

Abstract
2-hydroxyglutarate (2-HG) is a hypoxic metabolite with potentially important epigenetic signaling roles. The mechanisms underlying 2-HG generation are poorly understood, but evidence suggests a potential regulatory role for the sirtuin family of lysine deacetylases. Thus, we hypothesized that the acetylation status of the major 2-HG-generating enzymes (lactate dehydrogenase (LDH), isocitrate dehydrogenase (IDH) and malate dehydrogenase (MDH)) may govern their 2-HG generating activity. In-vitro acetylation of these enzymes, with confirmation by western blotting, mass spectrometry, reversibility by recombinant sirtuins, and an assay for global lysine occupancy, yielded no effect on 2-HG generating activity. In addition, while elevated 2-HG in hypoxia is associated with the activation of lysine deacetylases, we found that mice lacking mitochondrial SIRT3 exhibited hyperacetylation and elevated 2-HG. These data suggest there is no direct link between enzyme acetylation and 2-HG production. Furthermore, our observed effects of in-vitro acetylation on the canonical activities of IDH, MDH and LDH appeared to contrast with previous findings wherein acetyl-mimetic lysine mutations resulted in inhibition of these enzymes. Overall, these data suggest that a causal relationship should not be assumed between acetylation of metabolic enzymes and their activities, canonical or otherwise.
Keywords: Deacetylation, Sirtuins, Ischemia, Hypoxia, Epigenetics
INTRODUCTION
2-hydroxyglutarate (2-HG) is a non-canonical metabolite with potential signaling roles that is synthesized from the Krebs cycle metabolite α-ketoglutarate (α-KG). Among the enzymes thought to convert α-KG to 2-HG are cancer-linked forms of mitochondrial NADP+-dependent isocitrate dehydrogenase (IDH2) [1], as well as malate dehydrogenase type 2 (MDH2) [2] and lactate dehydrogenase (LDH) [3,4]. The mechanisms which trigger a neomorphic 2-HG synthetic activity are not fully elucidated.
It has been reported that a specific IDH2 mutation (R172K) found in glioma cells facilitates 2-HG production [1], leading to the early characterization of 2-HG as an oncometabolite. In addition, hypoxia [3–5] and acidic pH [6–8] trigger 2-HG production by MDH2 and LDH. Acidic pH is thought to protonate α-KG, enhancing its access to the substrate binding pocket of these enzymes [7]. However, it is unclear whether post-translational modification of these enzymes can also regulate 2-HG production.
Lysine acetylation, regulated by the sirtuin family of NAD+-dependent protein deacylases (SIRTs) [9], is emerging as an important regulatory mechanism for numerous metabolic processes [10–13]. Previously we demonstrated that an elevation in 2-HG levels observed in cardiac ischemic preconditioning (IPC) was associated with lysine deacetylation [14,15], and we and others showed that IPC-induced deacetylation occurs on LDH-A, IDH2 and MDH2 [15–17]. Intriguingly, both lysine deacetylation and the elevation in 2-HG seen in IPC were inhibited by the SIRT1 inhibitor splitomicin (Sp) [14,15]. As such, we hypothesized herein that the 2-HG generating capacity of these dehydrogenases may be regulated by lysine acetylation status.
METHODS
Reagents
All chemicals and reagents were obtained from Sigma (St. Louis MO) unless otherwise stated. LDH was from bovine heart (L3916, Sigma), while MDH2 (M2634, Sigma) and NADP+ dependent mitochondrial IDH2 (I2002, Sigma) were from porcine heart. HEK293 cells were from ATCC (Manassas VA).
In-vitro protein acetylation, quantitation & western blotting
Protein acetylation was performed similar to [18]. LDH, IDH2, or MDH2 or LDH (2 mg/ml) were incubated overnight at 37 °C in 50 mM Tris-HCl (pH 8) plus 1.5 mM acetyl coenzyme A, followed by neutralization of pH prior to further analysis. For MDH2, acetylated protein was further subjected to deacetylation by incubation with recombinant human SIRT3 (ab125810, AbCam, Cambridge MA).
To monitor protein acetylation, naïve and acetylated proteins were resolved on 12% SDS-PAGE gels, transferred to nitrocellulose and probed with pan anti-acetyl-lysine (K-Ac) or anti-SIRT3 antibodies (Cell Signaling, Danvers MA) at 1:1000 dilution, followed by HRP-linked secondary antibody (GE Biosciences, Pittsburgh PA) at 1:2500 and enhanced chemiluminescent detection (Pierce, Rockford IL). In addition for MDH2, acetylation at the previously published K239 site [17,19] was determined by mass spectrometry in the University of Rochester Proteomics and Mass Spectrometry Core Facility. In brief, proteins were reduced and alkylated (DTT, IAA), trypsinized (Pierce) desalted (C18 Tips, Pierce), dried and suspended in 0.1% TFA. Peptides were separated by C18 HPLC (Easy nLC-1000, Thermo Fisher), with a custom column and an elution gradient of 0.1% formic acid in water → 0.1% formic acid in acetonitrile over 30 min. LC effluent was diverted to a Q Exactive Plus mass spectrometer (Thermo Fisher), operated in data-dependent mode, with a full MS1 scan (400–1400 m/z, resolution 70,000 at m/z 200, AGC target of 106, max injection time 50 ms), followed by 8 data-dependent MS2 scans (resolution 35,000, AGC target of 105, max injection time 120 ms). Isolation width was 1.5 m/z, offset 0.3 m/z, and normalized collision energy 27. Raw data were searched using Mascot (Matrix Science) within Proteome Discoverer software (Thermo Fisher), using the SwissProt Sus scrofa database with <2 missed cleavages, MS1 tolerance 10 ppm, and MS2 tolerance 25 mmu. Carbamidomethyl was set as a fixed modification, with methionine oxidation, lysine acetylation and N-term acetylation as variable modifications. Percolator was used as the FDR calculator, filtering peptides with q-value > 0.01. Protein acetylation was also monitored using the trinitrobenzene sulfonic acid (TNBS) assay for free lysine ε-amino groups [33], following removal of excess acetyl-CoA from proteins by size-exclusion chromatography (BioRad Micro Bio-Spin™ P-6).
