

Abstract
Recent progress in human organoids has provided 3D tissue systems to model human development, diseases, as well as develop cell delivery systems for regenerative therapies. While direct differentiation of human embryoid bodies holds great promise for cardiac organoid production, intramyocardial cell organization during heart development provides biological foundation to fabricate human cardiac organoids with defined cell types. Inspired by the intramyocardial organization events in coronary vasculogenesis, where a diverse, yet defined, mixture of cardiac cell types self-organizes into functional myocardium in the absence of blood flow, we have developed a defined method to produce scaffold-free human cardiac organoids that structurally and functionally resembled the lumenized vascular network in the developing myocardium, supported hiPSC-CM development and possessed fundamental cardiac tissue-level functions. In particular, this development-driven strategy offers a robust, tunable system to examine the contributions of individual cell types, matrix materials and additional factors for developmental insight, biomimetic matrix composition to advance biomaterial design, tissue/organ-level drug screening, and cell therapy for heart repair.
Keywords: human cardiac organoid, human induced stem cell-derived cardiomyocyte, coronary vasculogenesis, development modeling, regenerative medicine
INTRODUCTION
With the advancement of human induced pluripotent stem cell technologies, the emerging human organoids have provided a powerful platform to model human organ development and pathologies, as well as provide cell delivery systems for regenerative therapy (1, 2). Organoids are 3D structures based on the self-organization of stem cell-derived organ-specific cell types to recapitulate major tissue/organ structures and functions. To this end, the development of a variety of functional human organoids (e.g., lung, liver, brain) has given novel insight into developmental and pathological processes (3–7). Despite the increasing progress in other organ systems, few studies have focused on the development of a human cardiac organoid (8, 9). Recapitulation of the 3D multicellular structure and function of the developing myocardium remains a challenge for the development and application of human cardiac organoids (10).
Significant advancements, however, have been made in the utilization of human induced pluripotent stem cell derived-cardiomyocytes (hiPSC-CMs) at the cellular and genetic level for the development of disease models and drug screening systems (11–13). In addition to 2D culture systems, significant resources have been devoted to developing 3D myocardial constructs to support cardiomyocyte development and improve viable transplantation approaches (14). Strategies have been engineered to target specific physiologically relevant features (e.g., multicellular, capillary-like diffusion, electrical stimulation) in the myocardium to improve cardiomyocyte functions and development (15–22). To this end, early cardiac tissue engineering efforts, such as the work of Kelm and others, demonstrated that cardiac spheroids made from a mixed cell population derived from neonatal/fetal rodent hearts supported myocardial extracellular matrix profile and cardiomyocyte sarcomere development (23, 24). This has been supported further by several hiPSC-CM-based studies showing that supporting cell types provide extracellular matrix microenvironment, incorporate angiogenic signaling, enhance microtissue assembly, and improve maturation of cardiac microtissues (25–31). Furthermore, the addition of vascular cell types to cardiac microtissues can model aspects of coronary angiogenesis and provide a functional benefit in heart repair by connecting to the host vasculature after transplantation (19, 23, 31, 32). By targeting specific hiPSC-CM functions/properties, tissue-engineered cardiac systems have been advantageous for the development of more sensitive drug testing models and improved cell delivery strategies. However, by using “output” structures/functions as the input design principle, these platforms can exclude tissue/organ-level intricacies that are critical for modeling cardiac development and pathologies.
While cardiac differentiation of embryoid bodies holds great potential for cardiovascular organoid production (8, 9), the intramyocardial organization events in coronary vasculogenesis provides the biological inspiration for a method of controlled, scaffold-free human cardiac organoid fabrication using defined cell types. In development, after the 4-chamber heart structure has formed, the formation of the epicardium involves a dynamic process of epithelial-mesenchymal transition of epicardium-derived cells that migrate into the myocardium and differentiate into vascular and cardiac interstitial cell types (33–35). Subsequently, the developing ventricular myocardium undergoes the intramyocardial organization events of coronary vasculogenesis where a diverse, yet defined, mixture of cardiac cell types (e.g., cardiomyocytes, cardiac fibroblasts, vascular cell types) organize into functional myocardium before anastomosis to the aorta (Figure 1A, 1B) (36–39). At this point in development, the cardiomyocytes also retain a spontaneous beating property, in part, due to incomplete sub-type specification of the cardiomyocytes, similar to hiPSC-CMs (40, 41). Inspired by these events and the developing/immature characterization of hiPSC-CMs, we have established a defined, scaffold-free fabrication strategy to assemble vascularized human cardiac organoids in vitro that recapitulate an array of structural, genetic, and functional cardiac tissue-level characteristics of the developing myocardium. Through incorporation of the events of coronary vasculogenesis as the major design principle in human cardiac organoids, the technology described here provides a powerful platform to incorporate and investigate the cellular, matrix/material and additional factors required to promote tissue/organ-level heart development. Furthermore, this platform lays down the foundation for the development of functional cardiac tissues for tissue-/organ-level drug screening and advanced cell therapy for heart repair.
Figure 1.
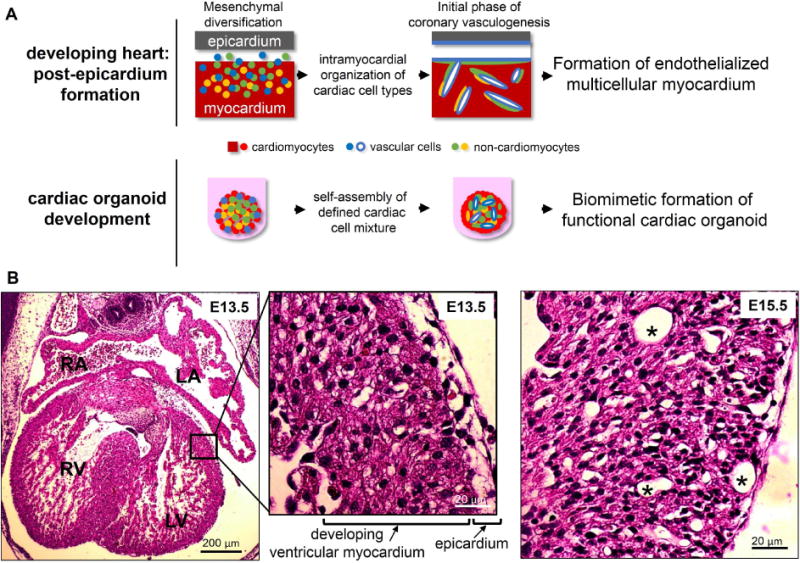
Developmental inspiration for organoid fabrication. (A) In the developing heart after the 4-chamber structure has formed, the epicardium and subsequent epithelial-to-mesenchymal transition give rise to a diverse mixture of cardiac cell types. This stage of intramyocardial organization marks the initial phase of coronary vasculogenesis. The self-assembly process surrounding this intramyocardial organization served as inspiration for analogous, self-assembled cardiac organoids from a defined mixture of cardiac cells prior to blood flow. (B) Hematoxylin and eosin-stained mouse embryonic heart sections at embryonic day 13.5 (E13.5) show the 4-chamber structure and the epicardium adjacent to the developing ventricular myocardium (inset). By E15.5 (right), evidence of preliminary coronary vessels (asterisk) can be seen in the developing ventricular myocardium.
RESULTS AND DISCUSSION
Developmental inspiration for cardiac organoid fabrication
The dynamic cellular organization events of coronary vasculogenesis during heart development (after the 4-chamber structure as formed) provided a developmental basis for the fabrication of cardiac organoids, represented in Figure 1A. To prepare human cardiac organoids, we utilized hiPSC-CMs, human ventricular cardiac fibroblasts (cFBs) and human umbilical vein endothelial cells (HUVECs) to represent the major cardiac cell types in the developing myocardium. Due to their immature characteristics, hiPSC-CMs served as an appropriate cell type to represent cardiomyocytes in the developing myocardium (40). While aligned cardiac microtissue research has significantly enhanced the understanding of stem cell-derived cardiomyocyte mechanical/electrical properties (e.g., contraction force, conduction velocity) (15, 22, 42–44), the cardiac organoid system does not incorporate an aligned cardiomyocyte design to resemble the developing heart at the stage of coronary vasculogenesis, in which significant anisotropic cardiomyocyte organization has not been established (Figure 1B). During the spheroid fabrication, the ratio between different cardiac cells was selected to approximate cell ratios in developing hearts (5:4:1, hiPSC-CMs:cFBs:HUVECs) (45–47). As an alternate rationale, human cardiac organoids were also fabricated using the ratio of cells corresponding to adult hearts (3:6:1, hiPSC-CMs:cFBs:HUVECs) (Figure 2A–C) (45–47). The experimental timeline can be seen in Figure S1.
Figure 2.
