JR. Anal Chem. 2015;87:99–118. doi: 10.1021/ac5040242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajabi K, Ashcroft AE, Radford SE. Methods. 2015;89:13–21. doi: 10.1016/j.ymeth.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Mendoza VL, Vachet RW. Mass Spectrom Rev. 2009;28:785–815. doi: 10.1002/mas.20203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanucara F, Holman SW, Gray CJ, Eyers CE. Nat Chem. 2014;6:281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- 12.Badman ER, Hoaglund-Hyzer CS, Clemmer DE. J Am Soc Mass Spectrom. 2002;13:719–723. doi: 10.1016/S1044-0305(02)00374-4. [DOI] [PubMed] [Google Scholar]
- 13.Breuker K, Oh H, Horn DM, Cerda BA, McLafferty FW. J Am Chem Soc. 2002;124:6407–6420. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 14.Breuker K, McLafferty FW. Angew Chem, Int Ed. 2005;44:4911–4914. doi: 10.1002/anie.200500668. [DOI] [PubMed] [Google Scholar]
- 15.Breuker K, Brüschweiler S, Tollinger M. Angew Chem Int Ed. 2011;50:873–877. doi: 10.1002/anie.201005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skinner OS, Breuker K, McLafferty FW. J Am Soc Mass Spectrom. 2013;24:807–810. doi: 10.1007/s13361-013-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. J Am Soc Mass Spectrom. 2010;21:1966–1968. doi: 10.1016/j.jasms.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Anal Chem. 2011;83:5598–5606. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warnke S, Baldauf C, Bowers MT, Pagel K, von Helden G. J Am Chem Soc. 2014;136:10308–10314. doi: 10.1021/ja502994b. [DOI] [PubMed] [Google Scholar]
- 20.Warnke S, von Helden G, Pagel K. Proteomics. 2015;15:2804–2812. doi: 10.1002/pmic.201400480. [DOI] [PubMed] [Google Scholar]
- 21.Nagornova NS, Rizzo TR, Boyarkin OV. J Am Chem Soc. 2010;132:4040–4041. doi: 10.1021/ja910118j. [DOI] [PubMed] [Google Scholar]
- 22.Nagornova NS, Rizzo TR, Boyarkin OV. Angew Chem Int Ed Engl. 2013;52:6002–6005. doi: 10.1002/anie.201301656. [DOI] [PubMed] [Google Scholar]
- 23.Papadopoulos G, Svendsen A, Boyarkin OV, Rizzo TR. Faraday Discuss. 2011;150:243–255. 257–292. doi: 10.1039/c0fd00004c. [DOI] [PubMed] [Google Scholar]
- 24.Stearns JA, Seaiby C, Boyarkin OV, Rizzo TR. Phys Chem Chem Phys. 2009;11:125–132. doi: 10.1039/b814143f. [DOI] [PubMed] [Google Scholar]
- 25.Kim TY, Valentine SJ, Clemmer DE, Reilly JP. J Am Soc Mass Spectrom. 2010;21:1455–1465. doi: 10.1016/j.jasms.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Choi CM, Simon AL, Chirot F, Kulesza A, Knight G, Daly S, MacAleese L, Antoine R, Dugourd P. J Phys Chem B. 2016;120:709–714. doi: 10.1021/acs.jpcb.5b11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulesza A, Daly S, Choi CM, Simon AL, Chirot F, MacAleese L, Antoine R, Dugourd P. Phys Chem Chem Phys. 2016;18:9061–9069. doi: 10.1039/c6cp00263c. [DOI] [PubMed] [Google Scholar]
- 28.Vonderach M, Ehrler OT, Matheis K, Karpuschkin T, Papalazarou E, Brunet C, Antoine R, Weis P, Hampe O, Kappes MM, Dugourd P. Phys Chem Chem Phys. 2011;13:15554–15558. doi: 10.1039/c1cp21528k. [DOI] [PubMed] [Google Scholar]
- 29.Cammarata MB, Brodbelt JS. Chem Sci. 2015;6:1324–1333. doi: 10.1039/c4sc03200d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cammarata MB, Thyer R, Rosenberg J, Ellington A, Brodbelt JS. J Am Chem Soc. 2015;137:9128–9135. doi: 10.1021/jacs.5b04628. [DOI] [PubMed] [Google Scholar]