Enzyme activities and LC-MS/MS metabolite assays
Activities of LDH, IDH or MDH were measured spectrophotometrically at 340 nm, representing either the consumption of NADH (for LDH and MDH) or the generation of NADPH (for IDH) [6]. In addition, aliquots from spectrophotometric cuvets were withdrawn for quantitation of selected reaction substrates or products by LC-MS/MS as previously reported [6]. Note: The LC-MS/MS method employed was not capable of discerning L vs. D enantiomers of 2-HG. As such we assume herein that 2-HG from LDH or MDH2 is L-2-HG, while that from IDH2 is D-2-HG [1,3,4].
Animals & primary cells
Male littermate wild type (WT) and Sirt3−/− mice on a C57BL/6J background were used at 2 mo. of age. All mice were maintained in a pathogen-free vivarium with a 12 hr. light/dark cycle and food and water available ad libitum. All procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the AAALAC-accredited University of Rochester Committee on Animal Resources.
For cellular hypoxia experiments, HEK293 cells were placed in a hypoxic chamber (pO2 <0.1%) for 20 hr. as previously described [6]. Primary adult mouse cardiomyocytes were isolated as previously described [20]. Cytosolic pH was measured by fluorescent microscopy using the pH-sensitive probe BCECF as previously described [6]. To model ischemia, cardiomyocytes were plated on laminin-coated 25 mm glass cover slips (25,000 cells per slide) in 35 mm dishes, followed by over placement of a second cover slip to exclude oxygen and limit extracellular volume. We previously reported on a similar method using cartridges within the Seahorse XF apparatus to achieve similar ends [20,21]. Cells in the center of the cover slip were imaged after 10 min.
Mitochondria were isolated from WT or Sirt3−/− hearts as previously described [22]. The activity of mitochondrial L-2-HG dehydrogenase was measured spectrophotometrically and by LC-MS/MS as previously described [6].
Statistics
Significance between groups was determined by ANOVA, assuming a normal distribution, followed by post-hoc Student’s t-test. Differences were considered statistically significant at p<0.05, and graphs show means ± standard errors.
RESULTS
We previously demonstrated a role for SIRT1 in the elevation of 2-HG observed in cardiac ischemic preconditioning (IPC) [14]. In addition, 2-HG is elevated in cells exposed to hypoxia [3,4]. However, since these are distinct experimental systems (intermittent whole organ ischemia vs. prolonged cellular hypoxia), it is not clear if SIRT1 is also required for the latter phenomenon. To investigate this we measured 2-HG levels in HEK293 cells exposed to hypoxia with or without the SIRT1 inhibitor splitomicin (Sp). As expected, hypoxia markedly increased 2-HG levels (Figure 1A), and notably this effect was blunted by Sp, suggesting a similar role for SIRT1 in both hypoxic situations.
Figure 1. Cellular Hypoxia, LDH Acetylation and Canonical vs. 2-HG Generating Activity.
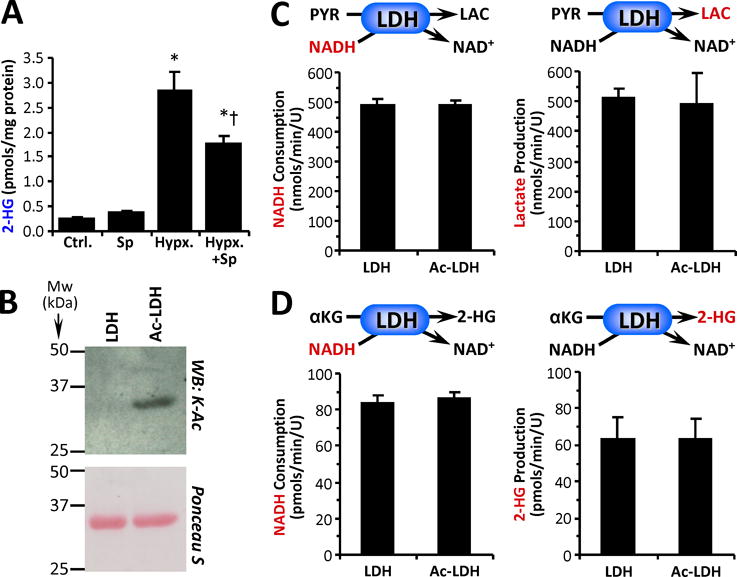
(A): HEK293 cells were exposed to hypoxia with or without the SIRT1 inhibitor splitomicin (Sp), followed by measurement of 2-HG levels. Data are means ± SEM, n=5. *p<0.05 vs. control (Ctrl.) †p<0.05 vs. hypoxia alone (Hypx.) (B): Purified LDH was acetylated in-vitro with acetyl-CoA (see methods), followed by western blot probe with anti-acetyl-lysine (K-Ac) antibody. Ponceau S stained membrane below indicates protein loading. Images representative of at least 4 independent experiments. (C): Canonical activity of naïve and acetylated LDH (Ac-LDH) with pyruvate and NADH as substrates was measured spectrophotometrically as NADH consumption (left panel) or by LC-MS/MS as lactate production (right panel). (D): Non-canonical activity of LDH with α-KG and NADH as substrates was measured spectrophotometrically as NADH consumption (left panel) or by LC-MS/MS as 2-HG production (right panel). All enzyme rate data are means ± SEM, n=4–7. Reactions monitored are shown above each graph, with the metabolite measured shown in red font.
