

Abstract
Antifreeze proteins are produced by extremophile species to control ice formation and growth, and they have potential applications in many fields. There are few examples of synthetic materials which can reproduce their potent ice recrystallization inhibition property. We report that self-assembled enantiomerically pure, amphipathic metallohelicies inhibited ice growth at just 20 μM. Structure–property relationships and calculations support the hypothesis that amphipathicity is the key motif for activity. This opens up a new field of metallo-organic antifreeze protein mimetics and provides insight into the origins of ice-growth inhibition.
Antifreeze (glyco)proteins [AF(G)Ps] from the blood of polar fish enable life to flourish in hostile, ice-rich environments.1 AF(G)Ps have several unique macroscopic properties including ice shaping and the non-colligative depression of the freezing point. Their property of ice recrystallization inhibition (IRI)—slowing of ice growth—is of great technological interest. Ice growth is a major cause of damage during the cryopreservation of tissues/cells2,3 and cryosurgery,4 and ice growth on wind turbines, on aeroplane wings, and in frozen food5 poses significant challenges.6,7 However, AF(G)Ps themselves are expensive and potentially immunogenic and cytotoxic,8 making synthetic mimics appealing for translation to application.9 The design of mimics is complicated by the lack of a full understanding of how AF(G)Ps function.
Ben and co-workers reported that short glycopeptides with a simplified amino acid sequence were potent IRIs but did not display any ice shaping or freezing point depression.10−12 This implied that a specific receptor–ligand type of interaction is not essential for IRI and that more diverse structures could potentially have useful activity, but most reports still required complex synthesis.12 More recently, surfactants,13 poly(vinyl alcohol),14−16 and poly(ampholyte)s17−19 have emerged with definite IRI activity. By slowing ice crystal growth, these IRI active compounds can dramatically improve the cryopreservation of cells20−22 and reduce the concentration of toxic organic solvents used on conventional cryopreservation.23,24
Structural analysis of AFPs shows that many are based on an amphiphilic α-helix25,26 (as well as other rigid structures including β-barrels) with facial amphiphilicity27 where hydrophilic/phobic domains are segregated but without promoting aggregation (unlike surfactants). For example, saffarin O has been found to mimic AFPs by self-assembly into an amphipathic fiber.28 This implies that synthetic systems with similar architectures might be interesting candidates as new IRIs. Helicates—multimetallic, multistrand coordination complexes—resemble α-helices in terms of their diameter and charge.29,30 We have recently designed and synthesized the first self-assembling enantiomerically pure metallohelical architectures,29 with a broad range of structurally dependent pharmacological properties including nucleic acid binding,31 enzyme inhibition,32 and anticancer33 antimicrobial activity34 and inhibiting amyloid fibril nucleation.35 Despite their structural complexity, these are readily synthesized from small building blocks on a practical scale.
Considering the above, we synthesized a range of our triplex metallohelices (Figure 1) to screen for IRI inspired by their rigidity, optical purity, water stability, and asymmetric amphipathic architectures. We report here the unprecedented IRI activity of these compounds, and we suggest, based on hydrophobicity calculations and structural analyses, that the activity stems from amphipathic charge distribution.
Figure 1.

Triplex metallohelices used in this study. The assemblies comprise three equivalent asymmetric ditopic ligand strands arranged head-to-head-to-tail and with a helical twist, thus rendering all the ligands chemically inequivalent.
The compounds shown in Figure 1 were chosen to probe various structural matters. Both enantiomers of each compound, i.e., assemblies with Δ and Λ helicity at the Fe(II) centers, were employed (see Supporting Information (SI)). IRI activity was measured using a modified “splat” assay. Briefly, a small droplet of the compound in buffer was dropped onto a microscope slide at −80 °C to seed a polynucleated ice wafer. These were annealed at −6 °C for 30 min, and the mean largest grain size (MLGS) was calculated relative to a PBS control. Smaller values indicated more inhibition.
A wide range of activities was observed (Figure 2) from the inactive to very active. A negative control, of poly(ethylene glycol) at equal mass concentrations gave MLGS values above 80%. Enantiomers of each compound display near identical activity, as would be expected in an otherwise achiral system. The compounds Δ- and Λ-1 were the most potent, displaying remarkable dose-dependent decreases in the MLGS, completely inhibiting all ice growth at 20 μM. This is more active than polyampholytes which are potent IRI cryopreservatives19,36 and comparable to small molecular alkyl glycosides developed by Ben et al.11 Budke et al. have undertaken a detailed comparison of various IRIs, for comparison.37 We did not see any evidence for dynamic ice shaping, but this is typically only seen at very high concentrations even for antifreeze proteins. The presence of additional polar groups on one face of the helix in enantiomers 2 and 3 attenuated the performance, and the addition of hydrophobic groups in 4 and 5, particularly in the case of the latter naphthyl derivative, led to reduction in activity. Perhaps most remarkably, the triplex enantiomers 6, which are isomers of 1, had essentially zero activity again highlighting the strong link between structure and function in these assemblies.