- 31.O’Brien JP, Li W, Zhang Y, Brodbelt JS. J Am Chem Soc. 2014;136:12920–12928. doi: 10.1021/ja505217w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ly T, Julian RR. J Am Chem Soc. 2010;132:8602–8609. doi: 10.1021/ja910665d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Julian RR. Int J Mass Spectrom. 2011;308:225–231. [Google Scholar]
- 34.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. J Am Chem Soc. 2013;135:12646–12651. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrison LJ, Rosenberg JA, Singleton JP, Brodbelt JS. J Am Soc Mass Spectrom. 2016;27:1443–1453. doi: 10.1007/s13361-016-1418-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robotham SA, Brodbelt JS. J Am Soc Mass Spectrom. 2015;26:1570–1579. doi: 10.1007/s13361-015-1186-y. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Cho EJ, Burstein G, Siegel D, Zhang Y. ACS Chem Biol. 2011;6:511–519. doi: 10.1021/cb100357t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Girod M, Sanader Z, Vojkovic M, Antoine R, MacAleese L, Lemoine J, Bonacic-Koutecky V, Dugourd P. J Am Soc Mass Spectrom. 2015;26:432–443. doi: 10.1007/s13361-014-1038-1. [DOI] [PubMed] [Google Scholar]
- 39.Bush MF, Hall Z, Giles K, Hoyes J, Robinson CV, Ruotolo BT. Anal Chem. 2010;82:9557–9565. doi: 10.1021/ac1022953. [DOI] [PubMed] [Google Scholar]
- 40.Bush MF, Campuzano IDG, Robinson CV. Anal Chem. 2012;84:7124–7130. doi: 10.1021/ac3014498. [DOI] [PubMed] [Google Scholar]
- 41.Mesleh MF, Hunter JM, Shvartsburg AA, Schatz GC, Jarrold MF. J Phys Chem. 1996;100:16082–16086. [Google Scholar]
- 42.Shvartsburg AA, Jarrold MF. Chem Phys Lett. 1996;261:86–91. [Google Scholar]
- 43.Blöchl PE. Phys Rev B. 1994;50:17953–17979. doi: 10.1103/physrevb.50.17953. [DOI] [PubMed] [Google Scholar]
- 44.Perdew JP, Burke K, Ernzerhof M. Phys Rev Let. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 45.Henkelman G, Uberuaga BP, Jónsson H. J Chem Phys. 2000;113:9901–9904. [Google Scholar]
- 46.Trygubenko SA, Wales DJ. J Chem Phys. 2004;120:2082–2094. doi: 10.1063/1.1636455. [DOI] [PubMed] [Google Scholar]
- 47.Florance HV, Stopford AP, Kalapothakis JM, McCullough BJ, Bretherick A, Barran PE. Analyst. 2011;136:3446–3452. doi: 10.1039/c1an15291b. [DOI] [PubMed] [Google Scholar]
- 48.May JC, McLean JA. Proteomics. 2015;15:2862–2871. doi: 10.1002/pmic.201400551. [DOI] [PubMed] [Google Scholar]
- 49.Morrison LJ, Brodbelt JS. Analyst. 2016;141:166–176. doi: 10.1039/c5an01819f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pérot M, Lucas B, Barat M, Fayeton JA, Jouvet C. J Phys Chem A. 2010;114:3147–3156. doi: 10.1021/jp908937s. [DOI] [PubMed] [Google Scholar]
- 51.Lepère V, Lucas B, Barat M, Fayeton JA, Picard VJ, Jouvet C, Çarçabal P, Nielsen I, Dedonder-Lardeux C, Grégoire G, Fujii A. J Chem Phys. 2007;127:134313. doi: 10.1063/1.2770458. [DOI] [PubMed] [Google Scholar]
- 52.Fan F, Mayo KH. J Biol Chem. 1995;270:24693–24701. doi: 10.1074/jbc.270.42.24693. [DOI] [PubMed] [Google Scholar]
- 53.Meredith JJ, Dufour A, Bruch MD. J Phys Chem B. 2009;113:544–551. doi: 10.1021/jp808379x. [DOI] [PubMed] [Google Scholar]
- 54.Sonnichsen FD, Van Eyk JE, Hodges RS, Sykes BD. Biochemistry. 1992;31:8790–8798. doi: 10.1021/bi00152a015. [DOI] [PubMed] [Google Scholar]
- 55.Kohtani M, Jones TC, Schneider JE, Jarrold MF. J Am Chem Soc. 2004;126:7420–7421. doi: 10.1021/ja048766c. [DOI] [PubMed] [Google Scholar]