LDH, IDH2 and MDH2 are enzymes capable of generating 2-HG [1,3,4,6,7] and are also thought to be targets for SIRT mediated deacetylation [15–17,19,23,24]. Thus, we performed an in-vitro analysis of the relationship between acetylation and 2-HG generation using purified enzymes (LDH, IDH2, MDH2). Lysine 5 within LDH-A was identified as an acetylation site [30], with acetyl-mimetic mutation of this residue (K5Q) resulting in enzyme inhibition. In contrast to these findings from mutational studies, when we acetylated purified LDH in-vitro (Figure 1B), we observed no effect on its canonical activity (pyruvate + NADH → lactate + NAD+) (Figure 1C). Furthermore, 2-HG generation from α-KG by LDH was not affected by acetylation (Figure 1D). Since Sp is thought to inhibit cytosolic SIRT1 [25–29], and LDH is cytosolic, we also tested whether Sp treatment of HEK293 cells had any effect on cytosolic LDH activity, but found no effect (control: 952 ± 23 vs. Sp: 987 ± 29 nmols/min/mg protein, means ± SEM, N=6, p=0.36). Together these data suggest that acetylation does not impact the enzymatic properties of LDH, and that SIRT1 may regulate hypoxic cellular 2-HG via other mechanisms (see below).
Next, we examined the effects on acetylation on IDH2. Purified NADP+-dependent mitochondrial IDH2 was acetylated in-vitro (Figure 2A), and its canonical reaction (i.e., NADP+ + isocitrate → α-KG + NADPH) was monitored both spectrophotometrically and by LC-MS/MS [6]. Acetylated IDH2 (Ac-IDH2) exhibited significantly higher activity than the naïve enzyme, measured either as NADPH or α-KG generation (Figure 2B). This result contrasts with a previous study [31], wherein an acetyl-mimetic IDH2 mutation (K413Q) demonstrated lower activity.
Figure 2. IDH2 Acetylation and Canonical vs. 2-HG Generating Activity.
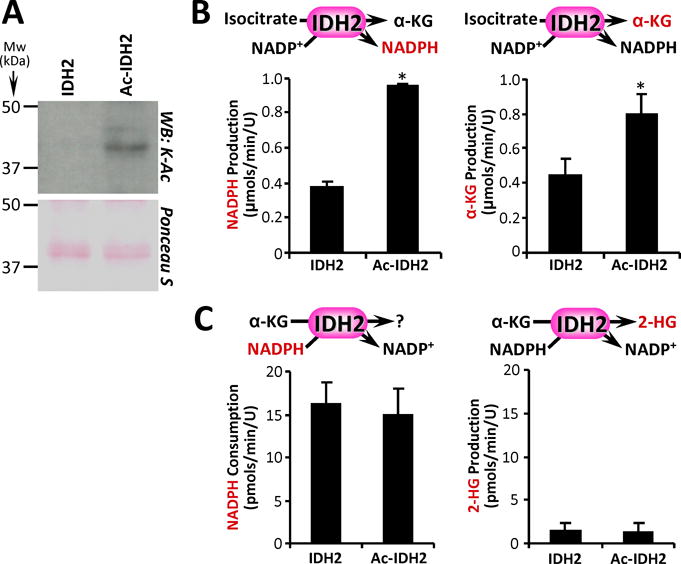
(A): Purified IDH2 was acetylated in-vitro with acetyl-CoA (see methods), followed by western blot probe with anti-acetyl-lysine (K-Ac) antibody. Ponceau S stained membrane below indicates protein loading. Images representative of at least 4 independent experiments. (B): Canonical activity of naïve and acetylated IDH2 (Ac-IDH2) with isocitrate and NADP+ as substrates was measured spectrophotometrically as NADPH production (left panel) or by LC-MS/MS as α-KG production (right panel). (C): Non-canonical reductive activity of IDH2 with α-KG and NADPH as substrates was measured spectrophotometrically as NADPH consumption (left panel) or by LC-MS/MS as 2-HG production (right panel). All enzyme rate data are means ± SEM, n=4. *p<0.05 between naïve and Ac-IDH2. Reactions monitored are shown above each graph, with the metabolite measured shown in red font.
In terms of 2-HG generation, Figure 2C shows that both naïve and Ac-IDH2 could use α-KG plus NADPH as substrates, but neither enzyme generated substantial corresponding amounts of 2-HG. Instead, IDH2 is known to operate in reverse, using α-KG in a reductive carboxylation reaction to produce isocitrate [6,32]. Together these data suggest that acetylation boosted the canonical activity of IDH2 (i.e., isocitrate → α-KG), with no effect on its non-canonical activities (2-HG generation or reductive carboxylation). The fact that acetylation did impact IDH2 canonical activity suggests the lack of an effect on non-canonical activities (negative result) was not simply due to confounding experimental issues, or an insufficient degree of acetylation (see below).
The mitochondrial enzyme MDH2 can also produce 2-HG [3,4,6,7], and notably lysine 239 of MDH2 is differentially acetylated in mice lacking the mitochondrial deacetylase SIRT3 [17]. Furthermore, an acetyl-mimetic K239Q mutant protein exhibits lower MDH activity [17], suggesting acetylation at this site is inhibitory. We acetylated purified MDH2 in-vitro [18] and verified this by western blot. As shown in Figure 2A (and in agreement with results from others [18]) this acetylation event was also reversed by incubation with human recombinant SIRT3 + NAD+. In addition, mass spectrometric analysis of naïve vs. acetylated MDH2 (Ac-MDH2) (Figure 3B/C/D) revealed a difference of 42 mass units in the y1+ ion for the peptide containing K239 (IQEAGTEVVK underlined in panel 2C), confirming acetylation at this site. Furthermore, a TNBS assay for free lysine ε-amino groups [33] revealed that the in-vitro acetylation method resulted in occupancy of 74.4 ± 2.7 % of lysines within MDH2.
Figure 3. MDH2 Acetylation and Canonical vs. 2-HG Generating Activity.