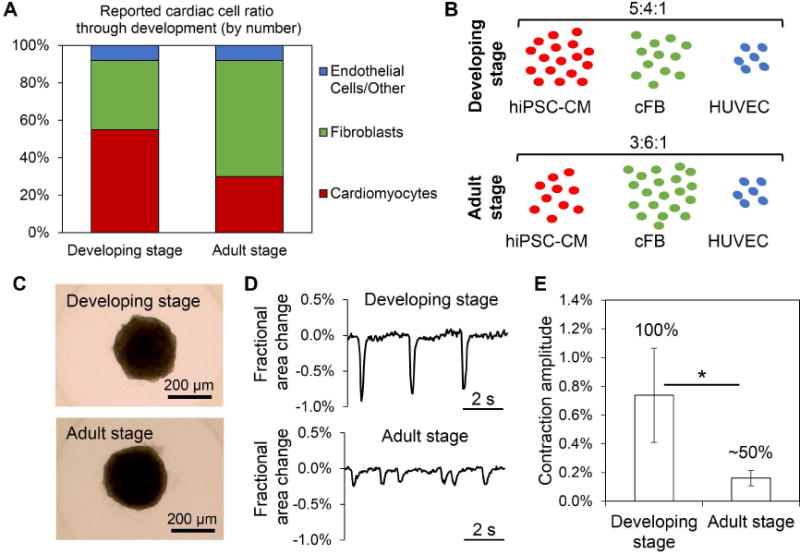
Examination of developmental inspiration for cardiac cell ratio for organoid fabrication. (A) The ratio of cardiac cell types (by number) changes throughout development, based on reported literature ref. 45–47. (B, C) A developing stage and adult stage cell ratios of cardiac cell types represent two organoid fabrication rationales to support cardiomyocyte development, seen in bright-field images at D0; n=human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM), human cardiac ventricular fibroblasts (cFB), human umbilical vein endothelial cells (HUVEC). (D) Representative contraction profiles for developing stage and adult stage ratio organoids were constructed from video analysis of spheroid fractional area change. (E) Analysis of average (±SEM) contraction amplitude (i.e., fractional area change) showed significant differences at D10; n=8–12 organoids. Developing stage ratio organoids showed 100% of organoids beating at D10, while adult stage ratio organoids had only ~50% beating. Asterisk represents significant difference, p<0.05.
By experimental Day 10 (D10), the developing stage ratio cardiac organoids showed significantly higher contraction amplitude (i.e., fractional area change) and regularity of beating than adult ratio organoids, which was attributed in part to the higher hiPSC-CM composition in the developing stage ratio organoids (Figure 2D, 2E). In addition, the difference in the contraction profiles was supported by enhanced sarcomere development (e.g., Z-line formation) in developing stage ratio organoids at D10 when compared to adult ratio organoids, which have minimal Z-line formation despite still having alpha sarcomeric actinin (α-SA)-positive regions (Figure S2). This suggested that the developing stage ratio provided a favorable microenvironment for hiPSC-CM development and supported the developmental basis for cardiac organoid fabrication. This finding is consistent with previous engineered findings showing that cardiac constructs containing a majority of cardiomyocytes of the construct supports increased hiPSC-CM functions (15, 17, 22, 48). The developing stage cell ratio for cardiac organoids was used for the remainder of the experiments.
To examine the effects of media components on organoid formation and maturation, DMEM/F12 media with varying amounts of FBS was prepared and compared to a ratiometric combination of cardiomyocyte media (DMEM/F12, 10% FBS), fibroblast media (FGM-3, defined growth factors, 10% FBS), and endothelial cell media (EGM-2, defined growth factors, 2% FBS) corresponding to the cell ratio (Figure S3). After 4 days of organoid formation, the cardiac organoids with the ratiometric combination showed significantly better viability index (i.e., reduced TUNEL-positive apoptotic nuclei) and was used for the remainder of the experiments.
Phenotypic cellular behavior in human cardiac organoids
Immunofluorescent analysis using von Willebrand factor (vWF) and CD31 endothelial markers of the cardiac organoids revealed limited staining and spreading of HUVECs at D0 through D10 (Figure 3A). Human adipose-derived stem cells (hADSCs) were added to the cardiac organoid system to promote endothelial function based on their pericyte-like function and pro-angiogenic properties (49–55). Cardiac organoids with hADSCs were fabricated with 50% hiPSC-CMs, 29% cFBs, 14% HUVECs, and 7% hADSCs. The relative amount of hADSCs reflects the limited amount of pericytes in the heart, supported by previous research in endothelial-ADSC co-culture systems (Figure 3B) (26, 49). The effects of hADSCs were noticeable from D0 by enhanced organization of vWF-positive cells (i.e., HUVECs) in a circular pattern surrounding the inner portion of the organoid (Figure 3A). By D10, the addition of hADSCs increased indications of enhanced endothelial function and/or viability, described by the increased covered area of vWF-positive structures, and promoted EC network formation in cardiac organoids compared to without hADSCs, as shown by vWF and CD31 staining (Figure 3A, 3C).
Figure 3.
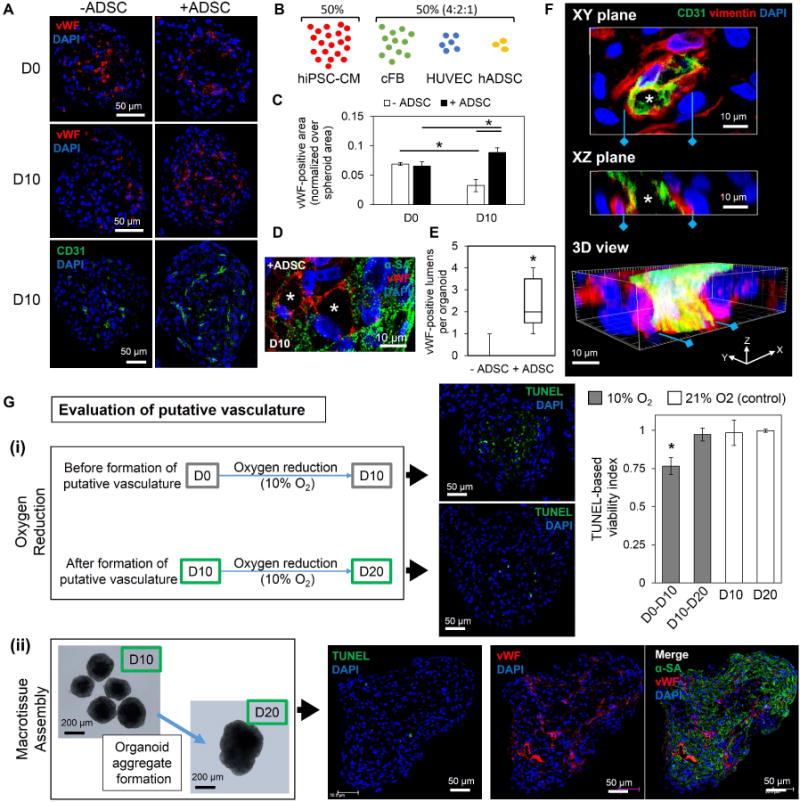
The addition of human adipose derived stem cells improves biomimetic formation of functional, lumenized vascular networks in cardiac organoids. (A) The addition of adipose-derived stem cells (+ADSC) led to increased vWF- and CD31-positive endothelial network formation by D10. Without human adipose-derived stem cells (-hADSC), cardiac organoids showed limited endothelial staining; n=3 organoids/group per endothelial stain. (B, C) The addition of hADSCs to the developmentally inspired cardiac organoid fabrication significantly improved the vWF-positive area over spheroid area by D10; n=3 organoids. (D) Lumen-like structures (asterisk) were observed in D10 cardiac organoids with hADSCs with vWF-positive (red) endothelial cells within α-SA-positive (green) hiPSC-CMs areas; blue-DAPI. (E) Significantly more vWF-positive lumen structures were observed per organoid in D10 organoids with hADSCs in comparison to without hADSCs; n=10 organoids per group. (F) 3D reconstruction of thick sections of D10 cardiac organoids with hADSCs show 3D structure of a CD31-positive (green) vessel with open lumens (asterisk) supported by vimentin-positive (red) support cells; blue-DAPI. (G) Evaluation of putative vasculature showed (i) that culturing cardiac organoids from D0–D10 (before putative vasculature formation) in an oxygen-reduced environment (10% O2) for 10 days resulted in a significant decrease in viability in the center, indicated by TUNEL apoptosis assay, compared to cardiac organoids with formed lumenized vascular networks (D10–D20) and D10 and D20 controls (21% O2); n=4–7 organoids. (ii) Macrotissue assembly using five D10 cardiac organoids cultured 10 days (D10–D20) showed minimal TUNEL expression and displayed extensive vWF-positive endothelial networks across the whole macrotissue (n=3 macrotissues). Cardiac organoids were fabricated using a developing stage cell ratio. Asterisk represents significant difference, p<0.05.
As in coronary vasculogenesis, where vascular cell types and cardiac interstitial cells form the endothelial plexus in the beating myocardium before connecting to the future coronary artery (36), lumen-like structures with vWF-positive cells were found within the cardiac organoids bordered by α-SA-positive hiPSC-CMs, although not every vWF-positive cell participated in the lumen formation (Figure 3D). Notably, cardiac organoids with hADSCs showed significantly more vWF-positive lumen structures than without hADSCs (Figure 3E). 3D reconstruction of thick cardiac organoid sections revealed CD31-positive vessel structures supported by vimentin-positive cells (Figure 3F). Serial images of confocal imaging of vWF-stained whole organoids further confirmed the formation of multiple areas with lumen-like regions in X, Y, and Z perspectives connected by vWF-positive ECs (Figure S4). The increase in vWF- and CD31-positive structures and lumen formation demonstrated the structural functionality of the HUVECs and was largely attributed to the pro-angiogenic role of hADSCs. This is supported by a growing amount of research indicating a perivascular origin and properties of mesenchymal stem cells (e.g., hADSCs), which is consistent with other studies showing the pro-angiogenic properties of hADSCs (49, 50, 52–54). In addition, the organizational changes seen after the addition of hADSCs is consistent with other multicellular research that indicates a higher order level of self-organization beyond differential cell adhesion dynamics (9, 56). Furthermore, vascular endothelial growth factor (VEGF) signaling plays an important role during coronary vasculogenesis (57). Gene expression of vascular endothelial growth factor A (VEGFA) and the main VEGF-A receptor 2 (VEGFR2) in cardiac organoids and spheroids of each individual cell type indicated the potential role in angiogenic signaling within cardiac organoids between the major VEGF providers (hiPSC-CMs, cFB, hADSCs) and VEGF receiver (HUVECs) (Figure S5).
