

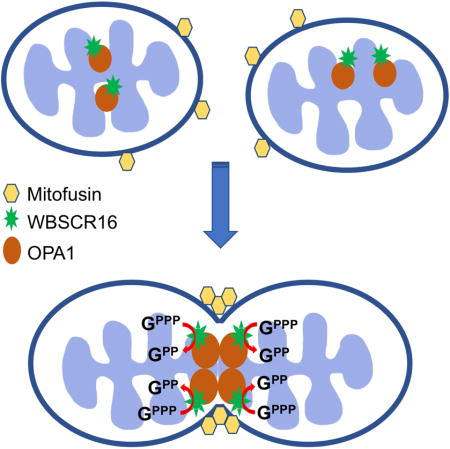
SUMMARY
Regulated inter-mitochondrial fusion/fission is essential for maintaining optimal mitochondrial respiration and control of apoptosis and autophagy. In mammals, mitochondrial fusion is controlled by outer membrane GTPases MFN1 and MFN2, and by inner membrane GTPase OPA1. Disordered mitochondrial fusion/fission contributes to various pathologies, and MFN2 or OPA1 mutations underlie neurodegenerative diseases. Here, we show that the WBSCR16 protein is primarily associated with the outer face of the inner mitochondrial membrane and is important for mitochondrial fusion. We provide evidence of a WBSCR16/OPA1 physical interaction in the intact cell and of a WBSCR16 function as an OPA1-specific guanine nucleotide exchange factor (GEF). Homozygosity for a Wbscr16 mutation causes early embryonic lethality, whereas neurons of mice heterozygous for the mutation have mitochondria with reduced membrane potential and increased susceptibility to fragmentation upon exposure to stress, suggesting roles for WBSCR16 deficits in neuronal pathologies.
Graphical abstract
INTRODUCTION
Mitochondria are organelles critical to cellular viability and function. They produce most cellular ATP, are integral to intracellular Ca++ signaling and are key modulators of apoptosis and autophagy (Jouaville et al., 1995; Pernas and Scorrano, 2016). Mitochondria normally undergo dynamic cycles of fusion and fission that form and remodel tubular networks, thus optimizing intracellular mitochondrial distribution, respiratory function, control of apoptosis and autophagy; and preventing genetic drift in mitochondrial DNA mutation distribution (Chan, 2006; Kasahara and Scorrano, 2014). Mitochondrial fusion is driven by 3 large dynamin-related GTPases: outer membrane GTPases MFN1 and MFN2, and inner membrane GTPase OPA1 (Chan, 2006). MFN2 and OPA1 mutations are causal in the neural degenerative diseases Charcot-Marie-Tooth neuropathy type 2A (neuropathy of long motor and sensory neurons) (Zuchner et al., 2004) and dominant optic atrophy (optic nerve degeneration, ataxia, deafness, and peripheral neuropathy), respectively (Delettre et al., 2000). Although mitochondria are important to the viability and functioning of most cells, the neurological phenotypes resulting from MFN2 and OPA1 mutations are thought to reflect the particularly high-energy needs of specialized neuronal functions. The latter include ionic pumps, channel activities, and synaptic transmission. These rely on optimal mitochondrial function, which in turn relies on fusion (Bossy-Wetzel et al., 2003).
Williams-Beuren syndrome (WBS) is a contiguous gene deletion syndrome in which ~28 genes are deleted from a WBS “critical region” (WBSCR) at chromosome 7q11.23 (Pober, 2010). WBS is characterized by cardiovascular, endocrine and neurological disorders, the latter of which includes cognitive impairment and visuospatial/visiomotor deficits (Pober, 2010). Various of the neurological deficits are thought to be due to hemizygosity for transcription factors encoded by WBSCR genes GTF2I and GTF2IRD1 (Schubert, 2009). However, roles that most WBSCR genes may play in neurological or other deficits are unknown.
Here we identify a spontaneously occurring mutation in gene Wbscr16, from the mouse chromosomal region corresponding to the human WBSCR. We show the protein product of this gene, WBSCR16, to be associated with the outer face of the mitochondrial inner membrane, to be in extremely close proximity to OPA1, indicative of physical interaction, and to be an OPA1-specific guanine nucleotide exchange factor (GEF) that affects mitochondrial fusion in a manner dependent upon the OPA1/MFN1 functional axis. Homozygosity for the spontaneous Wbscr16 mutation results in early embryonic lethality due to deficits in early placentation. Mice heterozygous for the mutation are shown to have neuronal mitochondria with reduced membrane potential and increased susceptibility to mitochondrial fragmentation in response to excitotoxic stress. Implications of the data are discussed.
RESULTS
Homozygosity for a Spontaneous Wbscr16 Mutation Causes Early Embryonic Lethality
Subsequent to 10 generations of backcrosses from a Black Swiss to a C57BL/6 (B6) background, mice null for gene Pcolce, involved in collagen biosynthesis, went from a phenotype of viability with mild skeletal abnormalities (Steiglitz et al., 2006) to an early embryonic lethal phenotype. This included fibrin clotting at the trophoblast component of the chorio-vitelline placenta of E7.5 embryos, conceptus growth arrest at E8.5 (Fig. 1 A and Fig. S1) and rapid death/resorption by E9.5. Numbers of trophoblast giant cells (TGCs) were greatly reduced and residual TGCs were morphologically abnormal with small nuclei (Fig. 1 B). Clotting likely resulted from deficits in TGCs, which normally prevent inappropriate clotting at the maternal-embryonic interface (Gu et al., 2002).
Figure 1. A Wbscr16 mutation causes early embryonic lethality.