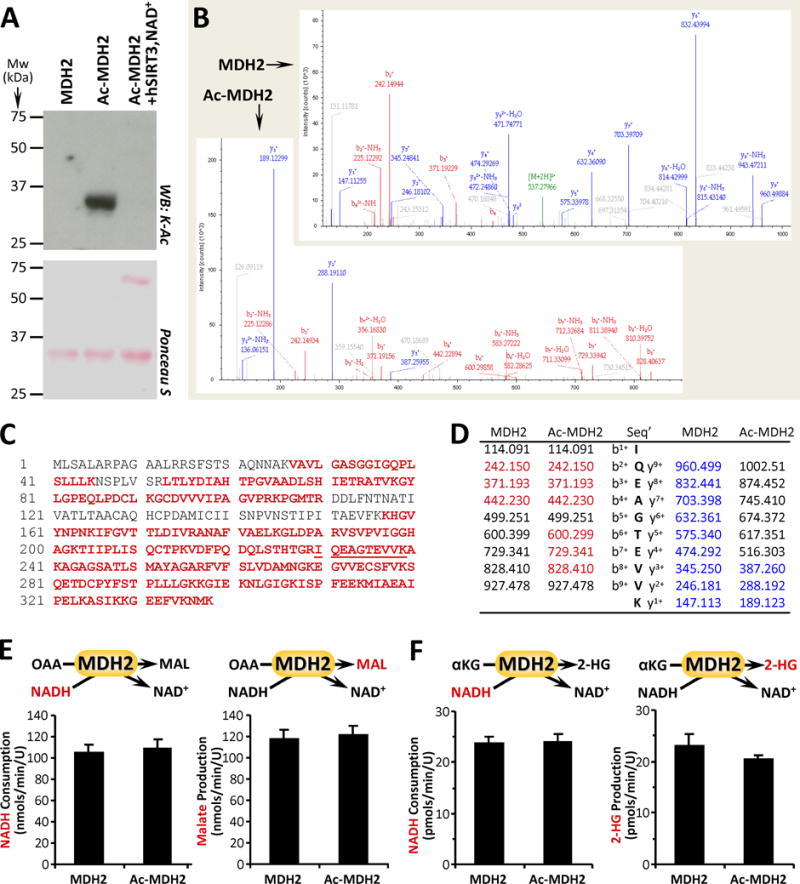
(A): Purified MDH2 was acetylated in-vitro with acetyl-CoA (see methods), followed by western blot probe with anti-acetyl-lysine (K-Ac) antibody. Ponceau S stained membrane below indicates protein loading. Images representative of at least 4 independent experiments. (B/C/D): Naïve and acetylated MDH2 (Ac-MDH2) were digested with trypsin and resultant peptides analyzed by mass spectrometry to identify acetylation sites. 76% sequence coverage was obtained (panel C, red font) including the peptide containing the proposed K239 acetylation site (underlined). Sample spectra are shown in B, with panel D listing identified peptides corresponding to bn+ ions (red) and yn+ ions (blue). A difference of 42 mass units between y+1 ion in MDH2 vs. Ac-MDH2 (189.123 – 147.113) indicates acetylation. (E): Canonical activity of naïve and acetylated MDH2 with OAA and NADH as substrates was measured spectrophotometrically as NADH consumption (left panel) or by LC-MS/MS as malate production (right panel). (F): Non-canonical activity of MDH2 with α-KG and NADH as substrates was measured spectrophotometrically as NADH consumption (left panel) or by LC-MS/MS as 2-HG production (right panel). All enzyme rate data are means ± SEM, n=4–6. Reactions monitored are shown above each graph, with the metabolite measured shown in red font.
Surprisingly, as shown in Figure 3E, the canonical MDH enzymatic reaction (i.e., OAA + NADH → malate + NAD+) was no different between naïve vs. Ac-MDH2 (Note: the normal forward reaction, malate → OAA, is energetically unfavorable, so the enzyme is routinely assayed in the reverse direction). Furthermore, with α-KG as a substrate the enzymatic reaction to generate 2-HG was also unaffected by acetylation status (Figure 3F). Overall, these data suggest that neither canonical nor alternative activities of MDH2 were impacted by acetylation.
To further investigate the possible role of lysine acetylation as a regulator of 2-HG generation, we examined mice with deletion of Sirt3 (Figure 4A), a mitochondrial SIRT known to deacetylate LDH, IDH2 and MDH2 [15–17,19,23,24]. As shown in Figures 4B/C, Sirt3−/− hearts exhibited the expected hyper-acetylation of cardiac mitochondrial proteins (vs. wild type, WT), but surprisingly this was accompanied by a 3.5-fold elevation in 2-HG. Such a result contrasts with the situation in both IPC [14] and cellular anoxia (Figure 1A), wherein deacetylation is associated with elevated 2-HG. Together these data support a disconnect between acetylation and 2-HG generation.
Figure 4. Acetylation, 2-HG Generation and pH in Sirt3−/− mice, and the Effects of SIRT Inhibition on Cytosolic pH.