To evaluate the functionality of the putative vasculature, cardiac organoids were placed in an oxygen-reduced environment (10%) before (D0) and after (D10) putative vasculature had formed for 10 days. Cardiac organoids cultured at 10% O2 from D0 (before putative vasculature formation) showed a significant increase in TUNEL expression at the organoid center when compared to cardiac organoids cultured at 10% O2 from D10 (Figure 3G(i), S6). This strongly indicates the formation of putative vasculature reduces cell apoptosis in the organoids center, suggesting their function to improve nutrient transport within the organoids. In addition, we examined whether the putative vasculature in the cardiac organoids can allow for the fabrication of large viable cardiac tissue engineering constructs, as oxygen diffusion in the engineered cardiac tissue is limited to a 100–200 μm range (58, 59). To this end, cardiac organoids were cultured for 10 days to form putative vasculature and then placed in groups of five to form organoid aggregates. Macrotissue assembly was observed until D20, where the aggregates had formed large macrotissues (>200 μm radius) with observable increased Z-dimension. TUNEL analysis showed no significant changes in viability by D20 and immunofluorescent staining revealed large regions of connected, vWF-positive endothelial cells (i.e., networks) across the organoid macrotissues (Figure 3G(ii), S7). The observed enhanced viability in both cases strongly indicates a functional benefit of putative vasculature, which is consistent with previous studies incorporating vascular networks in rodent-cell based and scaffold-based engineered cardiac constructs for improved viability/integration after transplantation and evident from cardiovascular tissue engineering studies showing large necrotic core in large scaffold-free constructs without pre-established vasculature (8, 60–62). This improvement may be attributed to the enhanced passive transport of nutrients whereby the presence of lumens creates a porous construct that increases diffusivity to the center of the micro-/macrotissues (63). These data support the functional benefit of recapitulating the lumenized vascular network of the developing myocardium in the cardiac organoids.
To explore the effects of endothelial cell source on the vasculature formation in the organoids, we substituted HUVECs with human adipose-derived microvascular endothelial cells (HAMECs) to fabricate cardiac organoids due to their microvascular/capillary origin in vivo (64). HAMEC cardiac organoids showed some evidence of network formation but overall showed less vWF-positive cells than HUVEC cardiac organoids on D10, while the cardiomyocyte organization was similar in both cases (Figure S8). This is consistent with reduced in vivo vascular network density observed in transplanted human dermis-derived microvascular endothelial cells compared to HUVECs (65). These results may reflect tissue-specific and/or developmental age properties of the cell source that influence their angiogenic/vasculogenic and structural functionality (66–69). However, given their facile, readily expandable culture properties and consistent vasculogenic characteristics in cardiac organoids, HUVECs were used for the remainder of the experiments.
As for the hiPSC-CM, cFB, and hADSC organization, D0 cardiac organoids with or without hADSCs showed heterogeneous populations of vimentin-positive cells (i.e., cFBs and hADSCs) throughout the microtissue (Figure 4A). The majority of α-SA-positive hiPSC-CMs with or without hADSCs organized in the outer regions of the cardiac organoids. In the native myocardium, adjacent fibroblast-cardiomyocyte interactions are critical for cardiac development (70). By D10, cardiac organoids with hADSCs displayed an increased density of vimentin-positive cells in the α-SA-positive (i.e., hiPSC-CMs) regions of the cardiac organoid (Figure 4A, 4B). Furthermore, the immunomodulatory potential of hADSCs was supported by a significant reduction in the pro-inflammatory interleukin IL6 gene expression in cardiac organoids, compared to organoids without hADSCs (Figure 4C) (71).
Figure 4.
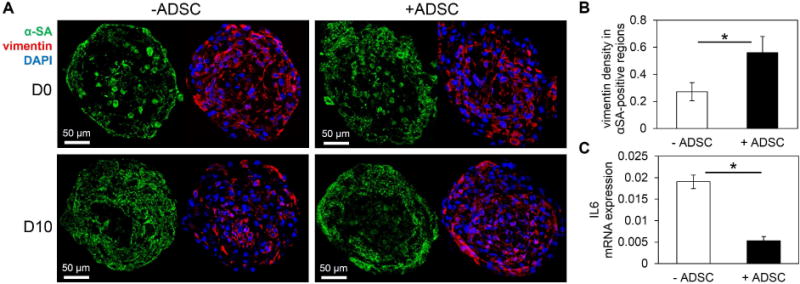
The addition of human adipose derived stem cells affects biomimetic cellular organization and introduces immunomodulatory effects. (A) α-SA-positive (green) hiPSC-CMs were mainly located in the outer region of spheroid cross-sections with or without hADSCs. The addition of hADSCs showed more homogeneous distribution of vimentin-positive (red) cells by D10; blue-DAPI. (B) The density of vimentin-positive staining area within α-SA regions significantly increased in cardiac organoids with hADSCs; n=3–4 organoids. (C) hADSCs demonstrated immunomodulatory role by significantly decreasing the expression of IL6 in cardiac organoids with hADSCs; n=3 experiments, 10–15 organoids/experiment. Cardiac organoids were fabricated using a developing stage cell ratio. Asterisk represents significant difference, p<0.05.
Extracellular matrix (ECM) protein production in human cardiac organoids
In addition to the phenotypic cellular distribution throughout the cardiac organoid, the extracellular matrix proteins in the cardiac organoids supported the development of an organotypic microenvironment. One limitation in hiPSC-CM spheroids (i.e., microtissues composed of 100% hiPSC-CMs) is the lack of collagen in the microtissue. While hiPSC-CM spheroids showed limited collagen I expression throughout the experiment with disconnected, dotted structures, the cardiac organoids expressed significantly more collagen I-positive, connected structures indicative of mature, stable collagen fibril formation (Figure 5A). This difference was attributed to the presence of cardiac fibroblasts in the organoids, as collagen is maintained by the cardiac fibroblasts in the native myocardium (70). As shown in the Figure 5A, collagen I expression in the organoids increased from D0 to D10, consistent with results from developing hearts (72).
Figure 5.
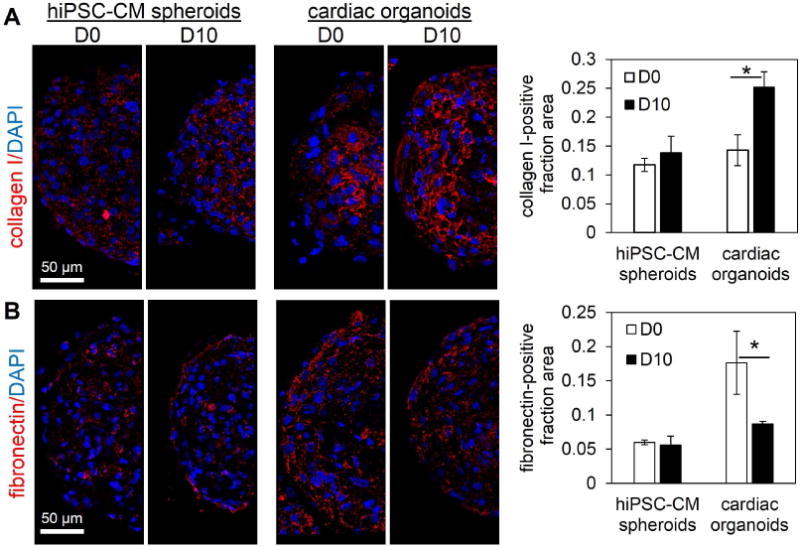
Human cardiac organoids display organotypic extracellular matrix (ECM) components. (A) Spheroids comprised of 100% hiPSC-CMs (hiPSC-CM spheroids) showed limited, dotted collagen I immunofluorescent staining, while cardiac organoids showed significantly increased, connected collagen I structures by D10. (B) hiPSC-CM spheroids showed overall low levels of fibronectin expression, while cardiac organoids decreased significantly by D10. Quantification of ECM components was based on the ratio of ECM-positive covered area divided by total spheroid area. hiPSC-CM-human induced pluripotent stem cell-derived cardiomyocyte. Cardiac organoids were fabricated using a developing stage cell ratio; n=3–6 microtissues. Asterisk represents significant difference, p<0.05.
Notably, in cardiac organoids, fibronectin-positive spheroid area decreased significantly from D0 to D10, while hiPSC-CM spheroids showed overall low levels of fibronectin-positive structures at D0 and D10 (Figure 5B). This may suggest the formation of non-pathological, ventricular-like extracellular microenvironment to support development of hiPSC-CMs, as fibronectin has been shown to be more abundant in atrial than ventricular myocardium, and increased ventricular fibronectin is associated with pathological cardiac hypertrophy (73, 74). The changes in fibronectin in cardiac organoids (high to low from D0 to D10) may also indicate the increased role of fibronectin during microtissue formation evidenced by the high levels at D0 and/or the observed role of fibronectin in myofibril organization in cardiac development (75–78). In addition, the basement membrane protein (laminin) is produced by hiPSC-CMs and showed strong immunofluorescent expression in both hiPSC-CM spheroids and cardiac organoids throughout the experimental timeline (Figure S9). Cardiac organoids provide a biomimetic matrix environment that supports cardiac function and can serve as a model for biomaterial/construct design in exploring the necessary material components for cardiac development.
Enhanced cardiomyocyte development within cardiac organoids
Previous work using hiPSC-CM spheroids has indicated conditioning approaches and extended culture are required to enhance the contractile properties toward physiological relevance (14, 79). We next explored the effect of cellular and matrix microenvironment as a biomimetic stimulus in the organoids to promote the contractile properties of hiPSC-CM. During the process of self-assembly, hiPSC-CM spheroid spontaneous beating started after two days of spheroid assembly (D-2), while cardiac organoid spontaneous beating started after four days of spheroid assembly (D0). hiPSC-CM spheroids at D0 showed more phenotypic banding of α-SA-positive Z-lines than cardiac organoids, however, by D10 the cardiac organoids showed more enhanced organization (e.g., Z-line width) of sarcomeres than hiPSC-CM spheroids (Figure 6A). Particularly, the presence of Z-line widths greater than 10 μm was observed more frequently among different cardiac organoids (13 out of 19; n=5 experiments) than in hiPSC-CM spheroids (1 out of 10; n=3 experiments) seen in Figure S10A, supporting the reproducibility of human cardiac organoids.