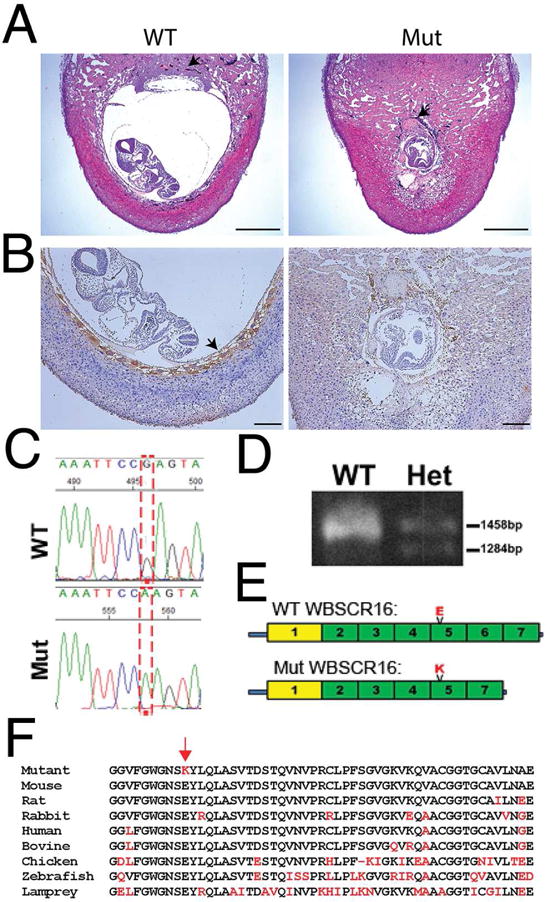
(A) H&E stained sections of implantation sites for Pcolce−/− E8.5 embryos found to be wild type (WT) or homozygous for a mutation (Mut), at the Wbscr16 locus. Both embryos were on a C57BL/6 background. Black arrows; ectoplacental cone (WT), fibrin/platelet clot replacing the ectoplacental cone (mutant). Scale bars 600 μm. (B) WT and mutant sections were immunostained (brown) for trophoblast giant cell (TGC) marker placental lactogen 1 (PL-1). Arrow, HRP signal for anti-PL-1 (WT). Brown color in mutant is mostly due to blood cells, but also to sparse HRP-stained TGCs with abnormally small nuclei. Scale bars 50 μm. (C) G to A transition (reverse strand). (D) PCR-amplified cDNA shows normal size Wbscr16 mRNA, plus a 164 base-smaller mRNA in heterozygotes not detected in WT. (E) Mutant WBSCR16 p.E308K substitution in RLD 5 and loss of RLD 6 by alternative splicing. Green, canonical RLDs; yellow, non-cannonical RLD. (F) RLD 5 aligned across vertebrate species (black, conserved residues; red, residues that do not match WT murine sequence). Red arrow, mutant Lys that substitutes for highly conserved Glu. See also Figures S1 and S2.
The unexpected early embryonic lethal phenotype from backcrossing suggested the existence of a locus (locusC) on the B6 background that strongly contributed to the phenotype, either by modifying Pcolce function or acting independently. One percent of Pcolce−/− mice on the B6 background were viable and fertile “escapers”, suggesting the occurrence of rare recombination events in a relatively small distance separating Pcolce and locusC, indicative of linkage. Gene mapping and deep sequencing (Experimental Procedures) showed the embryonic lethal phenotype to be due to a spontaneous mutation in gene Wbscr16, situated ~3.43 Mbp from Pcolce. Subsequent matings to separate the Pcolce-null and Wbscr16 mutant alleles confirmed homozygosity for the latter to be sufficient to produce early lethality.
The human homologue gene WBSCR16 is located at the telomeric end of the WBSCR, one allele is deleted in some WBS patients, and WBSCR16 is directly adjacent to large deleted sequences in other WBS patients (Merla et al., 2002; Schubert, 2009).
WBSCR16 protein is in the RCC1 (Regulator of Chromosome Condensation 1) family of proteins, characterized by RCC1-like domains (RLDs) (Hadjebi et al., 2008). WBSCR16 has seven RLDs, and the Wbscr16 mutation comprised an NM_033572.2:c.922G>A transition (Fig. 1 C), resulting in a p.Glu308Lys substitution in RLD 5 and splicing out of a 164-bp downstream exon, with consequent in-frame deletion of RLD 6 (Fig. 1, D and E). The substituted Lys is highly conserved across vertebrate species (Fig. 1 F). In the RCC1 family, WBSCR16 is most closely related by sequence homology and domain structure to RCC1 itself and to TD-60, both of which are demonstrated GEFs with nuclear localization and roles in mitosis (Hadjebi et al., 2008; Papini et al., 2015). Immunohistochemistry showed WBSCR16 to be broadly distributed in tissues at E8.5, and at particularly high levels in TGCs and ectoplacental cone (Fig. S2, A and B). Mutant WBSC16 was also detectable (Fig. S2, C and D), showing some degree of stability. Databases [e.g.(Su et al., 2002)] show WBSCR16 to be broadly expressed in tissues and cells, consistent with involvement in processes common to a broad range of cell types.
WBSCR16 is a Mitochondrial Protein Important to Fusion
WBSCR16 is expressed in HeLa cells [e.g. (Su et al., 2002)], which thus presented a system for determining WBSCR16 subcellular localization/function in cells that normally express this protein. Transfection of HeLa cells with plasmids expressing red fluorescent protein (RFP) – tagged wild type or mutant WBSCR16 showed both to localize to mitochondria (Fig. 2 A). Consistent with this finding, use of two independent software methodologies (Bannai et al., 2002; Fukasawa et al., 2015) predicted WBSCR16 to have a mitochondrial targeting sequence, containing the core sequence RLGRRLS (residues 11–20).
Figure 2. WBSCR16 is a mitochondrial protein that controls fusion.