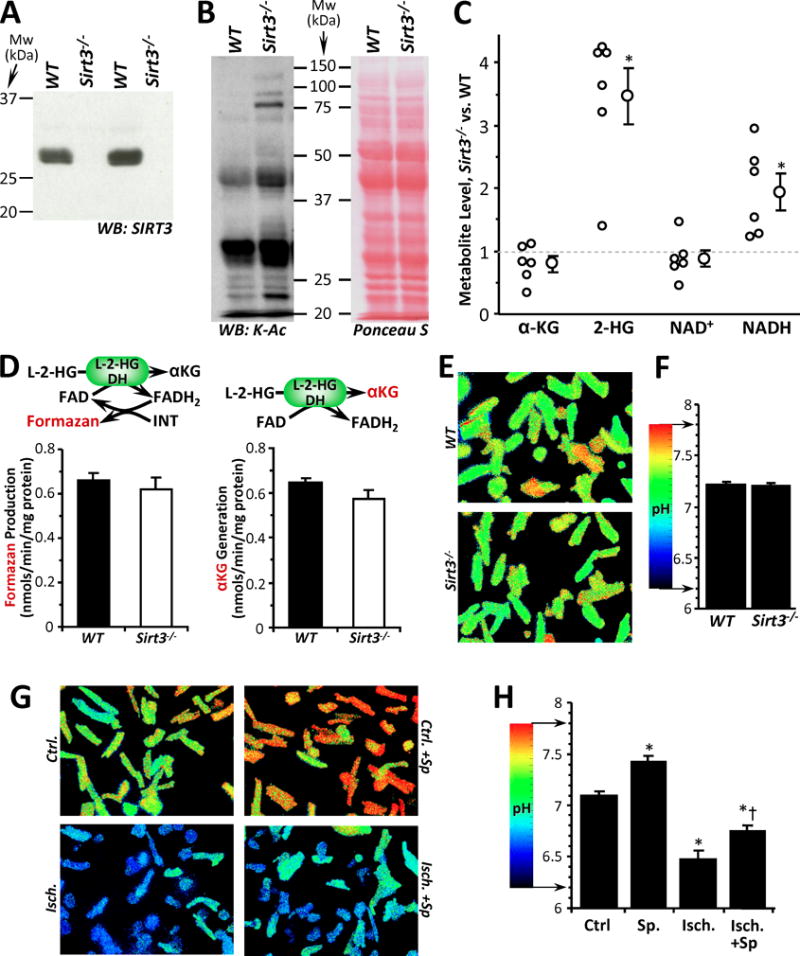
(A): Western blot shows absence of SIRT3 protein in cardiac tissue from Sirt3−/− mice (vs. WT). (B): Western blot shows protein acetylation (pan anti-acetyl-lysine antibody) in cardiac mitochondria from WT and Sirt3−/− mice. Ponceau S stained membrane indicates protein loading. Blots are representative of at least 4 independent experiments. (C): LC-MS/MS based metabolite profiling of WT vs. Sirt3−/− cardiac tissue. Data are expressed as the metabolite level in Sirt3−/− relative to paired WT samples, with individual data points (n=6 pairs) alongside means ± SEM. *p<0.05 between WT and Sirt3−/−. (D): L-2-HG dehydrogenase (L-2-HGDH) activity in cardiac mitochondria from WT and Sirt3−/− mice was measured spectrophotometrically via formazan-linked assay (left panel) or by LC-MS/MS as α-KG formation (right panel). Enzyme rate data are means ± SEM, n=5. Reactions monitored are shown above each graph, with the metabolite measured shown in red font. (E/F): Cytosolic pH measurements in primary cultured adult cardiomyocytes from WT and Sirt3−/− mice. Alongside representative images of cellular pH (E), graph shows quantitation of pH from 4 independent cell preparations (F). Data are means ± SEM. (G/H): Cytosolic pH measurements in primary cultured adult cardiomyocytes from WT mice subjected to simulated ischemia (see methods), with or without the SIRT1 inhibitor splitomicin (Sp). Alongside representative images of cellular pH (G), graph shows quantitation of pH from 4–6 independent cell preparations (H). Data are means ± SEM. *p<0.05 vs. control (Ctrl.) †p<0.05 vs. ischemia alone (Isch.).
Nevertheless, these data raise an intriguing question as to the mechanism of 2-HG elevation in Sirt3−/− mice. One possibility may be inhibition of the mitochondrial 2-HG catabolic enzyme L-2-HG dehydrogenase (L-2-HGDH) [34]. However, as shown in Figure 4D, we observed no difference in L-2-HGDH activity between Sirt3−/− and WT cardiac mitochondria.
Another potentially important regulator of 2-HG generation is acidic pH [6,7]. However, we did not observe any significant difference in pH between cardiomyocytes isolated from WT or Sirt3−/− hearts (Figures 4E and 4F). Notably (and in agreement with previous studies), Sirt3−/− hearts exhibited elevated NADH levels (Figure 4C), a phenomenon that has been attributed to inhibition of mitochondrial respiratory complex I (NADH ubiquinone oxidoreductase) [22,35]. It is thought that excessive NADH accumulation (a.k.a. reductive stress), is a key driver of the α-KG → 2-HG reaction [36], and together these findings suggest that a likely cause of elevated 2-HG in Sirt3−/− hearts is increased drive from NADH.
Finally, to uncover the basis for Sp sensitivity in the hypoxic increase in 2-HG ([14] and Figure 1A), we measured the effects of Sp on ischemia-induced acidification of primary mouse cardiomyocytes. As shown in Figures 4G and 4H, Sp elicited a slight alkalinization alone and blunted the acidification caused by ischemia. These data suggest that the role of SIRT1 in the elevation of 2-HG in hypoxia/ischemia, is likely at a position upstream of acidification.
DISCUSSION & CONCLUSIONS
The conclusions of this study are as follows: (i) both canonical enzyme activities and 2-HG generation by Ac-LDH and Ac-MDH2 were similar to their corresponding naïve enzymes; (ii) IDH2 (either naïve or acetylated) did not produce 2-HG, although Ac-IDH2 showed higher canonical activity (isocitrate → α-KG); (iii) 2-HG was elevated in Sirt3−/− hearts, concurrent with higher NADH levels; and (iv) the ability of the SIRT1 inhibitor splitomicin to blunt 2-HG elevation in hypoxia/ischemia may be due to an upstream role for SIRT1 in regulating acidosis. Overall, these data suggest that direct acetylation of the major identified 2-HG generating enzymes is not a mechanism that regulates 2-HG. Although our utilization of a range of experimental systems (HEK293 cells, purified enzymes, isolated mitochondria, pharmacologic inhibitors, Sirt3−/− mice, isolated cardiomyocytes) would normally be considered a caveat, the broad agreement of results obtained across these systems suggests conserved mechanisms.