Figure 6.
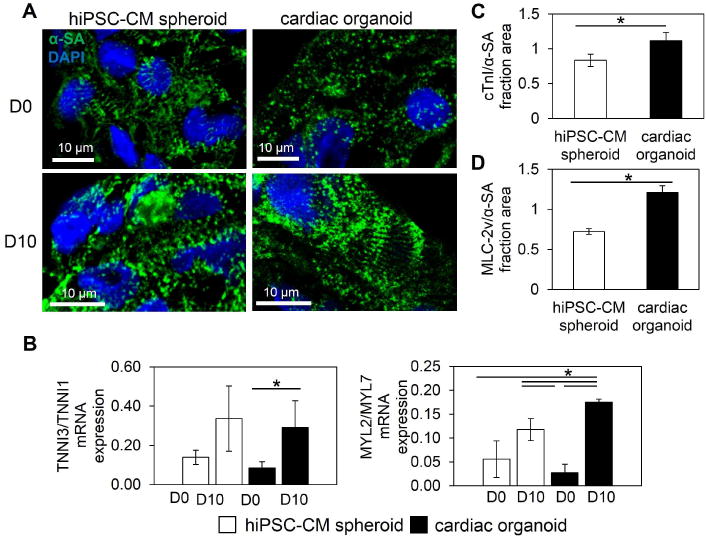
Human cardiac organoids support contractile development of hiPSC-CMs. (A) Cardiac organoids showed signs of improved sarcomere development (e.g., Z-line width) by D10 in comparison to hiPSC-CM spheroids. (B) Ratiometric gene expression of hiPSC-CM spheroids and cardiac organoids reveal improvements in hiPSC-CM development within microtissues. Gene expression of cardiac troponin I (TNNNI3) over slow, skeletal troponin I (TNNNI1) showed greater significant increases in cardiac organoids, indicating improved contractile maturation of hiPSC-CMs by D10. Ratiometric expression of myosin light chain ventricular isoform (MYL2) over atrial isoform (MYL7) showed more significant increases by D10 in cardiac organoids, indicating improved ventricular specification of hiPSC-CMs; n=3 experiments, 10–15 microtissues/experiment. (C,D) Shifts in gene ratios were supported by increased cardiac troponin I (cTnI)-positive and ventricular myosin light chain (MLC-2v)-positive fraction area over α-sarcomeric actinin (α-SA)-positive area (hiPSC-CM-specific areas), represented by low and high magnification immunofluorescent images; n=3 microtissues. Cardiac organoids were fabricated using a developing stage cell ratio. hiPSC-CM-human induced pluripotent stem cell-derived cardiomyocyte. Asterisk represents significant difference, p<0.05.
To further evaluate contractile improvements in cardiac organoids, contractile maturation and cardiomyocyte subtype specification (i.e., ventricular) were examined. As an indicator of contractile maturation (80) and to evaluate hiPSC-CM-specific development, ratiometric gene expression of adult cardiac troponin I and immature troponin I (i.e., TNNI3/TNNI1) revealed a significant increase in TNNI3/TNNI1 in cardiac organoids and an insignificant increase in hiPSC-CM spheroids from D0 to D10 (Figure 6B). This was further supported by significantly higher density of cardiac troponin I (cTnI) expression in the cardiac organoids than hiPSC-CM spheroids, when normalized to the α-SA-positive area of stained spheroid sections (Figure 6C, S10B). To examine the cardiac organoids for working myocardium development, ventricular myosin light chain (protein:MLC2v, gene:MYL2) and atrial myosin light chain (protein:MLC2a, gene:MYL7) were used for ratiometric gene expression. At D10 both cardiac organoids and hiPSC-CM spheroids showed increased ventricular/atrial (i.e., MYL2/MYL7) expression, though cardiac organoids showed a significantly greater shift toward ventricular gene expression (Figure 6B). In addition, the immunofluorescent expression of MLC2v showed increased staining density per α-SA-positive regions in cardiac organoids over hiPSC-CM spheroids (Figure 6D, S10C). Although both groups show spontaneous beating throughout the experiment, the observed enhanced sarcomere development, increased adult cardiac troponin I expression, and a greater ventricular shift in expression supports the development of hiPSC-CMs in cardiac organoids.
Interestingly, the contraction amplitude of cardiac organoids is significantly lower than hiPSC-CM spheroids (0.61 ± 0.25% vs. 1.98 ± 1.2%), despite having similar spontaneous beat rates (cardiac organoids: 36.9 ± 8.0 beats per minute (bpm) vs. hiPSC-CM spheroids: 33.8 ± 3.4 bpm) (Figure S11). The low contraction amplitude of cardiac organoids is possibly due to the increased extracellular matrix components (e.g., collagen) and interstitial cell types, resulting in a denser/stiffer construct that creates an increased load for cardiomyocyte contraction. This was supported by the improved contractile structure and gene profile in cardiac organoids despite low contraction amplitude, highlighting the importance of the multicellular and matrix environment for hiPSC-CM maturation and ventricular lineage specification seen by others (26, 81). It is worth noting that the observed relationship between cardiomyocyte function and changes in cellular/ECM components is equally important to cardiac development and maturation as it is to cardiac pathophysiology, such as in chronic cardiac fibrosis (82, 83). The previously mentioned data from Figure 2D and 2E (showing decreased contraction amplitude with increased fibroblast number) also supports these tissue-level dynamics and offers evidence of possible pathological applications within a tunable cardiac organoid system.
Human cardiac organoids display phenotypic responses to pharmacological compounds
For a functional cardiac organoid, the observed phenotypic structure, organization, and contractile development in D10 cardiac organoids must also be supported by demonstrated functions of fundamental characteristics of cardiac tissue. Given that calcium handling in cardiomyocytes is involved in electrical signal generation and propagation and is intimately involved in the contractile apparatus (40), the explored improvements in contractile characteristics of cardiac organoids suggested functioning calcium-handling properties. Membrane calcium channels (e.g., L-type calcium channel) contribute to the initiation of the calcium-induced calcium-release (CICR) phenomenon in cardiomyocytes. Verapamil is a multi-channel blocker that causes a decreased calcium transient peak due to a large calcium L-type channel blockade. However, it has served as a false-positive arrhythmogenic drug in hERG channel based arrhythmogenic drug screening (84). Administration of 500 μM of verapamil resulted in a significant decrease in peak fluorescence and no observed arrhythmogenic activity (Figure 7A). This response validates the phenotypic functionality of L-type calcium channels to support CICR dynamics for contraction. The initial inward flux of calcium then binds to ryanodine receptors on the sarcoplasmic reticulum that release the intracellular stores of calcium located in the sarcoplasmic reticulum to provide enough calcium for contraction. To examine the calcium release functionality of cardiac organoids, the ryanodine receptor was blocked using 50 μM ryanodine. Ryanodine receptor blockade resulted in a significant decrease in peak calcium transient (Figure 7B). Also, ryanodine administration significantly increased time to peak calcium, indicating proper function of the ryanodine receptor to regulate the intracellular flux of calcium required for contraction. These results demonstrated the cardiac organoids recapitulate important calcium handling properties of working human myocardium.
Figure 7.
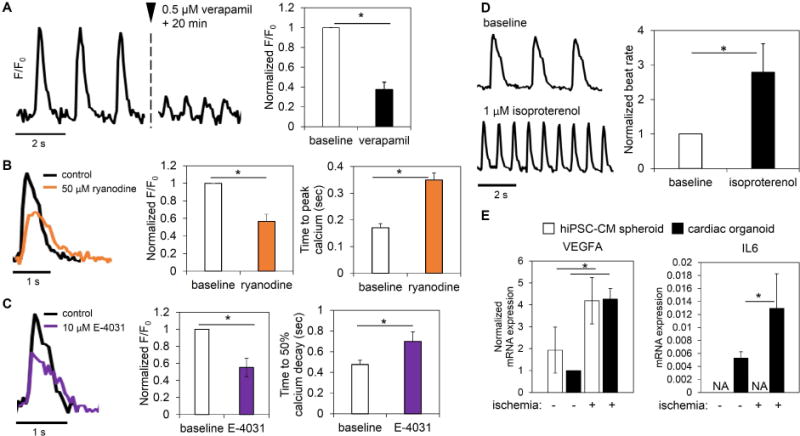
Human cardiac organoids display phenotypic channel functionality and organotypic responses to physiological/pathological stimuli. Cardiac organoids showed functional calcium handling. (A) Verapamil, which blocks the L-type calcium channel, displayed significant phenotypic reduction of calcium transient amplitude without inducing arrhythmia in cardiac organoids; n=4 organoids. (B) The use of ryanodine, known to block the ryanodine receptor of the sarcoplasmic reticulum and reduce endogenous calcium release, showed a significant decrease in calcium transient amplitude and a significant increase time to peak calcium; n=3 organoids. (C) Blockade of the hERG potassium channel using E-4031 showed a significant drop in calcium transient peak and a significant increase in time to 50% calcium decay; n=3 organoids. (D) A significant increase in beat rate after application of isoproterenol indicated intact β-adrenergic signaling in cardiac organoids; n=5 organoids. (E) Cardiac organoids showed organotypic transcription responses to ischemic stimuli; n=3 experiments; 10–15 microtissues/experiment. Cardiac organoids and hiPSC-CM spheroids showed increased VEGFA expression in response to ischemic stimulus. IL6 expression, a prominent interleukin in ischemic heart failure, increased in cardiac organoids in ischemia, while it was not detected in hiPSC-CM spheroids. hiPSC-CM-human induced pluripotent stem cell-derived cardiomyocytes. Cardiac organoids were fabricated using a developing stage cell ratio and were used at D10. Asterisk represents significant difference, p<0.05.