- 56.Antoine R, Broyer M, Chamot-Rooke J, Dedonder C, Desfrançois C, Dugourd P, Grégoire G, Jouvet C, Onidas D, Poulain P, Tabarin T, van der Rest G. Rapid Communications in Mass Spectrometry. 2006;20:1648–1652. doi: 10.1002/rcm.2489. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L, Cui W, Thompson MS, Reilly JP. J Am Soc Mass Spectrom. 2006;17:1315–1321. doi: 10.1016/j.jasms.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 58.Kamenski T, Heilmeier S, Meinhart A, Cramer P. Mol Cell. 2004;15:399–407. doi: 10.1016/j.molcel.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 59.Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Mol Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berlow RB, Dyson HJ, Wright PE. FEBS Lett. 2015;589:2433–2440. doi: 10.1016/j.febslet.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uversky VN. FEBS Lett. 2015;589:2498–2506. doi: 10.1016/j.febslet.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 62.Binolfi A, Theillet FX, Selenko P. Biochem Soc Trans. 2012;40:950–954. doi: 10.1042/BST20120096. [DOI] [PubMed] [Google Scholar]
- 63.Smith AE, Zhou LZ, Pielak GJ. Protein Sci. 2015;24:706–713. doi: 10.1002/pro.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bellina B, Brown JM, Ujma J, Murray P, Giles K, Morris M, Compagnon I, Barran PE. Analyst. 2014;139:6348–6351. doi: 10.1039/c4an01656d. [DOI] [PubMed] [Google Scholar]
- 65.Amer DAM, Irvine GB, El-Agnaf OMA. Exp Brain Res. 2006;173:223–233. doi: 10.1007/s00221-006-0539-y. [DOI] [PubMed] [Google Scholar]
- 66.Maries E, Dass B, Collier TJ, Kordower JH, Steece-Collier K. Nat Rev Neurosci. 2003;4:727–738. doi: 10.1038/nrn1199. [DOI] [PubMed] [Google Scholar]
- 67.Dores RM. Ann N Y Acad Sci. 2009;1163:93–100. doi: 10.1111/j.1749-6632.2009.04434.x. [DOI] [PubMed] [Google Scholar]
- 68.Shi H, Atlasevich N, Merenbloom SI, Clemmer DE. J Am Soc Mass Spectrom. 2014;25:2000–2008. doi: 10.1007/s13361-014-0834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Freitas MA, Hendrickson CL, Emmett MR, Marshall AG. Int J Mass Spectrom. 1999;185–187:565–575. [Google Scholar]
- 70.Reid GE, Wu J, Chrisman PA, Wells JM, McLuckey SA. Anal Chem. 2001;73:3274–3281. doi: 10.1021/ac0101095. [DOI] [PubMed] [Google Scholar]
- 71.Brutscher B, Brüschweiler R, Ernst RR. Biochemistry. 1997;36:13043–13053. doi: 10.1021/bi971538t. [DOI] [PubMed] [Google Scholar]
- 72.Shi H, Clemmer DE. J Phys Chem B. 2014;118:3498–3506. doi: 10.1021/jp4097327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koeniger SL, Clemmer DE. J Am Soc Mass Spectrom. 2007;18:322–331. doi: 10.1016/j.jasms.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 74.Ridgeway ME, Silveira JA, Meier JE, Park MA. Analyst. 2015;140:6964–6972. doi: 10.1039/c5an00841g. [DOI] [PubMed] [Google Scholar]
- 75.Cassady CJ, Carr SR. J Mass Spectrom. 1996;31:247–254. doi: 10.1002/(SICI)1096-9888(199603)31:3<247::AID-JMS285>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 76.Purves RW, Barnett DA, Ells B, Guevremont R. J Am Soc Mass Spectrom. 2000;11:738–745. doi: 10.1016/S1044-0305(00)00136-7. [DOI] [PubMed] [Google Scholar]
- 77.Robinson EW, Williams ER. J Am Socfor Mass Spectrom. 2005;16:1427–1437. doi: 10.1016/j.jasms.2005.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ferreon ACM, Gambin Y, Lemke EA, Deniz AA. Proc Nat Acad Sci. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Young LM, Saunders JC, Mahood RA, Revill CH, Foster RJ, Tu LH, Raleigh DP, Radford SE, Ashcroft AE. Nat Chem. 2015;7:73–81. doi: 10.1038/nchem.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Momma K, Izumi F. J Appl Crystallogr. 2011;44:1272–1276. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.