Our observation that IDH2 acetylation by a physiologically-relevant mechanism (acetyl-CoA) increases its canonical activity (Figure 2B) is an interesting result. Since acetyl-CoA is the upstream substrate for the Krebs cycle, it is possible that acetyl-CoA mediated IDH2 acetylation represents a novel feed-forward activation mechanism for the cycle. The importance of this relative to the already well-characterized regulation of this cycle by numerous allosteric mechanisms (e.g., Ca2+, NADH, phosphorylation) [37–39] remains to be seen. Furthermore, although we show here that IDH2 can be acetylated non-enzymatically, it has recently been noted that considerable variance can exist between the reactivity of specific lysines for in-vitro acetylation vs. their in-vivo enzymatic modification [40]. In this regard, it is not known whether the combination of acetylation events that occur under in-vitro conditions may adequately model the in-vivo situation, where mitochondrial protein acetylation is regulated not only by deacetylases such as SIRT3, but also by acetyl-transferases such as GCN5L1 [41].
While the application of quantitative proteomic labeling techniques to determine site-specific stoichiometry of lysine acetylation [42] may be informative, nevertheless our in-vitro acetylation method yielded ~75% global lysine site occupancy (for MDH2), suggesting that the lack of an effect of acetylation observed herein was not simply due to low site occupancy of lysines.
A consistent finding in the current study was contrast with previous observations using acetyl-mimetic mutations in dehydrogenase enzymes [17,30,31]. Specifically, acetyl-mimetic K5Q of LDH-A, K413Q of IDH2 and K239Q of MDH2 were all shown to result in lower activities for these enzymes. In contrast we found that acetylation stimulated IDH2 activity, while having no effect on LDH or MDH2. The acetylation methods used herein were physiologically relevant [18], and acetylation was verified by western blot, mass spectrometry, reversibility with a recombinant SIRT, and assessment of lysine occupancy by TNBS assay. Interpreting mutational studies can be problematic when mutations result in loss of function, and in the lysine acetylation field there are vanishingly few examples of acetyl-mimetic mutation resulting in stimulation of activity. As such, we speculate that the results of some mutational studies reporting enzyme inhibition may be artifactual, due to unexpected effects of mutations on protein folding or other enzyme properties. Indeed, it was shown that the acetyl-mimetic mutants K3Q and K118Q of MDH isoform 1 had no effect on activity, while the corresponding deacetylation mimetics K3R and K118R were inhibitory [43]. Thus, in particular for MDH, the relationship between acetylation and enzyme activity is not clear.
An important caveat in interpreting our findings is subcellular compartmentation. In cardiomyocytes SIRT1 is cytosolic [15,44], and as shown in Figure 4H the SIRT1 inhibitor Sp results in alkalinization of the cardiomyocyte cytosol. Sp also blunts the hypoxic increase in 2-HG (Figure 1A). However, acetylation of cytosolic LDH does not impact its 2-HG generation. Thus we conclude that SIRT1 may play a role in regulating hypoxic cellular 2-HG at a point upstream of acidification. In contrast, SIRT3 is mitochondrial and its genetic ablation had no effect on cytosolic pH and no effect on L-2HGDH activity. Furthermore, no effect of acetylation on 2-HG generation was seen for the mitochondrial enzymes IDH2 or MDH2. Thus, in the case of Sirt3−/−, we speculate that elevated 2-HG originates at the level of reductive stress (excess NADH), possibly due to inhibition of mitochondrial respiratory complex I [22,35]. Although it was recently reported that mitochondrial complex II (succinate ubiquinone oxidoreductase) is down-regulated in Sirt3−/− mice [45], it is unclear how such a phenomenon might be mechanistically linked to reductive stress or 2-HG generation.
Interestingly, the Sirt3−/− mouse exhibits acetylation of cytosolic LDH [17], and we showed that SIRT1 inhibition resulted in acetylation of numerous mitochondrial enzymes [15]. Thus, there appear to be signaling mechanisms by which inactivation (genetic or pharmacologic) of a SIRT in one cell compartment can impact acetylation in another compartment. Indeed, it was recently shown that SIRT1 can translocate from cytosol to mitochondria [47]. Furthermore, in the context of compartmentation it has also been reported that 2-HG can be transported into cardiomyocytes [46]. As such, it is intriguing that we found a small but non-significant increase in 2-HG levels in the serum of Sirt3−/− mice (1.68 ± 0.29 fold vs. control, mean ± SEM, n=6, p=0.078, paired t-test). This finding suggests an alternative hypothesis: the elevated 2-HG level observed in Sirt3−/− hearts may originate elsewhere in the body. However, regardless the compartment, it is clear from our results that direct regulation of 2-HG generating enzymes by SIRT1 or SIRT3 mediated deacetylation is unlikely to be a mechanism regulating 2-HG levels.
It was recently proposed that conversion of α-KG to 2-HG is a quantitatively significant mechanism by which “cells handle a mounting pool of reducing equivalents” such as excess NADH [36]. 2-HG was also proposed to represent a “redox reservoir”, to enable cells to handle reductive stress [36]. However, the canonical activities of LDH and MDH2 are at least 3-orders of magnitude greater than the rates at which these enzymes reduce α-KG to 2-HG [2,6]. In conditions of hypoxia and reductive stress, the 2-HG reaction is quantitatively insignificant as a consumer of NADH, relative to the role of LDH (pyruvate + NADH → lactate + NAD+). In hypoxia, 2-HG generation is also several orders of magnitude smaller than another important redox sink, namely the generation of succinate from fumarate by reversal of respiratory complex II [48–50]. Furthermore, 2-HG is not a true redox reservoir because the reverse reaction to regenerate α-KG from 2-HG (i.e., the reaction catalyzed by L-2-HGDH) uses FAD as an electron acceptor and is not known to regenerate NADH. These quantitative limitations on the role of 2-HG as a redox sink do not diminish the potential importance of 2-HG as a hypoxic signaling molecule [6,51,52].
Finally, the recent finding that 2-HG can inhibit α-ketoglutarate dehydrogenase (α-KGDH) activity [46] suggests that 2-HG and the TCA cycle may exhibit a complex cross-talk, wherein inhibition of α-KGDH would elevate α-KG levels, providing more substrate for 2-HG generation, resulting in a feed-forward activation for 2-HG generation. In conclusion, while direct enzyme acetylation does not appear to be an important 2-HG regulatory mechanism, the role of other protein post-translational modifications and signaling pathways that regulate this metabolite may be worthy of further investigation.