To counter the flux of calcium during contraction, potassium channels, such as the hERG potassium channel, play a large role in repolarization phase of the cardiac action potential (40). To examine the repolarization functionality of cardiac organoids, hERG potassium channels were blocked with 10 μM E-4031. By blocking a major potassium channel, repolarization is slowed, indicated by an extension in calcium transient decay time. Blockade of the hERG channel resulted in significantly decreased peak calcium and a significant phenotypic extension of the time to 50% calcium decay (Figure 7C). Together with calcium handling results, these data showed the cardiac organoids supported the proper development of tissue-level channel functions.
Human cardiac organoids display phenotypic functionality under physiological and pathological stimuli
A series of physiologically relevant stimuli showed indications of a wider range of tissue-level functionality of cardiac organoids beyond drug screening and electrical property development. Beta-adrenergic signaling is a physiological process of beat rate regulation by the autonomic nervous system by catecholamines (85). Using video analysis of the beating rate of cardiac organoids, a beta-adrenergic agonist, isoproterenol (1 μM), showed a significant increase in beat rate (Figure 7D). This validates the intact beta-adrenergic signaling pathways in hiPSC-CMs within the cardiac organoids for physiologically relevant stimulation.
Despite immature characteristics of cardiac organoids compared to adult ventricular tissue, the presence of fundamental physiological cardiac functions provided a basis for exploring critical pathways involved in cardiac pathologies, supported by recent progress using hiPSC-CMs for cardiac pathophysiology insights (12, 86–89). Particularly, cardiac organoids have potential to serve as an in vitro cardiac model for ischemic heart failure, which makes up the majority of cardiovascular-related disorders (90). To evaluate the initial response to a pathological stimulus, cardiac organoids and hiPSC-CM spheroids were cultured for 6 hrs in ischemia. While the VEGFA expression in cardiac organoid is lower than the hiPSC-CM spheroids in a normoxia environment, ischemic stimulus led to a significant increase in VEGF-A gene expression in the cardiac organoids to similar levels to that of hiPSC-CM spheroids (Figure 7E). Spheroids of each cell type all showed increases in VEGFA expression under ischemic conditions (Figure S12A). hiPSC-CMs and cFBs showed the highest levels of ischemic VEGFA expression, suggesting their dominant role in VEGF signaling during ischemic stress. This trend is analogous to increases in VEGF expression in ischemic cardiac tissue and further supports the biomimetic regulation of VEGFA expression in cardiac organoids (91). Furthermore, the addition of non-cardiomyocytes into the cardiac organoids system incorporated an extended inflammatory response, namely, IL-6 gene expression, a prominent interleukin up-regulated in heart failure. After ischemic insult, IL6 expression increased significantly in cardiac organoids and was absent in CM spheroids (Figure 7E). This was attributed the presence of cFB in the organoids as single cell spheroid evaluation showed cFBs with the highest levels of IL-6 gene expression in response to ischemic stimuli (Figure S12B). Drug-based and biomimetic stimulation of cardiac organoids can provide a fundamental and potentially clinically relevant platform to explore the critical pathways involved in cardiac pathophysiology.
CONCLUSION
With inspiration from major events in coronary vasculogenesis and a defined method of microtissue fabrication, we have developed a robust platform to fabricate vascularized human cardiac organoids that recapitulated the lumenized vascular network of the developing myocardium, supported hiPSC-CM development and demonstrated fundamental cardiac tissue-level functions. In contrast to the common organoid fabrication approach of directed differentiation of embryoid bodies (EBs), the dynamic cellular organization surrounding coronary vasculogenesis provided the developmental basis for the use of defined cell types to develop a tunable cardiac organoid system. This platform provides a consistent and efficient method of human cardiac organoid fabrication (e.g., 100% beating, consistent cellular organization, and minimal size variability) that supports high throughput applications (e.g., drug testing) in comparison to cardiovascular EB differentiation, where there is high variability in size and morphology with only ~2%–15% of EBs showing contractile beating (8). Furthermore, while hiPSC-CMs have been extensively used to model cardiac disorders, other cardiac cell types, such as cardiac fibroblasts and endothelial cells, are involved in many cardiac pathologies (92, 93). The use of a defined cardiac cell mixture for cardiac organoid formation is advantageous by providing an adaptable system to incorporate and investigate the contribution of each cardiac cell type, matrix materials, additional factors, and other physiological system relationships (e.g., immunological cell types) to cardiac developmental/pathological dynamics at a tissue/organ level.
In addition, similar to developing cardiomyocytes, the spontaneous beating property of hiPSC-CMs supports contractile development and provided the source of pacing in the cardiac organoids. To obtain adult stage ventricular maturation, in which cardiomyocytes do not beat spontaneously, exogenous pacing may be required. Interestingly, recent optimizations in stem cell-derived cardiomyocyte media suggest that mature contractile characteristics, such as positive force-frequency, can be achieved through the supply of defined growth factors (22). Looking toward future applications, cardiac organoids with lumenized vascular networks provide a tunable foundation to develop future pre-vascularized, injectable vehicles for regenerative cell therapy to treat heart failure. In summary, the human cardiac organoid technology presented here provides a robust platform for the development of the next generation of hiPSC-based cardiac organoids in pursuit of developmental and pathological insight for regenerative medicine applications.
MATERIAL AND METHODS
Cell culture
Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) (iCell Cardiomyocytes, Cellular Dynamics International-CDI, Madison, WI, USA) were cultured according to the manufacturer’s protocol. Briefly, hiPSC-derived cardiomyocytes were plated on 0.1% gelatin coated 6-well plates in iCell Cardiomyocyte Plating Medium (CDI) and incubated at 37 °C in 5% CO2 for 4 days. Two days after plating, the plating medium was removed and replaced with 4 mL of iCell Cardiomyocytes Maintenance Medium (CDI). After 4 days of monolayer pre-culture, cells were detached using trypLE Express (Gibco Life Technologies, Grand Island, NY) and prepared for spheroid/organoid fabrication. Human cardiac ventricular fibroblasts (cFBs) (Lonza, Basel, Switzerland) were cultured in FGM-3 media (Lonza) were used at passage 3–4 for spheroid/organoid fabrication. Human umbilical vein endothelial cells (HUVECs) (Lonza) were cultured in EGM-2 media (Lonza) and were used at passage 2–3 for spheroid/organoid fabrication. Human adipose-derived microvascular endothelial cells (HAMECs) (kind gift from Dr. Michael Yost) and were cultured in EGM-2 media (Lonza) and were used at passage 5–6 for organoid fabrication. Human adipose-derived stem cells (hADSCs) (Lonza) were cultured in low glucose Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin, 1% glutamine and 1% antimycin (Gibco Life Technologies, Grand Island, NY). hADSCs were used at passage 3–4 for spheroid/organoid fabrication. For cFBs, hADSCs, and HUVECs, media was changed every 2 days and were passaged using trypLE Express (Gibco) at >80% confluency. An array of media were evaluated to best support consistent organoid formation and culture, shown in Figure S1. The optimal culture media for viable cardiac organoids was comprised of a ratiometric combination of cell-specific media reflecting the cell ratio of the organoid. In organoid media, CDI hiPSC-CM Maintenance Media (supplied without glucose) was substituted with glucose-containing DMEM/F12 media with 10% FBS and 1% non-essential amino acids (Gibco).
Organoid and spheroid fabrication
As described in our previous publication (79), the agarose hydrogel molds were prepared using 2% agarose (Sigma Aldrich, St. Louis, MO) and master micro-molds from Microtissues, Inc (Providence, RI) as negative replicates to create non-adhesive agarose hydrogels molds containing 35 microwells with hemispheric bottoms (800 μm diameter, 800 μm deep) to facilitate the formation of spherical microtissues. Molds were soaked in cell/organoid specific media prior to microtissue fabrication. Working cell suspensions of each cell type were used at ~4.0 × 106 cells/mL to make organoid cell ratio mixtures and mixed with 1 volume media for a final concentration of ~2.0 × 106 cells/mL. Approximately 75 μl of the cell suspension was pipetted into each agarose mold. After the cells settled into the recesses of the mold (10 min), additional media was added to submerge the molds in an 8-well plate and exchanged every 2 days for the length of the experiment (10 days). Day 0 (D0) of the experiment was marked after 4 days of spheroid assembly. A scheme of the experimental timeline can be found in Figure S1. TUNEL staining for apoptotic nuclei revealed that D0 cardiac organoids maintained high viability throughout the microtissue (Figure S3).
Contraction analysis of beating spheroids
Videos of spontaneously beating spheroids from each group were recorded at ~37 °C for each conditi on using a Carl Zeiss Axiovert A1 Inverted Microscope and Zen 2011 software (Zeiss, Göttingen, Germany). Videos were converted to a series of TIFF format pictures by Adobe Premiere (Adobe, San Jose, CA). Threshold edge-detecting in ImageJ software (National Institutes of Health) was used on high contrast spheroid picture series and graphed to realize beating profiles of fractional area change (i.e., contraction amplitude), from which beats per minute and contraction amplitude was calculated. Contraction amplitudes were calculated as the percent change in fractional area change amplitude between contraction and relaxation.