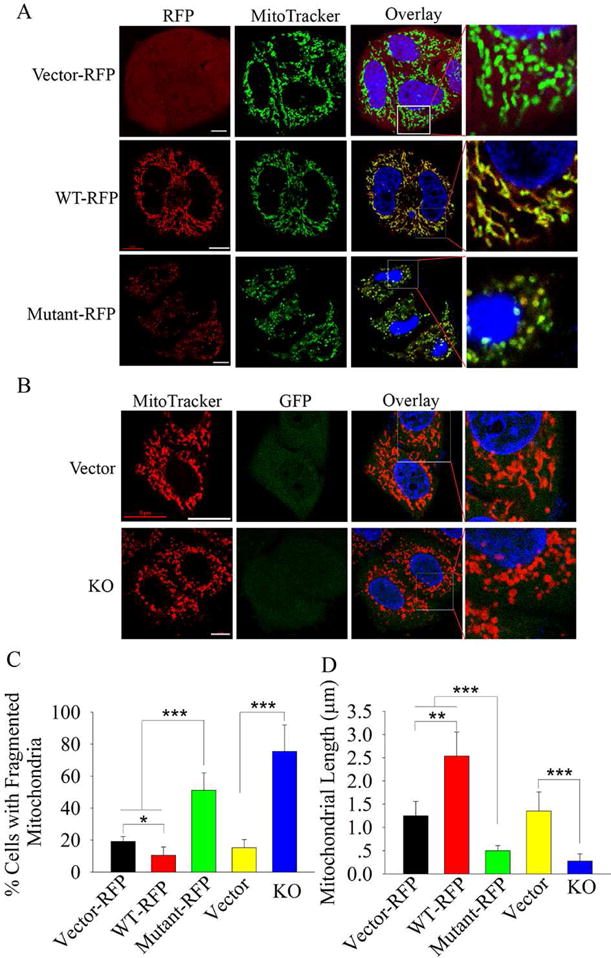
(A) HeLa cells transfected with vectors expressing RFP-tagged wild type or mutant WBSCR16 were stained with MitoTracker Green, which co-localized with both wild type and mutant WBSCR16 (overlay). (B) HeLa cells were transfected with empty vector (Vector) or vector for WBSCR16 CRISPR-Cas9 knockout (KO). Vector-expressed GFP identified successfully transfected cells. Enlargements of boxed areas of overlay results (column 4) more clearly show mitochondrial fragmentation upon WBSCR16 mutant overexpression (A) or WBSCR16 KO (B). Scale bars 5 μm. Quantification is shown for percentage of cells with fragmented mitochondria (C) and for mitochondrial length (D). For panel C, > 20 cells were scored from each set of cells transfected with a particular vector. For panel D, 10 mitochondria were measured per cell, for a total of > 20 cells per set of cells transfected with a particular vector.
Importantly, mutant WBSCR16 overexpression disrupted the fused mitochondria and tubular mitochondrial networks normally found in growing cells into individual, fragmented mitochondria (Fig. 2 A, C and D). We speculate this fission to be due to swamping of binding sites/partners of endogenous WT WBSCR16 by overexpressed, nonfunctional mutant WBSCR16. CRISPR/Cas-9 knockout of endogenous WBSCR16 also resulted in mitochondrial fragmentation (Fig. 2 B, C, and D), confirming loss of WBSCR16 function to be sufficient for this effect. Fragmentation was rescued by transfection of knockout cells with wild type, but not mutant WBSCR16-expressing vector (Fig. S3), further documenting ability of WBSCR16 to affect mitochondrial fusion. WBSCR16-knockout or mutant WBSCR16 overexpression also markedly decreased mitochondrial dynamics, such that mitochondrial motility and the frequency of fusion events were markedly decreased (Fig. S4, movies S1 and S2). Fragmentation and diminished motility and frequency of fusion are similar to previously described effects from loss of mitochondrial fusion GTPases MFN1, MFN2 (Chen et al., 2003), and OPA1 (Griparic et al., 2004).
WBSCR16 knockout also markedly decreased HeLa cell growth (Fig. 3 A), due to increased apoptosis (Fig. 3 B and C) and to blockage at the G1-S transition (Fig. 3 E and F, Table S1), which was associated with decreased cyclin E levels (Fig. 3 D). All these effects can occur secondarily to mitochondrial fragmentation (Kasahara and Scorrano, 2014; Mitra et al., 2009).
Figure 3. WBSCR16 knockout leads to markedly decreased cell growth, increased apoptosis, decreased cyclin E levels and blockage at the G1-S transition.
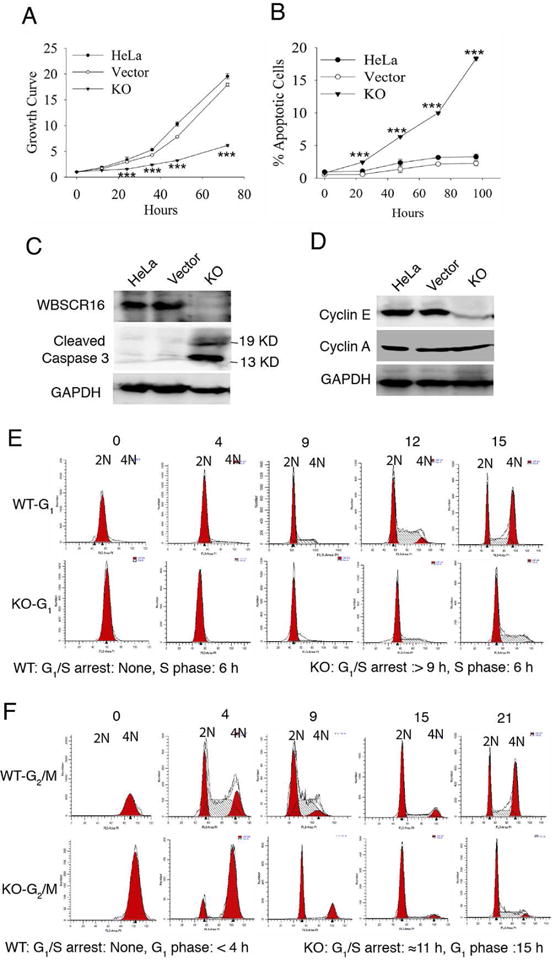
(A) MTT assay growth curves for untransfected HeLa cells, empty vector-transfected HeLa cells, or CRISPR-Cas9 WBSCR16 knockout (KO) HeLa cells. Ordinate axis: 1 is OD at time 0, with increasing numbers denoting fold increases thereafter. (B) FACS analysis shows increased apoptotic cells (sub-G1 DNA content) upon WBSCR16 CRISPR/Cas9 knockout. ***, P<0.005. Immunoblots show (C) increased cleaved caspase 3 and (D) reduced cyclin E in WBSCR16 CRISPR/Cas9 KO cells, compared with untransfected cells and cells transfected with empty vector. GAPDH, loading control. Cyclin A, additional loading control in D, showing cyclin E effects to be specific. (E) G1-phase HeLa cells, transfected with empty vector (WT) entered S-phase immediately. In contrast, G1-phase HeLa cells subjected to CRISPR-Cas9 WBSCR16 KO took at least 9 h to enter S-phase, with some entering after 12 h (blocked 9–12 h at the G1-S transition). Once in S, both WT and KO cells took 6 h to enter G2/M. (F) Both WT and WBSCR16-KO G2/M-phase cells entered G1-phase immediately. WT cells then transitioned quickly to S after G1, but KO cells were blocked in G1 for ~11 h, entering G1 only at the 15-h time point. Thus, experiments with G1-phase (E) and G2/M-phase (F) cells both indicate a block at G1-S transition in consequence of WBSCR16 ablation. See also Table S1.