Acknowledgments
This work was funded by a grant from the US National Institutes of Health (RO1 HL-071158) to PSB. We thank Dr. George Porter Jr. for Sirt3−/− mice.
Footnotes
DATA SHARING
The full original data set for this work is available on the FigShare website (https://doi.org/10.6084/m9.figshare.5170531).
DISCLOSURES
The authors declare no conflicts of interest.
References
- 1.Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M. l-2-Hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis. 2007;30:681–689. doi: 10.1007/s10545-007-0487-0. [DOI] [PubMed] [Google Scholar]
- 3.Intlekofer AM, Dematteo RG, Venneti S, Finley LWS, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015;22:304–311. doi: 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-Mediated Increases in l -2-hydroxyglutarate Coordinate the Metabolic Response to Reductive Stress. Cell Metab. 2015;22:291–303. doi: 10.1016/j.cmet.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armitage EG, Kotze HL, Allwood JW, Dunn WB, Goodacre R, Williams KJ. Metabolic profiling reveals potential metabolic markers associated with Hypoxia Inducible Factor-mediated signalling in hypoxic cancer cells. Sci Rep. 2015;5:15649. doi: 10.1038/srep15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS. Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling. J Biol Chem. 2016;291:20188–20197. doi: 10.1074/jbc.M116.738799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, Salah S, Gunner MR, Chodera JD, Cross JR, et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol. 2017;13:494–500. doi: 10.1038/nchembio.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teng X, Emmett MJ, Lazar MA, Goldberg E, Rabinowitz JD. Lactate Dehydrogenase C Produces S-2-Hydroxyglutarate in Mouse Testis. ACS Chem Biol. 2016;11:2420–7. doi: 10.1021/acschembio.6b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson KA, Green MF, Huynh FK, Wagner GR, Hirschey MD. SnapShot: Mammalian Sirtuins. Cell. 2014;159:956–956.e1. doi: 10.1016/j.cell.2014.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang JY, Hirschey MD, Shimazu T, Ho L, Verdin E. Mitochondrial sirtuins. Biochim Biophys Acta. 2010;1804:1645–1651. doi: 10.1016/j.bbapap.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 11.Correia M, Perestrelo T, Rodrigues AS, Ribeiro MF, Pereira SL, Sousa MI, Ramalho-Santos J. Sirtuins in metabolism, stemness and differentiation. Biochim Biophys Acta. 2017;1861:3444–3455. doi: 10.1016/j.bbagen.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Van de Ven RAH, Santos D, Haigis MC. Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol Med. 2017;23:320–331. doi: 10.1016/j.molmed.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye X, Li M, Hou T, Gao T, Zhu W, Yang Y. Sirtuins in glucose and lipid metabolism. Oncotarget. 2015;8:1845–1859. doi: 10.18632/oncotarget.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nadtochiy SM, Urciuoli W, Zhang J, Schafer X, Munger J, Brookes PS. Metabolomic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation. J Mol Cell Cardiol. 2015;88:64–72. doi: 10.1016/j.yjmcc.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine deacetylation in ischaemic preconditioning: the role of SIRT1. Cardiovasc Res. 2011;89:643–649. doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parker BL, Shepherd NE, Trefely S, Hoffman NJ, White MY, Engholm-Keller K, Hambly BD, Larsen MR, James DE, Cordwell SJ. Structural basis for phosphorylation and lysine acetylation cross-talk in a kinase motif associated with myocardial ischemia and cardioprotection. J Biol Chem. 2014;289:25890–906. doi: 10.1074/jbc.M114.556035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49:186–99. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner GR, Payne RM. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288:29036–45. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Still AJ, Floyd BJ, Hebert AS, Bingman CA, Carson JJ, Gunderson DR, Dolan BK, Grimsrud PA, Dittenhafer-Reed KE, Stapleton DS, et al. Quantification of mitochondrial acetylation dynamics highlights prominent sites of metabolic regulation. J Biol Chem. 2013;288:26209–19. doi: 10.1074/jbc.M113.483396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadtochiy SM, Madukwe J, Hagen F, Brookes PS. Mitochondrially targeted nitro-linoleate: a new tool for the study of cardioprotection. Br J Pharmacol. 2014;171:2091–2098. doi: 10.1111/bph.12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo S, Olm-Shipman A, Walters A, Urciuoli WR, Devito S, Nadtochiy SM, Wojtovich AP, Brookes PS. A cell-based phenotypic assay to identify cardioprotective agents. Circ Res. 2012;110:948–57. doi: 10.1161/CIRCRESAHA.111.263715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM. SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts. Am J Physiol Heart. 2014;306:1602–9. doi: 10.1152/ajpheart.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sol EM, Wagner SA, Weinert BT, Kumar A, Kim HS, Deng CX, Choudhary C. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase sirt3. PLoS One. 2012;7:e50545. doi: 10.1371/journal.pone.0050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci. 2013;110:6601–6. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breitenstein A, Stein S, Holy EW, Camici GG, Lohmann C, Akhmedov A, Spescha R, Elliott PJ, Westphal CH, Matter CM, et al. Sirt1 inhibition promotes in vivo arterial thrombosis and tissue factor expression in stimulated cells. Cardiovasc Res. 2011;89:464–72. doi: 10.1093/cvr/cvq339. [DOI] [PubMed] [Google Scholar]