Immunofluorescent analysis
Freshly collected spheroids were flash frozen in Tissue-Tek OCT compound (Sakura, Torrance, CA). Embedded spheroids were cryosectioned into 7 μm sections onto glass slides. The sections were fixed with cooled acetone (−20 °C) for 10 min. After washing (2 times at 5 min) in PBS with 0.1% Triton X-100 (PBST) (Sigma), blocking buffer was made with 10% serum corresponding to host species of secondary antibody in PBST and added to sections for 1 hr at room temperature. Sections were incubated with primary antibody diluted in PBST (1:200) overnight at 4 °C or 2 hrs at room temperature: rabbit anti-laminin (Sigma), rabbit-anti collagen type I (Abcam), rabbit anti-fibronectin (Abcam), mouse anti-alpha sarcomeric actinin (Abcam), rabbit anti-cardiac troponin I – cTnI (Abcam), rabbit anti-MYL2 (MLC-2v) (Abcam), rabbit anti-vimentin (Abcam), mouse anti-CD31 (BD Biosciences, San Jose, CA), rabbit anti-von Willebrand factor (vWF) (Abcam). After washing in PBST (2 times at 5 min), sections were incubated with complement secondary antibodies diluted in PBST for 1 hr at room temperature: goat anti-mouse Alexa Fluor 546 (Thermo), goat anti-rabbit Alexa Fluor 647 (Jackson ImmunoResearch, West Grove, PA). After washing in PBST (2 times at 5 min), nuclei were counterstained with DAPI (Molecular Probes/Invitrogen, Eugene, OR) diluted in PBST for 15 min at room temperature. Following the final wash procedure (PBST, 2 times at 5 min), glass cover slips were added to the slides using Fluoro-Gel (Electron Microscopy Sciences, Hatfield, PA). TCS SP5 AOBS laser scanning confocal microscope (Leica Microsystems, Inc., Exton, PA) was used for imaging. Imaris software (Bitplane, Zurich, Switzerland) was used to reconstruct z-stacked confocal images for 3D visualization. Fluorescent protein expression was calculated as the antibody-positive fluorescence area coverage: endothelial density was calculated as vWF-positive covered area divided by spheroid area, lumens per organoid a was calculated as the number of vWF-positive ring-shaped structures with adjacent DAPI-stained nuclei per organoid, vimentin density was the ratio of vimentin-positive area within the α-SA-positive area, ECM fraction area was calculated as the ECM-stain positive area divided by the spheroid area, and the relative contractile protein amount was calculated as the ratio of cTnI or MLC-2v area to total α-SA covered area. Each analysis consisted of high resolution images at 400× total magnification of 3–5 spheroids. Hematoxylin and eosin-stained embryonic mouse heart sections were a kind gift from the lab of Dr. Russell Norris.
TUNEL staining for apoptosis
Roche In Situ Cell Death Detection Kit (Sigma) was used to visualize the viability of cells in frozen sections of cardiac organoids based on the Roche protocol. Briefly, cardiac organoid frozen sections were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Following washing in PBS for 30 minutes, samples were incubated in a permeabilization solution (0.1% Triton X-100 and 0.1% sodium citrate in PBS) for 2 minutes on ice. Then 50 μl of the TUNEL reaction mixture were added to samples and incubated at 37 °C for 1 hr. After washing in PBS (2 times at 5 min), nuclei were counterstained with DAPI (Molecular Probes/Invitrogen) diluted in PBS for 15 min at ambient temperature. Following the final wash procedure (PBS, 2 times at 5 min), glass cover slips were added to the slides using Fluoro-Gel (Electron Microscopy Sciences). TCS SP5 AOBS laser scanning confocal microscope (Leica Microsystems) was used for imaging. TUNEL-based viability index was calculated as [1 – (TUNEL-positive area/DAPI-positive area)].
Gene expression with qRT-PCR
Total RNA was isolated according to the kit and protocol of an RNeasy Micro Kit (Qiagen, Vinlo, Netherlands) with the addition of the Homogenizer Columns (Omega Biotek, Norcross, GA) during the homogenization step for spheroids. For each group, a minimum of 10 spheroids were used for RNA isolation. At least 25 ng of total RNA for each group was subjected to cDNA synthesis using the Bio-Rad (Hercules, USA) iScript cDNA synthesis kit. qRT-PCR step was performed using validated Life Technologies Taqman primers (Thermo Fisher Scientific) in 10 μl reactions for targeted genes: ACTB (Hs01060665_g1), GAPDH (Hs02758991_g1), IL6 (Hs00985639_m1), KDR (VEGFR2, Hs00911700_m1), MYL2 (Hs00166405_m1), MYL7 (Hs01085598_g1), TNNI1 (Hs00913333_m1), TNNI3 (Hs00165957_m1), VEGFA (Hs00900055_m1). Data was normalized as the change in cycle threshold (Ct) to the geometric mean of GAPDH and ACTB (dCt) and analyzed using, mRNA expression = 2ˆ(-dCt). Data was then averaged across n=3 experiments.
Calcium transient imaging
Life Technologies’s Fluo-4 Direct Calcium Assay Kit (Life Technologies, Carlsbad, CA) was used to label calcium in the whole organoids based on the manufacturer protocol. Briefly, organoids stained with a working solution of 1:1 calcium dye solution to media and incubated at 37 °C, 5% CO2, 20% O2 for 30 min. Carl Zeiss Axiovert A1 Inverted Microscope completed with GFP fluorescence imaging capacity (Zeiss) was used to collect the videos of the calcium transient of whole spheroids with a capture rate of 20 frames per second. Finally, we used Zen 2011 software (Zeiss) to convert videos to a series of TIFF format pictures. Mean gray value of the whole organoid calcium transient (controlled for area for pre- and post-treatment) was measured using ImageJ software (National Institutes of Health) to construct calcium transient profiles. Normalized F/F0 was calculated as the peak calcium fluorescence (F) divided by the start fluorescence level (F0), then divided by the F/F0 of the baseline transient before treatment. Time to peak calcium (sec) was calculated as the difference in time between the peak calcium and the start of the calcium transient. Time to 50% calcium decay (sec) was calculated as the difference in time between the start of the calcium transient and the half-peak (F50) of the calcium decay.
Evaluation of putative vasculature
Oxygen reduction studies were performed at 10% O2 in a hypoxia chamber (C-Chamber, BioSpherix, Parish, NY) for a duration of 10 days. Media was exchanged every 2 days. Microtissues were embedded in OCT, cryosectioned, and Roche In Situ Cell Death Detection Kit (Sigma) was used to visualize the viability of cells in frozen sections of cardiac organoids based on the Roche protocol. Macrotissue assembly was performed in agarose-coated (non-adhesive) 96-well plates. Five cardiac organoids (developing stage cell ratio) were placed in each well and culture for 10 days until the macrotissues had an appreciable Z-dimension, observed by pipette agitation. Organoid aggregate macrotissues were embedded in OCT and cryosectioned for immunofluorescent analysis. Media was exchanged every day.
Pharmacological, physiological, and pathological stimulus
Pharmacological compounds were used to block channels of human cardiac organoids and were performed on single organoids in ultra low-adhesive GravityTRAP Plates (InSphero, Schlieren, Switzerland) at 37 °C. Freshly prepared stock solutions of verapamil (Sigma), ryanodine, and E-4031 were diluted in the calcium buffer provided in the calcium staining kit (Life Technologies). Verapamil (0.5 μM), ryanodine (50 μM), and E-4031 (10 μM) added to the organoids and calcium signal was video captured after 20 mins. Isoproterenol (1 μM) was added for 10 mins before bright field videos were captured. For ischemic stimulus, microtissues were rinsed in low glucose DMEM (1 g/L) (Gibco) and placed in agarose molds in serum-free, low glucose (1 g/L) DMEM and 1% O2 (BioSpherix) for 6 hrs to examine the initial response. Controls were cultured at normoxia in cell/organoid-specific media.
Statistics Analysis
Differences between experimental groups were analyzed on JMP Pro 12 Statistical software (SAS, Cary, NC) using Student’s t-test, matched pairs comparison, one-way ANOVA, and two-way ANOVA with Tukey’s post-hoc test, and p<0.05 was considered significantly difference for all statistical tests.