WBSCR16 Affects Mitochondrial Fusion Via an OPA1/MFN1 Functional Axis and Interacts with OPA1 on the Outer Face of the Inner Membrane
To determine if WBSCR16 might act as a GEF to directly affect fusion via the mitochondrial GTPases that control this process, RFP-tagged WBSCR16 was overexpressed in mouse embryo fibroblasts (MEFs) defective for mitochondrial fusion due to absence of MFN1 (Mfn1−/−), MFN2 (Mfn2−/−), both MFN1 and 2 (Mfn1/2−/−), or OPA1 (Opa1−/−) (Chen et al., 2003; Song et al., 2007). WBSCR16 overexpression rescued mitochondrial fusion only in Mfn2−/− MEFs (Fig. 4, A and B). Thus, WBSCR16 depends on MFN1 and OPA1, but not MFN2 to induce mitochondrial fusion. OPA1 is known to require MFN1, but not MFN2, to promote mitochondrial fusion in a “functional axis” that appears to coordinate inner and outer membrane fusion (Cipolat et al., 2004), and the rescue experiments suggested WBSCR16 to operate via this OPA1-MFN axis (Cipolat et al., 2004). Interestingly, WBSCR16 overexpression in wild type MEFs, and even in Mfn2−/− MEFs, resulted in cells with considerably higher mitochondrial tubular network content (hyperfusion) than observed in control empty vector-transfected wild type MEFs (Fig. 4, C and D). Thus, increased intracellular WBSCR16 levels is sufficient to induce a hyperfused mitochondrial state in cells.
Figure 4. WBSCR16 relies on OPA1 and MFN1, but not MFN2 to drive fusion.
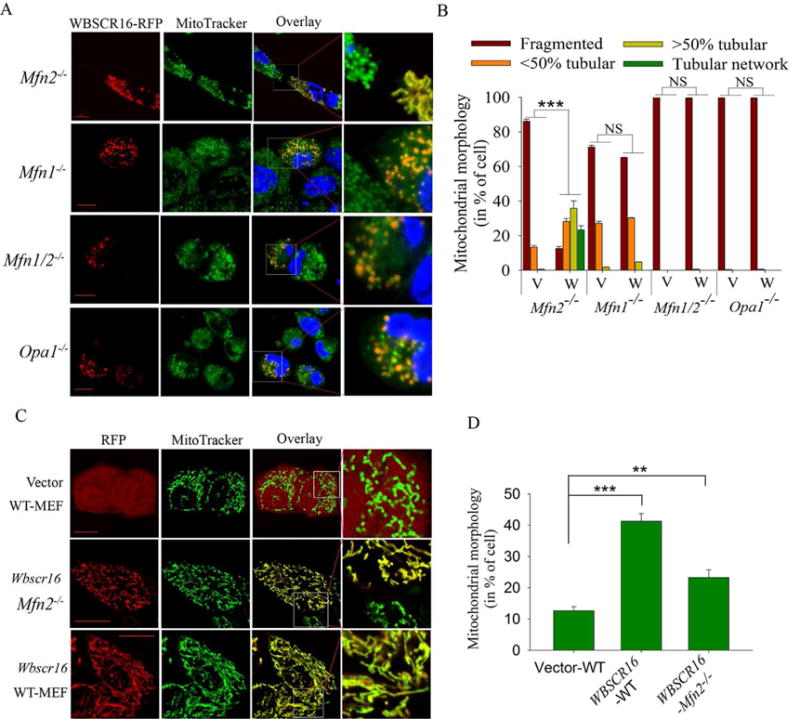
Visualization (A) and quantification (B) of transient WBSCR expression effects in Mfn2−/−, Mfn1−/−, Mfn1/2−/−, and Opa1−/− MEFs. (A) RFP-tagged WBSCR16 (column 1), MitoTracker Green (column 2) and overlay of the two signals (column 3). Boxed areas of overlay column are enlarged in column 4. (B) Quantification of results in >300 RFP-expressing cells per MEF genotype transfected with WBSCR16 (W) or empty vector (V). ***, P< 10−8. Visualization of representative cells (C) and quantification of % cells exhibiting a hyperfused tubular mitochondrial network (>70% of mitochondria per cell are fused within the tubular network) (D) for WT-MEFs transfected with empty vector (expressing only RFP) or WT or Mfn2−/− MEFs transfected for WBSCR16 overexpression. **, P<0.01; ***, P<0.005. Scale Bars 5 μm. In B and D, a minimum of 300 RFP-expressing cells were examined per group. Organelle morphology was microscopically assessed in at least 3 independent experiments.
Immunoprecipitations of endogenous MEF mitochondrial proteins showed WBSCR16 and OPA1 to be bound within mitochondria (Fig. 5, A and B), with OPA1 detected as multiple bands, consistent with the alternative splicing and proteolytic processing involved in its biosynthesis (Civiletto et al., 2015). However, WBSCR16 and MFN1 were not detectably bound (Fig. 5, A and C), consistent with the possibility that WBSCR16 may operate in the OPA1-MFN1 functional axis via direct interactions with OPA1, but not MFN1. Binding between OPA1 and MFN2 was not detected (Fig. 5D). Consistent with the possibility that WBSCR16-OPA1 interactions are direct, in vitro pull-down experiments employing recombinant proteins found WBSCR16 to be capable of binding OPA1 (Fig. S5). Interestingly, mutant WBSCR16 did not bind OPA1 in such assays (Fig. S5). Similarly, pull-down experiments from lysates of HeLa cells transfected for expression of RFP-wild type or -mutant WBSCR16 confirmed that WT, but not mutant WBSCR16 binds OPA1 in the cell. Such results are consistent with the conclusion that the WBSCR16 mutation produces loss of function related to inability to interact with OPA1.