- 26.Chapalamadugu KC, Panguluri SK, Bennett ES, Kolliputi N, Tipparaju SM. High level of oxygen treatment causes cardiotoxicity with arrhythmias and redox modulation. Toxicol Appl Pharmacol. 2015;282:100–7. doi: 10.1016/j.taap.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stavrou EX, Fang C, Merkulova A, Alhalabi O, Grobe N, Antoniak S, Mackman N, Schmaier AH. Reduced thrombosis in Klkb1−/− mice is mediated by increased Mas receptor, prostacyclin, Sirt1, and KLF4 and decreased tissue factor. Blood. 2015;125:710–719. doi: 10.1182/blood-2014-01-550285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein S, Lohmann C, Schäfer N, Hofmann J, Rohrer L, Besler C, Rothgiesser KM, Becher B, Hottiger MO, Borén J, et al. SIRT1 decreases Lox-1-mediated foam cell formation in atherogenesis. Eur Heart J. 2010;31:2301–9. doi: 10.1093/eurheartj/ehq107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein S, Schäfer N, Breitenstein A, Besler C, Winnik S, Lohmann C, Heinrich K, Brokopp CE, Handschin C, Landmesser U, et al. SIRT1 reduces endothelial activation without affecting vascular function in ApoE−/− mice. Aging. 2010;2:353–60. doi: 10.18632/aging.100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY, et al. Lysine-5 Acetylation Negatively Regulates Lactate Dehydrogenase A and Is Decreased in Pancreatic Cancer. Cancer Cell. 2013;23:464–476. doi: 10.1016/j.ccr.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 Protein Deacetylates Isocitrate Dehydrogenase 2 (IDH2) and Regulates Mitochondrial Redox Status. J Biol Chem. 2012;287:14078–14086. doi: 10.1074/jbc.M112.355206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wise DR, Ward PS, Shay JES, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci. 2011;108:19611–6. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cayot P, Tainturier G. The Quantification of Protein Amino Groups by the Trinitrobenzenesulfonic Acid Method: A Reexamination. Anal Biochem. 1997;249:184–200. doi: 10.1006/abio.1997.2161. [DOI] [PubMed] [Google Scholar]
- 34.Rzem R, Achouri Y, Marbaix E, Schakman O, Wiame E, Marie S, Gailly P, Vincent MF, Veiga-da-Cunha M, Van Schaftingen E. A Mouse Model of L-2-Hydroxyglutaric Aciduria, a Disorder of Metabolite Repair. PLoS One. 2015;10:e0119540. doi: 10.1371/journal.pone.0119540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–52. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loscalzo J. Adaptions to Hypoxia and Redox Stress: Essential Concepts Confounded by Misleading Terminology. Circ Res. 2016;119:511–3. doi: 10.1161/CIRCRESAHA.116.309394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofmeyr JHS, Rohwer JM, Snoep JL. Conditions for effective allosteric feedforward and feedback in metabolic pathways. Syst Biol. 2006;153:327–31. doi: 10.1049/ip-syb:20060019. [DOI] [PubMed] [Google Scholar]
- 38.Bouskila M, Hunter RW, Ibrahim AFM, Delattre L, Peggie M, van Diepen JA, Voshol PJ, Jensen J, Sakamoto K. Allosteric Regulation of Glycogen Synthase Controls Glycogen Synthesis in Muscle. Cell Metab. 2010;12:456–466. doi: 10.1016/j.cmet.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka T, Sue F, Morimura H. Feed-forward activation and feed-back inhibition of pyruvate kinase type L of rat liver. Biochem Biophys Res Commun. 1967;29:444–449. doi: 10.1016/0006-291x(67)90477-9. [DOI] [PubMed] [Google Scholar]
- 40.Baeza J, Smallegan MJ, Denu JM. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem Biol. 2015;10:122–8. doi: 10.1021/cb500848p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J. 2012;443:655–61. doi: 10.1042/BJ20120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baeza J, Dowell JA, Smallegan MJ, Fan J, Amador-Noguez D, Khan Z, Denu JM. Stoichiometry of Site-specific Lysine Acetylation in an Entire Proteome. J Biol Chem. 2014;289:21326–21338. doi: 10.1074/jbc.M114.581843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim EY, Kim WK, Kang HJ, Kim JH, Chung SJ, Seo YS, Park SG, Lee SC, Bae KH. Acetylation of malate dehydrogenase 1 promotes adipogenic differentiation via activating its enzymatic activity. J Lipid Res. 2012;53:1864–1876. doi: 10.1194/jlr.M026567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem. 2007;282:6823–32. doi: 10.1074/jbc.M609554200. [DOI] [PubMed] [Google Scholar]
- 45.Parodi-Rullán RM, Chapa-Dubocq X, Rullán PJ, Jang S, Javadov S. High Sensitivity of SIRT3 Deficient Hearts to Ischemia-Reperfusion Is Associated with Mitochondrial Abnormalities. Front Pharmacol. 2017;8:275. doi: 10.3389/fphar.2017.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karlstaedt A, Zhang X, Vitrac H, Harmancey R, Vasquez H, Wang JH, Goodell MA, Taegtmeyer H. Oncometabolite d-2-hydroxyglutarate impairs α-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci. 2016;113:10436–41. doi: 10.1073/pnas.1601650113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson JW, Dave KR, Saul I, Narayanan SV, Perez-Pinzon MA. Epsilon PKC increases brain mitochondrial SIRT1 protein levels via heat shock protein 90 following ischemic preconditioning in rats. PLoS One. 2013;8:e75753. doi: 10.1371/journal.pone.0075753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanadi DR, Fluharty AL. On The Mechanism Of Oxidative Phosphorylation. Vii. The Energy-Requiring Reduction Of Pyridine Nucleotide By Succinate And The Energy-Yielding Oxidation Of Reduced Pyridine Nucleotide By Fumarate. Biochemistry. 2:523–8. doi: 10.1021/bi00903a023. [DOI] [PubMed] [Google Scholar]
- 50.Hochachka PW, Owen TG, Allen JF, Whittow GC. Multiple end products of anaerobiosis in diving vertebrates. Comp Biochem Physiol B. 1975;50:17–22. doi: 10.1016/0305-0491(75)90292-8. [DOI] [PubMed] [Google Scholar]
- 51.Su X, Wellen KE, Rabinowitz JD. Metabolic control of methylation and acetylation. Curr Opin Chem Biol. 2016;30:52–60. doi: 10.1016/j.cbpa.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]