Figure 2.
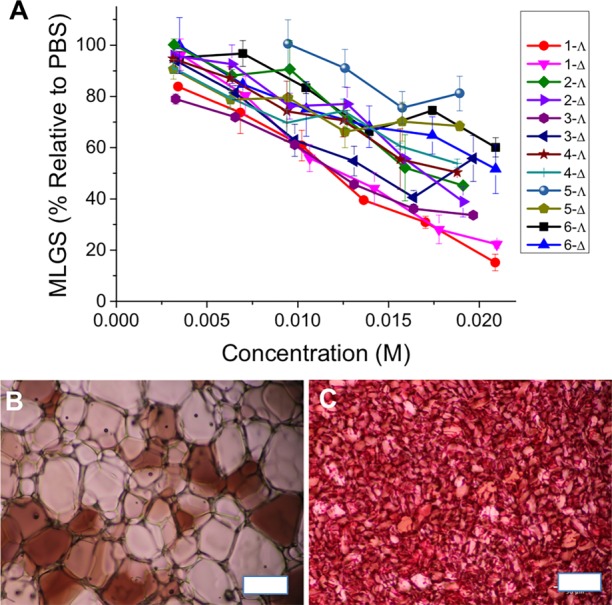
IRI activity of metallohelix library. (A) IRI activity concentration dependence of metallohelices. (B) Example ice wafer of PBS buffer control. (C) Example wafer for 1. Error bars represent minimum of three repeats, MLGS = mean largest grain size relative to phosphate buffer saline control. Images are of equal magnification, scale bars are 100 μm.
Our hypothesis is that spatially segregated hydrophobic and hydrophilic domains are essential for IRI activity and are a common structural feature of all AFPs and AFGPs, suggesting that specific ice-binding faces are not essential for IRI. Considering compounds 1–3, we see that the presence of specific functional groups on one face of the complex is not the source of activity as 1 (Y = H) is the most potent. However, the physical properties of these compounds are very different from the constituent ligands; for example without metal coordination the ligands are essentially insoluble in water. This fits our hypothesis—essentially we have a water-soluble compound constructed entirely from hydrophobic ligands. We thus examined the possibility that the IRI activity might arise from anisotropy in distribution of the charge arising from Fe2+.
All the compounds in this study feature π-stacking interactions between coordinated pyridine ligands and pendant (non-conjugated) phenyl groups (C-atoms colorized pink in Figure 3a,b,e,f). Examination of several molecular structures of compounds containing this motif (see SI),34 revealed that the regions near the faces of the pendant phenyl groups are essentially free of counter-anions and polar solvents. The pendant arenes are thus able to shield the ligand-delocalized positive charge from external species. Therefore, this substantial anisotropy (amphipathicity) in the surface hydrophobicity of the triplex may be the underlying cause of the observed IRI behavior.
Figure 3.