Figure 5. WBSCR16 and OPA1 are bound and co-localize, and are in close proximity, on the outer face of the mitochondrial inner membrane.
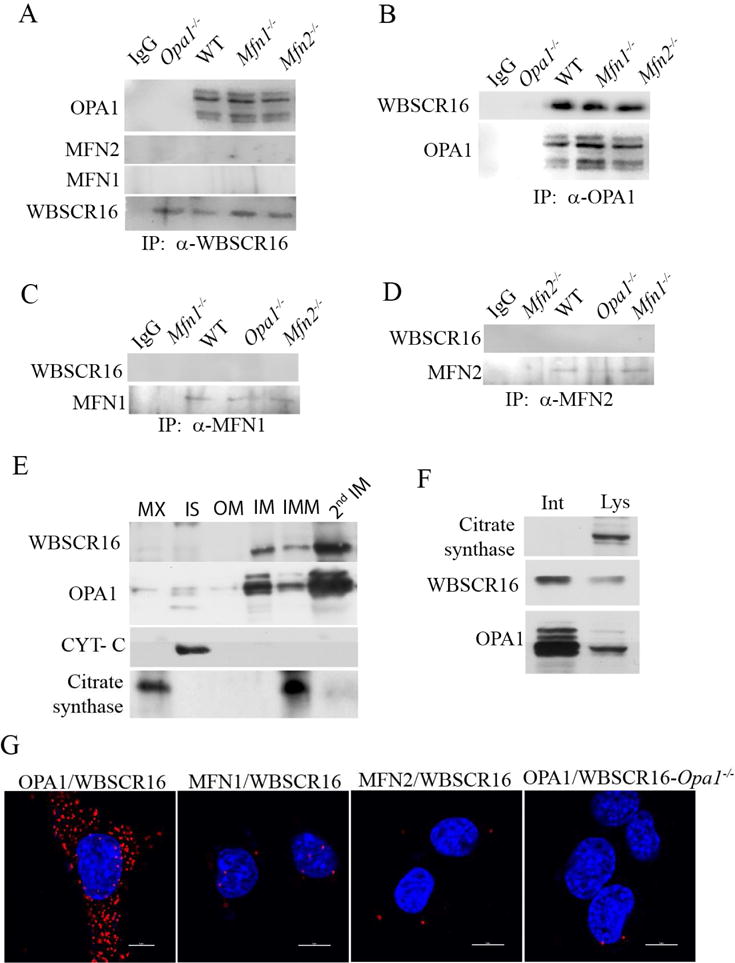
(A) Immunoblots show that immunoprecipitation of endogenous WBSCR from mitochondrial lysates co-precipitates OPA1, but not MFN1 or MFN2. Similarly, immunoprecipitation of (B) OPA1, but not MFN1 (C), or MFN2 (D) co-precipitated WBSCR16. (E) Mouse liver mitochondria were subfractionated and immunoblotting was performed to detect WBSCR16, OPA1, cytochrome c (marker of the intermembrane space, IS), and citrate synthase (matrix marker). OM, outer membrane; IM, inner membrane; IMM, pellet comprising matrix and some intact IM. Subsequent to additional sonication of the IMM pellet to disrupt remaining intact inner membrane, a second gradient was run to separate more highly purified matrix (MX), and inner membrane (2nd IM). (F) Mitoplasts were subjected to immunoprecipitation with antibodies for citrate synthase, WBSCR16, or OPA1 while intact (Int) or subsequent to lysis (Lys). (G) Generation of a signal in an in situ proximity ligation assay (PLA) shows close, <40 nm proximity of OPA1 and WBSCR16 40 nm, but absence of signal above background shows absence of close proximity for WBSCR16 with MFN1 or MFN2 in wild type MEFs (first three panels). 4th panel, a negative control in which PLA probing for WBSCR16 and OPA1 was employed on Opa1−/− MEFs.
The long, uncleaved form of OPA1, a form necessary for mitochondrial fusion, is an integral protein of the inner membrane (IM), with a short N-terminal transmembrane domain that penetrates the IM and protrudes into the matrix, and a more C-terminal GTPase domain exposed to the intermembrane space (IS) (MacVicar and Langer, 2016). To determine if WBSCR16 is also associated with the IM, mitochondrial subfractionation experiments were performed. Consistent with the possibility of in vivo OPA1-MFN1 interactions, most OPA1 and WBSCR16 were found within the IM fraction (Fig. 5 E). To determine if WBSCR16 was located on the IM outer or inner face, the outer membranes of purified mitochondria were stripped away with digitonin, and the resulting mitoplasts (matrix bound by intact inner membranes) were incubated with antibodies to WBSCR16, OPA1, or citrate synthase (a matrix protein), followed by immunoprecipitation of proteins exposed on the IM outer face. This initial immunoprecipitation was followed by washing of the same intact mitoplasts, and then lysis. Incubation of the lysate with antibodies to WBSCR16, OPA1 and citrate synthase, was then followed by immunoprecipitation, to obtain of the fractions of the 3 proteins associated with the IM inner face and matrix. The majority of WBSCR16 and, as expected, OPA1 were located on the IM outer face (Fig. 5F). Thus, most WBSCR16 is appropriately placed in vivo to interact with the OPA1 GTPase domain. As expected, all citrate synthase, which is localized to the IM inner face and in the matrix, was only detected upon mitoplast lysis. It is of interest that some WBSCR16, based on the subfractionation results of figure 5E and the mitoplast results of figure 5F, seemed localized to the IM inner face.
We next employed an in situ proximity ligation assay (PLA) in which separate probes are used for two proteins within a cell and a fluorescent signal is obtained only if the two proteins are in close enough proximity for direct protein-protein interactions (< 40 nm)(Ivanusic et al., 2014; Jarvius et al., 2007; Soderberg et al., 2006). The in situ PLA produced compelling results showing WBSCR16 and OPA1 to be in extremely close proximity in situ, within the cell (Fig. 5G), indicative of physical interaction between the two proteins. In contrast, in situ PLA did not detect co-localization/proximity of WBSCR16 with MFN1 or MFN2 (Fig. 5G).