Supplementary Material
Acknowledgments
The work is supported by the National Institutes of Health (8P20 GM103444, U54 GM104941), the startup funds from Clemson University, the National Science Foundation (NSF - EPS-0903795), the NIH Cardiovascular Training Grant (T32 HL007260), and US Department of Veterans Affairs Merit Review (I01 BX002327). This study used the services of the Morphology, Imaging, and Instrumentation Core, which is supported by NIH-NIGMS P30 GM103342 to the South Carolina COBRE for Developmentally Based Cardiovascular Diseases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clevers H. Modeling Development and Disease with Organoids. Cell. 2016;165(7):1586–97. doi: 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- 2.Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345(6194):1247125. doi: 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- 3.Baker LA, Tiriac H, Clevers H, Tuveson DA. Modeling pancreatic cancer with organoids. Trends Cancer. 2016;2(4):176–90. doi: 10.1016/j.trecan.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499(7459):481–4. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 5.Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526(7574):564–8. doi: 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- 6.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–9. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dye BR, Dedhia PH, Miller AJ, Nagy MS, White ES, Shea LD, et al. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. eLife. 2016;5 doi: 10.7554/eLife.19732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shkumatov A, Baek K, Kong H. Matrix rigidity-modulated cardiovascular organoid formation from embryoid bodies. PloS one. 2014;9(4):e94764. doi: 10.1371/journal.pone.0094764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takebe T, Enomura M, Yoshizawa E, Kimura M, Koike H, Ueno Y, et al. Vascularized and Complex Organ Buds from Diverse Tissues via Mesenchymal Cell-Driven Condensation. Cell stem cell. 2015;16(5):556–65. doi: 10.1016/j.stem.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Gunter J, Wolint P, Bopp A, Steiger J, Cambria E, Hoerstrup SP, et al. Microtissues in Cardiovascular Medicine: Regenerative Potential Based on a 3D Microenvironment. Stem Cells Int. 2016;2016:9098523. doi: 10.1155/2016/9098523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsa E, Ahrens JH, Wu JC. Human Induced Pluripotent Stem Cells as a Platform for Personalized and Precision Cardiovascular Medicine. Physiol Rev. 2016;96(3):1093–126. doi: 10.1152/physrev.00036.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsa E, Burridge PW, Yu KH, Ahrens JH, Termglinchan V, Wu H, et al. Transcriptome Profiling of Patient-Specific Human iPSC-Cardiomyocytes Predicts Individual Drug Safety and Efficacy Responses In Vitro. Cell stem cell. 2016 doi: 10.1016/j.stem.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127(16):1677–91. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogle BM, Bursac N, Domian I, Huang NF, Menasche P, Murry CE, et al. Distilling complexity to advance cardiac tissue engineering. Science translational medicine. 2016;8(342) doi: 10.1126/scitranslmed.aad2304. 342ps13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mannhardt I, Breckwoldt K, Letuffe-Breniere D, Schaaf S, Schulz H, Neuber C, et al. Human Engineered Heart Tissue: Analysis of Contractile Force. Stem cell reports. 2016;7(1):29–42. doi: 10.1016/j.stemcr.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards DJ, Tan Y, Coyle R, Li Y, Xu R, Yeung N, et al. Nanowires and Electrical Stimulation Synergistically Improve Functions of hiPSC Cardiac Spheroids. Nano letters. 2016;16(7):4670–8. doi: 10.1021/acs.nanolett.6b02093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eng G, Lee BW, Protas L, Gagliardi M, Brown K, Kass RS, et al. Autonomous beating rate adaptation in human stem cell-derived cardiomyocytes. Nature communications. 2016;7:10312. doi: 10.1038/ncomms10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thavandiran N, Dubois N, Mikryukov A, Masse S, Beca B, Simmons CA, et al. Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(49):E4698–707. doi: 10.1073/pnas.1311120110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens KR, Kreutziger KL, Dupras SK, Korte FS, Regnier M, Muskheli V, et al. Physiological function and transplantation of scaffold-free and vascularized human cardiac muscle tissue. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(39):16568–73. doi: 10.1073/pnas.0908381106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ribas J, Sadeghi H, Manbachi A, Leijten J, Brinegar K, Zhang YS, et al. Cardiovascular Organ-on-a-Chip Platform for Drug Discovery and Development. Applied In Vitro Toxicology. 2016;2(2):82–96. doi: 10.1089/aivt.2016.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathur A, Loskill P, Shao K, Huebsch N, Hong S, Marcus SG, et al. Human iPSC-based Cardiac Microphysiological System For Drug Screening Applications. Scientific reports. 2015;5:8883. doi: 10.1038/srep08883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiburcy M, Hudson JE, Balfanz P, Schlick SF, Meyer T, Chang Liao ML, et al. Defined Engineered Human Myocardium with Advanced Maturation for Applications in Heart Failure Modelling and Repair. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.024145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelm JM, Djonov V, Hoerstrup SP, Guenter CI, Ittner LM, Greve F, et al. Tissue-transplant fusion and vascularization of myocardial microtissues and macrotissues implanted into chicken embryos and rats. Tissue Eng. 2006;12(9):2541–53. doi: 10.1089/ten.2006.12.2541. [DOI] [PubMed] [Google Scholar]
- 24.Kelm JM, Ehler E, Nielsen LK, Schlatter S, Perriard JC, Fussenegger M. Design of artificial myocardial microtissues. Tissue Eng. 2004;10(1–2):201–14. doi: 10.1089/107632704322791853. [DOI] [PubMed] [Google Scholar]
- 25.Hussain A, Collins G, Yip D, Cho CH. Functional 3-D cardiac co-culture model using bioactive chitosan nanofiber scaffolds. Biotechnology and bioengineering. 2013;110(2):637–47. doi: 10.1002/bit.24727. [DOI] [PubMed] [Google Scholar]
- 26.Burridge PW, Metzler SA, Nakayama KH, Abilez OJ, Simmons CS, Bruce MA, et al. Multi-cellular interactions sustain long-term contractility of human pluripotent stem cell-derived cardiomyocytes. American journal of translational research. 2014;6(6):724–35. [PMC free article] [PubMed] [Google Scholar]
- 27.Kaneko T, Nomura F, Yasuda K. On-chip constructive cell-network study (I): contribution of cardiac fibroblasts to cardiomyocyte beating synchronization and community effect. J Nanobiotechnology. 2011;9:21. doi: 10.1186/1477-3155-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nichol JW, Engelmayr GC, Jr, Cheng M, Freed LE. Co-culture induces alignment in engineered cardiac constructs via MMP-2 expression. Biochemical and biophysical research communications. 2008;373(3):360–5. doi: 10.1016/j.bbrc.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts MA, Tran D, Coulombe KL, Razumova M, Regnier M, Murry CE, et al. Stromal Cells in Dense Collagen Promote Cardiomyocyte and Microvascular Patterning in Engineered Human Heart Tissue. Tissue engineering Part A. 2016;22(7–8):633–44. doi: 10.1089/ten.tea.2015.0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saini H, Navaei A, Van Putten A, Nikkhah M. 3D cardiac microtissues encapsulated with the co-culture of cardiomyocytes and cardiac fibroblasts. Advanced healthcare materials. 2015;4(13):1961–71. doi: 10.1002/adhm.201500331. [DOI] [PubMed] [Google Scholar]
- 31.Kelm JM, Diaz Sanchez-Bustamante C, Ehler E, Hoerstrup SP, Djonov V, Ittner L, et al. VEGF profiling and angiogenesis in human microtissues. J Biotechnol. 2005;118(2):213–29. doi: 10.1016/j.jbiotec.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 32.Garzoni LR, Rossi MI, de Barros AP, Guarani V, Keramidas M, Balottin LB, et al. Dissecting coronary angiogenesis: 3D co-culture of cardiomyocytes with endothelial or mesenchymal cells. Exp Cell Res. 2009;315(19):3406–18. doi: 10.1016/j.yexcr.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Smart N, Dube KN, Riley PR. Coronary vessel development and insight towards neovascular therapy. Int J ExpPathol. 2009;90(3):262–83. doi: 10.1111/j.1365-2613.2009.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dyer L, Pi X, Patterson C. Connecting the coronaries: how the coronary plexus develops and is functionalized. Dev Biol. 2014;395(1):111–9. doi: 10.1016/j.ydbio.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Olivey HE, Svensson EC. Epicardial-myocardial signaling directing coronary vasculogenesis. Circulation research. 2010;106(5):818–32. doi: 10.1161/CIRCRESAHA.109.209197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reese DE, Mikawa T, Bader DM. Development of the coronary vessel system. Circulation research. 2002;91(9):761–8. doi: 10.1161/01.res.0000038961.53759.3c. [DOI] [PubMed] [Google Scholar]
- 37.de Bakker BS, de Jong KH, Hagoort J, Oostra RJ, Moorman AF. Towards a 3-dimensional atlas of the developing human embryo: the Amsterdam experience. ReprodToxicol. 2012;34(2):225–36. doi: 10.1016/j.reprotox.2012.05.087. [DOI] [PubMed] [Google Scholar]
- 38.de Boer BA, van den Berg G, de Boer PA, Moorman AF, Ruijter JM. Growth of the developing mouse heart: an interactive qualitative and quantitative 3D atlas. Dev Biol. 2012;368(2):203–13. doi: 10.1016/j.ydbio.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Wong MD, van Eede MC, Spring S, Jevtic S, Boughner JC, Lerch JP, et al. 4D atlas of the mouse embryo for precise morphological staging. Development. 2015;142(20):3583–91. doi: 10.1242/dev.125872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van den Heuvel NH, van Veen TA, Lim B, Jonsson MK. Lessons from the heart: mirroring electrophysiological characteristics during cardiac development to in vitro differentiation of stem cell derived cardiomyocytes. Journal of molecular and cellular cardiology. 2014;67:12–25. doi: 10.1016/j.yjmcc.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 41.Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circulation research. 2014;114(3):511–23. doi: 10.1161/CIRCRESAHA.114.300558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madden L, Juhas M, Kraus WE, Truskey GA, Bursac N. Bioengineered human myobundles mimic clinical responses of skeletal muscle to drugs. eLife. 2015;4 doi: 10.7554/eLife.04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nature methods. 2013;10(8):781–7. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackman CP, Carlson AL, Bursac N. Dynamic culture yields engineered myocardium with near-adult functional output. Biomaterials. 2016;111:66–79. doi: 10.1016/j.biomaterials.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adler CP. Relationship between deoxyribonucleic acid content and nucleoli in human heart muscle cells and estimation of cell number during cardiac growth and hyperfunction. Recent Adv Stud Cardiac Struct Metab. 1975;8:373–86. [PubMed] [Google Scholar]