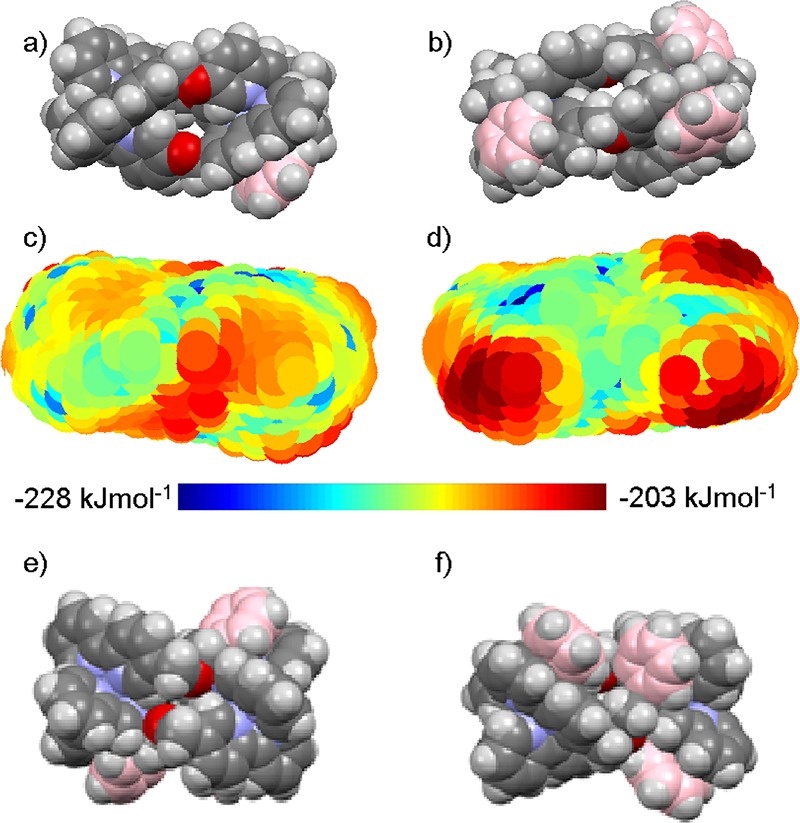
Charge distribution and π-stacking in the metallohelices. (a,b) Views of DFT-calculated structure of active compound Λ-1; (c,d) corresponding hydrophobicity plots (water O-atoms colored according to energy scale); and (e,f) views of the molecular structure from X-ray crystallography of the Zn(II) perchlorate analogue of inactive compound 6.
For peptidic α-helices, hydrophobicity is calculated or estimated by residue-based analysis, but the systems presented here require a bespoke computational approach. Using a computed model of the most active compound Δ-1 we calculated the bonding energy (AM1) of a single water molecule at multiple positions on the surface of the complex (see ESI for details).38,39Figure 3a,b shows two different faces of Δ-1 (DFT calculated structure), and the equivalent views from the hydrophobicity calculations are shown in Figure 3c,d. In the latter, water O-atoms are plotted in calculated positions and colored according to the relative energy, from hydrophilic (blue) to hydrophobic (red). The hydration energy values span 25 kJ mol–1. Figure 3a,c are viewed toward a hydrophilic face with two partially exposed metal atoms whose charge is delocalized over coordinated ligands. Figure 3b,d, displaying the opposite face of the complex, depicts three distinct hydrophobic regions corresponding to the three π-stacked arenes as well as a more hydrophilic central region associated with the edges of charge-bearing arenes and ether α-CH2 groups. In contrast, for the least active compound Δ-6, the most hydrophilic (Figure 3e) and hydrophobic (Figure 3f) faces are actually rather similar, the latter comprising end-on π-stacks and an additional charge-exposed region (bottom left). We propose that this accounts for the dramatic difference in IRI performance between these two isomeric compounds. Finally, we determined the crystal structure of 6 as a perchlorate salt which shows that in the solid state the cation is quite uniformly surrounded by anions and solvent, i.e., no apparent charge anisotropy (see SI).
The triplex metallohelices compare favorably with previously reported IRI-active small molecules, being more active than carbohydrate based surfactants with the exception of n-octyl-β-d-galactopyranoside and a lysine-based cationic surfactant for which similar levels of activity was reported.11 They are also more active than most reported polymeric inhibitors,17,19,36 apart from poly(vinyl alcohol) which has unusually high activity.40 While PVA is a flexible polymer, it is also a surfactant, and hence does have amphipathic characteristics. Triplex metallohelices are synthetically more accessible than glycopeptides, and have the benefit over polymeric inhibitors in that they have no molecular weight dispersity. Further, our hypothesis regarding charge anisotropy can be tested readily by synthesizing a range of systems designed to modulate this, e.g., with different heterocycles and pendant arenes, and provides a scaffold to obtain further structure–activity relationships which may help us to understand the fundamental origins of ice growth inhibition and lead to the design of practical synthetic IRIs.
In conclusion, this Communication demonstrates a new concept in ice recrystallization inhibition based upon self-assembled, enantiomerically pure metallohelical architectures. A wide range of activities is observed, the most active compound displaying a distinctly amphipathic architecture arising from asymmetric folding of the helix as encoded by the chiral ligands. We propose, on the basis that natural antifreeze proteins have a similar amphipathic structure, that this is the physical origin of the IRI activity, but it is not clear if a helix is essential. More generally, our observations indicate that rather than using traditional functionalization in order to generate metallohelices that mimic peptide α-helices, we should consider ways in which charge anisotropy and thus hydrophobicity can be controlled.
Acknowledgments
We thank EPSRC for a studentship for D.E.M. (EP/F500378/1). MIG holds an ERC Starter Grant (CRYOMAT 638661). We thank the National Crystallographic Service for recording the data for AF19.41
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05822.