WBSCR16 is an OPA1-specific GEF
Importantly, a direct in vitro GEF assay, involving ability to load a GTP analogue (mant-GTP) onto GTPases clearly showed WBSCR16 to act as a strong and rapid GEF for OPA1, in a concentration-dependent manner (Fig. 6, A and D). Thus, WBSCR16 is capable of catalyzing the GTP loading that leads to GTP-dependent conformational changes of OPA1 necessary to fusion (DeVay et al., 2009). In contrast, WBSCR16 had no detectable GEF activity towards MFN1 or MFN2 (Fig. 6 B–D). Small GTPase CDC42 and its cognate GEF Dbs were used as a positive control in the assay (Fig. 6 E), while Dbs and small GTPase Rac1, for which Dbs is not a GEF, were used as a negative control (Fig. 6F). WBSCR16 did not show GEF activity for either small GTPase CDC42 or Rac1 (Fig. S6, A and B).
Figure 6. WBSCR16 is a GEF for OPA1.
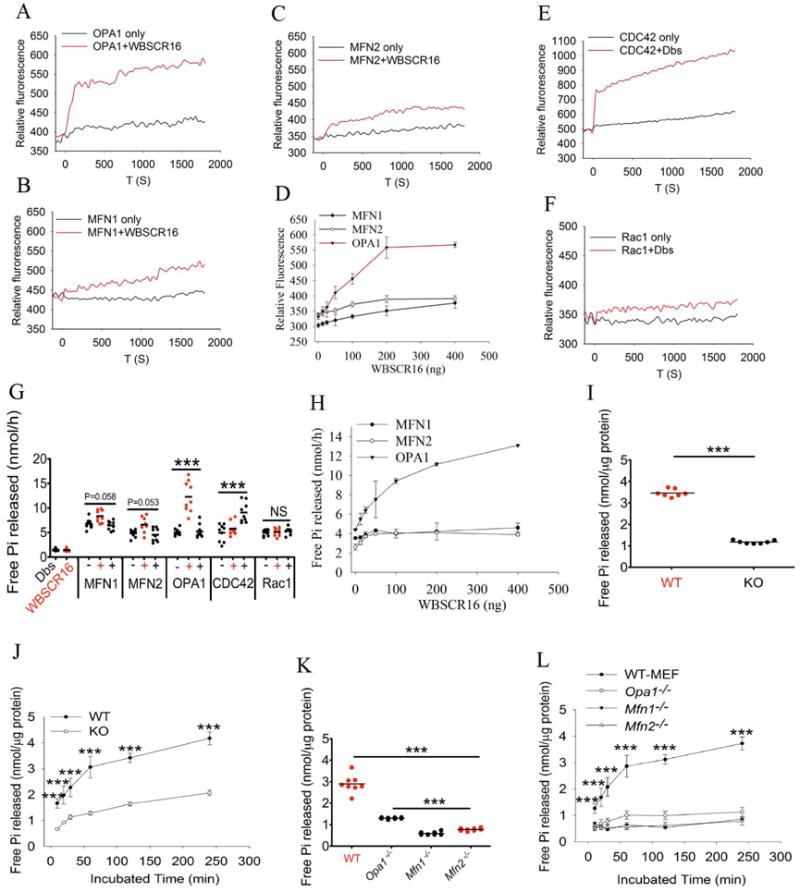
A GEF activity fluorescent assay (loading of mant-GTP) shows definitive WBSCR16 GEF activity for OPA1 (A), but not for MFN1 (B) or MFN2 (C). (D) WBSCR16 has concentration-dependent GEF activity for OPA1. Positive and negative controls for the assay employed Dbs, a GEF for CDC42 (E), but not for Rac1 (F). (G) WBSCR16 GEF activity is rate limiting for GTP hydrolysis by OPA1, but not by MFN1 or MFN2, or by small GTPases CDC42 or Rac1 in an in vitro assay. (H) WBSCR16 GEF effect on OPA1 GTP hydrolysis is concentration-dependent. Assays in panels A–H employed purified recombinant proteins (see Supplemental experimental procedures). (I) Endogenous GTPase activity is reduced in mitochondria from CRISPR/Cas-9 knockout WBSCR16-ablated HeLa cells. (J) Time course of GTP hydrolysis assay for isolated HeLa cell mitochondria. (K) Reduction of GTP hydrolysis in mitochondria isolated from Opa1−/−, Mfn1−/−, and Mfn2−/− MEFs. (L) Time course of GTP hydrolysis assay for isolated MEF mitochondria.
Since it has very recently been reported that WBSCR16 can affect intramitochondrial translation (Arroyo et al., 2016), we tested whether WBSCR16 might have GEF activity for the intramitochondrial GTPases mtEF-Tu and mtEF-G1, which act as translation elongation factors (Smits et al., 2010). However, WBSCR16 did not show GEF activity for either of the two GTPase-elongation factors in the mant-GTP loading assay (Fig. S6, C and D).
We next tested whether the GTP loading, GEF, activity of WBSCR16 genuinely increases the rate of GTPase hydrolysis by OPA1 (i.e. whether the GEF activity of WBSCR16 is rate limiting for OPA1 GTPase activity), which is necessary for inner membrane fusion (Escobar-Henriques and Anton, 2013). In an in vitro assay, GTP hydrolysis by OPA1 was markedly increased by WBSCR 16 in a concentration-dependent manner, whereas WBSCR16 had no effect on GTP hydrolysis by MFN1, MFN2, or by small GTPase controls CDC42 or Rac 1 (Fig. 6 G and H). In the context of the results of the mant-GTP loading assay (Fig. 6A), the most straightforward interpretation of the in vitro GTP hydrolysis results is that WBSCR16 is a GEF that acts to load OPA1 with GTP, thus providing more opportunity for intrinsic OPA1 GTPase activity. Subsequent to the in vitro GTP hydrolysis results performed with purified proteins, we tested whether mitochondria isolated from HeLa cells in which WBSCR16 had been ablated by CRISPR/Cas-9 had detectably reduced GTP hydrolysis activity. Importantly, GTP hydrolysis activity in WBSCR16-ablated mitochondria was substantially reduced relative to control HeLa cell mitochondria (Fig. 6 I and J), with the magnitude of lost GTP hydrolysis activity approaching that observed in mitochondria isolated from Opa1−/−, Mfn1−/−, or Mfn2−/− MEFs (Fig. 6 K and L). Thus, WBSCR16, like each of the three fusion GTPases, appears to be responsible for a substantial fraction of the GTP hydrolysis occurring within intact mitochondria, strongly supporting an in vivo role for WBSCR16 as a mitochondrial GEF.