- 46.Adler CP, Friedburg H. Myocardial DNA content, ploidy level and cell number in geriatric hearts: post-mortem examinations of human myocardium in old age. Journal of molecular and cellular cardiology. 1986;18(1):39–53. doi: 10.1016/s0022-2828(86)80981-6. [DOI] [PubMed] [Google Scholar]
- 47.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. American journal of physiology Heart and circulatory physiology. 2007;293(3):H1883–91. doi: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 48.Bian W, Badie N, Himel HDt, Bursac N. Robust T-tubulation and maturation of cardiomyocytes using tissue-engineered epicardialmimetics. Biomaterials. 2014;35(12):3819–28. doi: 10.1016/j.biomaterials.2014.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rubina K, Kalinina N, Efimenko A, Lopatina T, Melikhova V, Tsokolaeva Z, et al. Adipose stromal cells stimulate angiogenesis via promoting progenitor cell differentiation, secretion of angiogenic factors, and enhancing vessel maturation. Tissue engineering Part A. 2009;15(8):2039–50. doi: 10.1089/ten.tea.2008.0359. [DOI] [PubMed] [Google Scholar]
- 50.Strassburg S, Nienhueser H, Bjorn Stark G, Finkenzeller G, Torio-Padron N. Co-culture of adipose-derived stem cells and endothelial cells in fibrin induces angiogenesis and vasculogenesis in a chorioallantoic membrane model. Journal of tissue engineering and regenerative medicine. 2016;10(6):496–506. doi: 10.1002/term.1769. [DOI] [PubMed] [Google Scholar]
- 51.Gimble JM, Katz AJ, Bunnell BA. Adipose-derived stem cells for regenerative medicine. Circulation research. 2007;100(9):1249–60. doi: 10.1161/01.RES.0000265074.83288.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell stem cell. 2008;3(3):301–13. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Caplan AI. The new MSC: MSCs as pericytes are sentinels and gatekeepers. J Orthop Res. 2017 doi: 10.1002/jor.23560. [DOI] [PubMed] [Google Scholar]
- 54.Caplan AI. All MSCs are pericytes? Cell stem cell. 2008;3(3):229–30. doi: 10.1016/j.stem.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 55.Zhang W, Kong CW, Tong MH, Chooi WH, Huang N, Li RA, et al. Maturation of human embryonic stem cell-derived cardiomyocytes (hESC-CMs) in 3D collagen matrix: Effects of niche cell supplementation and mechanical stimulation. ActaBiomater. 2017;49:204–17. doi: 10.1016/j.actbio.2016.11.058. [DOI] [PubMed] [Google Scholar]
- 56.Steinberg MS. Differential adhesion in morphogenesis: a modern view. CurrOpin Genet Dev. 2007;17(4):281–6. doi: 10.1016/j.gde.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 57.Tomanek RJ, Ratajska A, Kitten GT, Yue X, Sandra A. Vascular endothelial growth factor expression coincides with coronary vasculogenesis and angiogenesis. DevDyn. 1999;215(1):54–61. doi: 10.1002/(SICI)1097-0177(199905)215:1<54::AID-DVDY6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 58.Radisic M, Malda J, Epping E, Geng W, Langer R, Vunjak-Novakovic G. Oxygen gradients correlate with cell density and cell viability in engineered cardiac tissue. Biotechnology and bioengineering. 2006;93(2):332–43. doi: 10.1002/bit.20722. [DOI] [PubMed] [Google Scholar]
- 59.Folkman J, Hochberg M. Self-regulation of growth in three dimensions. J Exp Med. 1973;138(4):745–53. doi: 10.1084/jem.138.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caspi O, Lesman A, Basevitch Y, Gepstein A, Arbel G, Habib IH, et al. Tissue engineering of vascularized cardiac muscle from human embryonic stem cells. Circulation research. 2007;100(2):263–72. doi: 10.1161/01.RES.0000257776.05673.ff. [DOI] [PubMed] [Google Scholar]
- 61.Kreutziger KL, Muskheli V, Johnson P, Braun K, Wight TN, Murry CE. Developing vasculature and stroma in engineered human myocardium. Tissue engineering Part A. 2011;17(9–10):1219–28. doi: 10.1089/ten.tea.2010.0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoehr A, Hirt MN, Hansen A, Seiffert M, Conradi L, Uebeler J, et al. Spontaneous Formation of Extensive Vessel-Like Structures in Murine Engineered Heart Tissue. Tissue engineering Part A. 2016;22(3–4):326–35. doi: 10.1089/ten.tea.2015.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rouwkema J, Koopman B, Blitterswijk C, Dhert W, Malda J. Supply of nutrients to cells in engineered tissues. Biotechnol Genet Eng Rev. 2010;26:163–78. doi: 10.5661/bger-26-163. [DOI] [PubMed] [Google Scholar]
- 64.Monsuur HN, Weijers EM, Niessen FB, Gefen A, Koolwijk P, Gibbs S, et al. Extensive Characterization and Comparison of Endothelial Cells Derived from Dermis and Adipose Tissue: Potential Use in Tissue Engineering. PloS one. 2016;11(11):e0167056. doi: 10.1371/journal.pone.0167056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Melero-Martin JM, Khan ZA, Picard A, Wu X, Paruchuri S, Bischoff J. In vivo vasculogenic potential of human blood-derived endothelial progenitor cells. Blood. 2007;109(11):4761–8. doi: 10.1182/blood-2006-12-062471. [DOI] [PubMed] [Google Scholar]
- 66.Langley RR, Ramirez KM, Tsan RZ, Van Arsdall M, Nilsson MB, Fidler IJ. Tissue-specific microvascular endothelial cell lines from H-2K(b)-tsA58 mice for studies of angiogenesis and metastasis. Cancer Res. 2003;63(11):2971–6. [PubMed] [Google Scholar]
- 67.Craig LE, Spelman JP, Strandberg JD, Zink MC. Endothelial cells from diverse tissues exhibit differences in growth and morphology. Microvasc Res. 1998;55(1):65–76. doi: 10.1006/mvre.1997.2045. [DOI] [PubMed] [Google Scholar]
- 68.Langenkamp E, Molema G. Microvascular endothelial cell heterogeneity: general concepts and pharmacological consequences for anti-angiogenic therapy of cancer. Cell and tissue research. 2009;335(1):205–22. doi: 10.1007/s00441-008-0642-4. [DOI] [PubMed] [Google Scholar]
- 69.Jackson CJ, Nguyen M. Human microvascular endothelial cells differ from macrovascular endothelial cells in their expression of matrix metalloproteinases. Int J Biochem Cell Biol. 1997;29(10):1167–77. doi: 10.1016/s1357-2725(97)00061-7. [DOI] [PubMed] [Google Scholar]
- 70.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovascular research. 2005;65(1):40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 71.Puissant B, Barreau C, Bourin P, Clavel C, Corre J, Bousquet C, et al. Immunomodulatory effect of human adipose tissue-derived adult stem cells: comparison with bone marrow mesenchymal stem cells. Br J Haematol. 2005;129(1):118–29. doi: 10.1111/j.1365-2141.2005.05409.x. [DOI] [PubMed] [Google Scholar]
- 72.Williams C, Quinn KP, Georgakoudi I, Black LD., 3rd Young developmental age cardiac extracellular matrix promotes the expansion of neonatal cardiomyocytes in vitro. ActaBiomater. 2014;10(1):194–204. doi: 10.1016/j.actbio.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Konstandin MH, Volkers M, Collins B, Quijada P, Quintana M, De La Torre A, et al. Fibronectin contributes to pathological cardiac hypertrophy but not physiological growth. Basic research in cardiology. 2013;108(5):375. doi: 10.1007/s00395-013-0375-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mamuya WS, Brecher P. Fibronectin expression in the normal and hypertrophic rat heart. J Clin Invest. 1992;89(2):392–401. doi: 10.1172/JCI115598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sevilla CA, Dalecki D, Hocking DC. Regional fibronectin and collagen fibril co-assembly directs cell proliferation and microtissue morphology. PloS one. 2013;8(10):e77316. doi: 10.1371/journal.pone.0077316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sevilla CA, Dalecki D, Hocking DC. Extracellular matrix fibronectin stimulates the selfassembly of microtissues on native collagen gels. Tissue engineering Part A. 2010;16(12):3805–19. doi: 10.1089/ten.tea.2010.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shiraishi I, Takamatsu T, Fujita S. Three-dimensional observation with a confocal scanning laser microscope of fibronectinimmunolabeling during cardiac looping in the chick embryo. AnatEmbryol (Berl) 1995;191(3):183–9. doi: 10.1007/BF00187817. [DOI] [PubMed] [Google Scholar]
- 78.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. MolBiol Cell. 2002;13(10):3546–59. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan Y, Richards D, Xu R, Stewart-Clark S, Mani SK, Borg TK, et al. Silicon nanowire-induced maturation of cardiomyocytes derived from human induced pluripotent stem cells. Nano letters. 2015;15(5):2765–72. doi: 10.1021/nl502227a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bedada FB, Chan SS, Metzger SK, Zhang L, Zhang J, Garry DJ, et al. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem cell reports. 2014;3(4):594–605. doi: 10.1016/j.stemcr.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Herron TJ, Rocha AM, Campbell KF, Ponce-Balbuena D, Willis BC, Guerrero-Serna G, et al. Extracellular Matrix-Mediated Maturation of Human Pluripotent Stem Cell-Derived Cardiac Monolayer Structure and Electrophysiological Function. Circ Arrhythm Electrophysiol. 2016;9(4) doi: 10.1161/CIRCEP.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circulation Heart failure. 2011;4(1):44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 83.Deb A, Ubil E. Cardiac fibroblast in development and wound healing. Journal of molecular and cellular cardiology. 2014;70:47–55. doi: 10.1016/j.yjmcc.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J. 2014;167(3):292–300. doi: 10.1016/j.ahj.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 85.Madamanchi A. Beta-adrenergic receptor signaling in cardiac function and heart failure. Mcgill J Med. 2007;10(2):99–104. [PMC free article] [PubMed] [Google Scholar]
- 86.Matsa E, Burridge PW, Wu JC. Human stem cells for modeling heart disease and for drug discovery. Science translational medicine. 2014;6(239) doi: 10.1126/scitranslmed.3008921. 239ps6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20(6):616–23. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dell’Era P, Benzoni P, Crescini E, Valle M, Xia E, Consiglio A, et al. Cardiac disease modeling using induced pluripotent stem cell-derived human cardiomyocytes. World J Stem Cells. 2015;7(2):329–42. doi: 10.4252/wjsc.v7.i2.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sala L, Bellin M, Mummery CL. Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: has the time come? Br J Pharmacol. 2016 doi: 10.1111/bph.13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 91.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circulation research. 2004;94(12):1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 92.Hsieh PC, Davis ME, Lisowski LK, Lee RT. Endothelial-cardiomyocyte interactions in cardiac development and repair. Annu Rev Physiol. 2006;68:51–66. doi: 10.1146/annurev.physiol.68.040104.124629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5(1):15. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.