Synthetic details and characterization plus analysis of the 3-D shape of the complexes (PDF)
Author Contributions
§ D.E.M. and R.A.V. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Davies P. L.Trends in Biochemical Sciences; Elsevier: Amsterdam, November 2014; pp 548–555. [DOI] [PubMed] [Google Scholar]
- Mazur P. In Life in the Frozen State; Fuller B., Lane N., Benson E. E., Eds.; CRC Press: Boca Raton, 2005; Vol. 17, pp 301–302. [Google Scholar]
- Mazur P. Science (Washington, DC, U. S.) 1970, 168 (3934), 939–949. 10.1126/science.168.3934.939. [DOI] [PubMed] [Google Scholar]
- Koushafar H.; Pham L.; Lee C.; Rubinsky B. J. Surg. Oncol. 1997, 66 (2), 114–121. . [DOI] [PubMed] [Google Scholar]
- Sidebottom C.; Buckley S.; Pudney P.; Twigg S.; Jarman C.; Holt C.; Telford J.; McArthur A.; Worrall D.; Hubbard R.; Lillford P. Nature 2000, 406 (6793), 256. 10.1038/35018639. [DOI] [PubMed] [Google Scholar]
- Breton G.; Danyluk J.; Ouellet F.; Sarhan F. Biotechnological applications of plant freezing associated proteins. Biotechnol. Annu. Rev. 2000, 6, 59–101. 10.1016/S1387-2656(00)06019-1. [DOI] [PubMed] [Google Scholar]
- Venketesh S.; Dayananda C. Crit. Rev. Biotechnol. 2008, 28 (1), 57–82. 10.1080/07388550801891152. [DOI] [PubMed] [Google Scholar]
- Liu S.; Wang W.; von Moos E.; Jackman J.; Mealing G.; Monette R.; Ben R. N. Biomacromolecules 2007, 8 (5), 1456–1462. 10.1021/bm061044o. [DOI] [PubMed] [Google Scholar]
- Gibson M. I. Polym. Chem. 2010, 1 (8), 1141–1152. 10.1039/c0py00089b. [DOI] [Google Scholar]
- Trant J. F.; Biggs R. A.; Capicciotti C. J.; Ben R. N. RSC Adv. 2013, 3 (48), 26005–26009. 10.1039/c3ra43835j. [DOI] [Google Scholar]
- Capicciotti C. J.; Leclere M.; Perras F. A.; Bryce D. L.; Paulin H.; Harden J.; Liu Y.; Ben R. N. Chem. Sci. 2012, 3 (5), 1408–1416. 10.1039/c2sc00885h. [DOI] [Google Scholar]
- Tam R. Y.; Rowley C. N.; Petrov I.; Zhang T.; Afagh N. A.; Woo T. K.; Ben R. N. J. Am. Chem. Soc. 2009, 131 (43), 15745–15753. 10.1021/ja904169a. [DOI] [PubMed] [Google Scholar]
- Capicciotti C. J.; Malay D.; Ben R. N. Recent Dev. Study Recryst. 2013, 177–224. 10.5772/54992. [DOI] [Google Scholar]
- Congdon T.; Notman R.; Gibson M. I. Biomacromolecules 2013, 14 (5), 1578–1586. 10.1021/bm400217j. [DOI] [PubMed] [Google Scholar]
- Inada T.; Modak P. R. Chem. Eng. Sci. 2006, 61 (10), 3149–3158. 10.1016/j.ces.2005.12.005. [DOI] [Google Scholar]
- Phillips D. J.; Congdon T. R.; Gibson M. I. Polym. Chem. 2016, 7 (9), 1701–1704. 10.1039/C5PY01948F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan R.; Jain M.; Matsumura K. J. Biomater. Sci., Polym. Ed. 2013, 24 (August), 1767–1780. 10.1080/09205063.2013.801703. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Cameron N. R.; Gibson M. I. Chem. Commun. 2015, 51 (65), 12977–12980. 10.1039/C5CC04647E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs C.; Lipecki J.; Gibson M. I. Biomacromolecules 2017, 18 (1), 295–302. 10.1021/acs.biomac.6b01691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. E.; Lovett J. R.; Armes S. P.; Gibson M. I. Angew. Chem., Int. Ed. 2016, 55 (8), 2801–2804. 10.1002/anie.201511454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. Nat. Commun. 2014, 5, 3244. 10.1038/ncomms4244. [DOI] [PubMed] [Google Scholar]