Heterozygous Wbscr16 Mutant Neurons have Mitochondrial Deficits
In initial assays into the extent to which partial loss of WBSCR16 function might affect the function of neuronal mitochondria, we examined primary cultures of hippocampal neurons isolated from newborn mice heterozygous for the mutation described above. Mitochondria of the heterozygous mutant neurons had reduced membrane potential (Fig. 7, A and B) and were more susceptible to fragmentation in response to glutamate-induced excitotoxicity (Fig. 7, C and D), than were the mitochondria of wild type control neurons.
Figure 7. Wbscr16 heterozygous mutant neurons have reduced membrane potential and increased fragmentation in response to glutamate excitotoxicity.
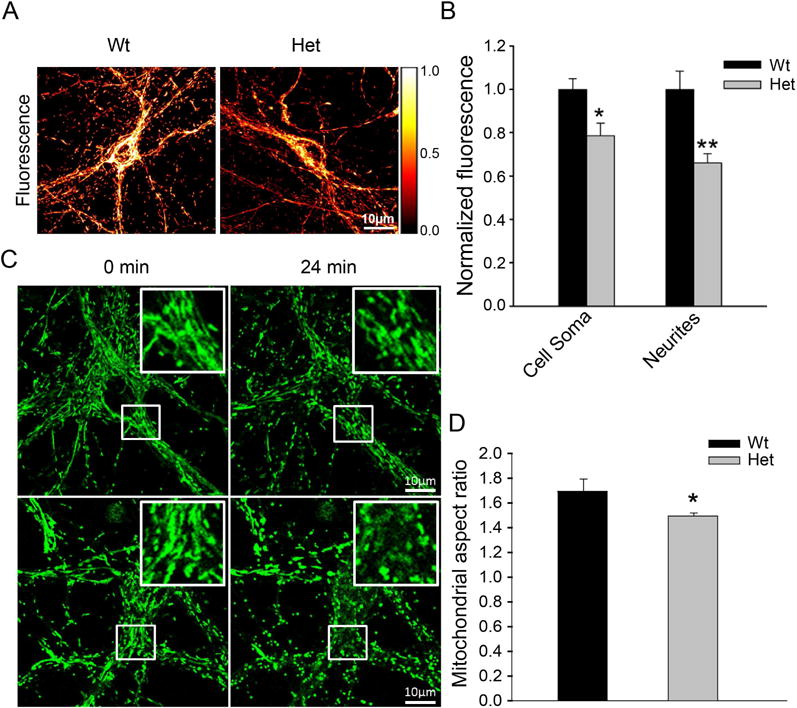
Representative images (A) and quantitative analysis (B) of fluorescence intensity of wild type (WT) and heterozygous mutant Wbscr16 (Het) neuronal cell soma and neurites, on 12–14 day in vitro, incubated with potentiometric dye TMRM. In panel A, a pseudocolor intensity scale represents white as maximum and black as lowest intensities. For quantitative analysis (B), N=32 and 40 for WT and Het, respectively, from 3 experiments. *, P<0.05; **, P<0.01. (C) Mitochondria in WT and Het neurons were labeled with MitoTracker Green and imaged for morphology before and after 50 μM glutamate exposure. Representative images show WT and Het neurons before (0 min) and 24 min after glutamate exposure. Inserts: enlarged images of boxed regions of neuronal cell soma. Scale bars 10 μm. (D) Quantification is shown for neuronal mitochondrial aspect ratios for WT (n=7) and Het (n=9), upon glutamate exposure. Data obtained from 5 different experiments. *, P<0.05 (P=0.017).
DISCUSSION
The present study identifies WBSCR16 as a component important to mitochondrial fusion. Time-lapse confocal microscopy showed a clear reduction in mitochondrial fusion events in the absence of WBSCR16 function, whereas WBSCR16 overexpression, as previously shown for OPA1 overexpression (Cipolat et al., 2004), produced mitochondrial hyperfusion, consistent with a direct role in fusion. Genetic rescue experiments in Opa1−/−, Mfn1−/−, Mfn2−/− and Mfn1/2−/− MEFs showed WBSCR16 induction of mitochondrial fusion to be independent of MFN2, but to be dependent upon the MFN1/OPA1 functional axis. Additionally, most WBSCR16 was shown, like OPA1, to localize to the outer face of the inner membrane, while the two proteins coimmunoprecipitated from mitochondrial lysates, indicating in vivo complex formation. Beyond these findings, in situ proximity ligation analysis (PLA) showed WBSCR16 and OPA1 to be in extremely close proximity to each other within intact cells, indicative of physical interaction. WBSCR16 was furthermore shown to provide highly OPA1-specific, dose-dependent GTP-loading (GEF) activity, and this GEF activity was shown to be rate limiting for OPA1 GTPase activity, which is known to induce the OPA1 conformational changes that cause inner membrane fusion (Escobar-Henriques and Anton, 2013). Thus, strong evidence is provided for a model in which WBSCR16 can contribute to regulation of mitochondrial fusion via its role as a GEF involved in regulating OPA1 activity.
Loss of WBSCR16 function is also shown to inhibit cell growth, due to increased apoptosis and blockage at the G1-S interface. As OPA1 controls both mitochondrial fusion and widening of cristae junctions, deficits in either of which can lead to apoptosis (Frezza et al., 2006; Kasahara and Scorrano, 2014), loss of WBSCR16 function may affect apoptosis secondary to effects on OPA1. It is of interest that blockage at the G1-S interface in cells with WBSCR16 deficits was associated with decreased cyclin E levels, as formation of “hyperfused” mitochondrial networks has been shown to be needed to generate increased cyclin E levels necessary for G1-to-S progression (Mitra et al., 2009). Thus, the G1-S block of WBSCR16-ablated cells may well occur secondary to mitochondrial fusion deficits. Our finding that WBSCR16 overexpression is sufficient to induce a mitochondrial hyperfusion suggests that WBSCR16 is not only necessary for optimal mitochondrial fusion, but that it may be a key inducer of mitochondrial fusion in cells, in response to environmental cues.