- Deller R. C.; Pessin J. E.; Vatish M.; Mitchell D. A.; Gibson M. I. Biomater. Sci. 2016, 4, 1079–1084. 10.1039/C6BM00129G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capicciotti C. J.; Kurach J. D. R.; Turner T. R.; Mancini R. S.; Acker J. P.; Ben R. N. Sci. Rep. 2015, 5, 9692. 10.1038/srep09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller R. C.; Vatish M.; Mitchell D. A.; Gibson M. I. ACS Biomater. Sci. Eng. 2015, 1 (9), 789–794. 10.1021/acsbiomaterials.5b00162. [DOI] [PubMed] [Google Scholar]
- Kwan A. H.; Fairley K.; Anderberg P. I.; Liew C. W.; Harding M. M.; Mackay J. P. Biochemistry 2005, 44, 1980–1988. (Figure 1) 10.1021/bi047782j. [DOI] [PubMed] [Google Scholar]
- Sicheri F.; Yang D. S. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1996, 52 (3), 486–498. 10.1107/S0907444995015253. [DOI] [PubMed] [Google Scholar]
- Mitchell D. E.; Gibson M. I. Biomacromolecules 2015, 16 (10), 3411–3416. 10.1021/acs.biomac.5b01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drori R.; Li C.; Hu C.; Raiteri P.; Rohl A.; Ward M. D.; Kahr B. J. Am. Chem. Soc. 2016, 138 (40), 13396–13401. 10.1021/jacs.6b08267. [DOI] [PubMed] [Google Scholar]
- Howson S. E.; Scott P. Dalton Trans. 2011, 40 (40), 10268–10277. 10.1039/c1dt11423a. [DOI] [PubMed] [Google Scholar]
- Lehn J. M.; Rigault A.; Siegel J.; Harrowfield J.; Chevrier B.; Moras D. Proc. Natl. Acad. Sci. U. S. A. 1987, 84 (9), 2565–2569. 10.1073/pnas.84.9.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malina J.; Scott P.; Brabec V. Dalt. Trans. 2015, 44 (33), 14656–14665. 10.1039/C5DT02018B. [DOI] [PubMed] [Google Scholar]
- Malina J.; Scott P.; Brabec V. Nucleic Acids Res. 2015, 43 (11), 5297–5306. 10.1093/nar/gkv438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaner R. A.; Allison S. J.; Faulkner A. D.; Phillips R. M.; Roper D. I.; Shepherd S. L.; Simpson D. H.; Waterfield N. R.; Scott P. Chem. Sci. 2016, 7 (2), 951–958. 10.1039/C5SC03677A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howson S. E.; Bolhuis A.; Brabec V.; Clarkson G. J.; Malina J.; Rodger A.; Scott P. Nat. Chem. 2012, 4 (1), 31–36. 10.1038/nchem.1206. [DOI] [PubMed] [Google Scholar]
- Yu H.; Li M.; Liu G.; Geng J.; Wang J.; Ren J.; Zhao C.; Qu X. Chem. Sci. 2012, 3 (11), 3145–3153. 10.1039/c2sc20372c. [DOI] [Google Scholar]
- Mitchell D. E.; Cameron N. R.; Gibson M. I. Chem. Commun. 2015, 51 (65), 12977–12980. 10.1039/C5CC04647E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budke C.; Dreyer A.; Jaeger J.; Gimpel K.; Berkemeier T.; Bonin A. S.; Nagel L.; Plattner C.; Devries A. L.; Sewald N.; Koop T. Cryst. Growth Des. 2014, 14 (9), 4285–4294. 10.1021/cg5003308. [DOI] [Google Scholar]
- Schmidt M. W.; Baldridge K. K.; Boatz J. A.; Elbert S. T.; Gordon M. S.; Jensen J. H.; Koseki S.; Matsunaga N.; Nguyen K. A.; Su S.; Windus T. L.; Dupuis M.; Montgomery J. A. J. Comput. Chem. 1993, 14 (11), 1347–1363. 10.1002/jcc.540141112. [DOI] [Google Scholar]
- Granovsky A. A.Firefly, version 8;; http://classic.chem.msu.su/gran/firefly/index.html.
- Congdon T.; Notman R.; Gibson M. I. Biomacromolecules 2013, 14 (5), 1578–1586. 10.1021/bm400217j. [DOI] [PubMed] [Google Scholar]
- Coles S. J.; Gale P. A. Chem. Sci. 2012, 3 (3), 683–689. 10.1039/C2SC00955B. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.