OPA1 is a member of the dynamin family of large GTPases. Previously, some members of the dynamin family that had been tested were found to have relatively low affinity for GDP, suggesting little need for GEF-facilitated nucleotide exchange (Praefcke and McMahon, 2004). However, clear-cut evidence is presented here that WBSCR16 exerts strong GEF activity for the loading of GTP onto OPA1, and that this GEF activity is rate-limiting for OPA1 GTPase activity. Thus, at least one dynamin-like GTPase is amenable to GEF-facilitated nucleotide exchange.
As this manuscript was being completed, a genome-wide screen by Arroyo et al (Arroyo et al., 2016) identified WBSCR16, among other genes, as essential for oxidative phosphorylation (OXPHOS), with suggested roles in intramitochondrial protein translation, in the human K562 cell line. We suggest that WBSCR16 effects on OXPHOS may be secondary to effects on mitochondrial fusion, as proper OXPHOS system assembly depends upon balanced mitochondrial fusion/fission, and as deficits in OPA1 or MFN2 function cause reduced mitochondrial membrane potential and OXPHOS enzyme activities (Chen and Chan, 2005; Liesa et al., 2009). In addition, it has been suggested that OPA1 plays roles in OXPHOS independent of its role in fusion, via physical interactions with respiratory chain complexes, assembly of respiratory chain supercomplexes, and cristae junction modeling (Bertholet et al., 2016). Thus, WBSCR16 may affect both fusion and OXPHOS via interactions with OPA1. In regard to possible WBSCR16 roles in intramitochondrial protein translation, it remains to be determined whether such effects may also be secondary to its effects on OPA1/mitochondrial fusion. We have shown that WBSCR16 did not act in in vitro assays as a GEF for the intramitochondrial translation elongation factor GTPases mtEF-Tu or mtEF-G1. However, we have not excluded the possibility that WBSCR16 might affect the activity of these two elongation factors in the presence of additional proteins in situ. Importantly, there is also no reason to exclude the possibility that WBSCR16 may affect mitochondrial fusion via OPA1, and affect OXPHOS and/or protein translation independently of OPA1. Certainly, although we found most WBSCR16 to be associated with the outer face of the inner membrane, we also detected some WBSCR16 associated with the inner face of the inner membrane as well.
Mitochondrial functional deficits can contribute to various human diseases, and deficits in mitochondrial fusion contribute to chronic neurodegenerative conditions that include Charcot-Marie-Tooth neuropathy type 2A, dominant optic atrophy, Parkinson’s disease and various dementias (Bertholet et al., 2016). Initial studies here of neurons from heterozygous mutant Wbscr16 mice shows them to have mitochondria with lowered membrane potential and increased susceptibility to fragmentation in response to glutamate excitotoxicity, a stressor implicated in the pathophysiology of various human neurological disorders (Pivovarova and Andrews, 2010). Thus, WBSCR16 is a candidate gene in which functional deficits may contribute to neuronal pathologies. In WBS itself, most contiguous gene deletions comprise ~1.5 megabase pairs (Mbp) that do not include WBSCR16 (Schubert, 2009). However, WBSCR16, and a set of adjacent genes, are included in some larger and atypical deletions [(Fusco et al., 2014; Schubert, 2009) and unpublished data]. Efforts are underway to determine phenotypic differences in WBS patients in which WBSCR16 has or has not been deleted.
Interestingly, OPA1 overexpression can ameliorate abnormal phenotypes in some mouse models of mitochondrial disease (Civiletto et al., 2015). Thus, as WBSCR16 is a GEF that enhances OPA1 function (and which can induce mitochondrial fusion upon overexpression), the potential exists that induction of elevated WBSCR16 levels in vivo may ameliorate phenotypes associated with mitochondrial functional deficits, as an approach towards therapeutic interventions.
EXPERIMENTAL PROCEDURES
Genetic Mapping, Deep Sequencing, and Animal Care
Extensive backcrossing the Pcolce–null allele onto a B6 background showed that a single copy of the Black Swiss allele at locusC was sufficient to confer viability on Pcolce−/− embryos. LocusC was mapped by identifying genome regions with decreased average homozygosity for B6 SNPs. SNPs between the parental Black Swiss and B6 backgrounds were identified by us via the Affymetrix Mouse Diversity Array. Decreased heterozygosity at two SNPs and 2-point linkage analysis indicated that locusC could be located between 45 and 80 cM on Chr 5. Pcolce is within this region (at 76 cM). To verify Pcolce − locusC linkage, a Black Swiss Pcolce+/+ male was crossed with B6 Pcolce+/− females to produce Pcolce+/− progeny in which the Pcolce null allele and surrounding region were contributed by the B6 parent. Pcolce+/− F1 mice were then backcrossed to B6 Pcolce+/− heterozygotes. The 6% of resulting N2 neonates that were Pcolce−/− was significantly less than the 20% expected if locusC had segregated independently of Pcolce, thus verifying linkage within 45 – 80 cM. Ten generations of backcrossing Black Swiss Pcolce−/− mice with newly purchased (Harlan Labs) B6 mice yielded a proper Mendelian ratio of viable Pcolce−/− B6 neonates, confirming the lethal polymorphism at locusC to be a spontaneous mutation from the original Pcolce−/− Black Swiss X B6 crosses rather than a B6 modifier gene.
Further mapping narrowed the region containing the causative mutation to a 4.6 Mb span 1.5 Mb upstream of Pcolce. Deep sequencing and aligning results to the reference mouse genome found an NM_033572.2:c.922G>A transition in Wbscr16 and no other polymorphisms in coding or noncoding sequences within the 4.6 Mb span. Subsequent matings to separate the Pcolce-null and Wbscr16 mutant alleles confirmed homozygosity for the latter to be sufficient to produce the early lethal phenotype. See Supplemental Experimental Procedures for mapping and deep sequencing details.
All mice were housed and treated in accordance with NIH guidelines, using protocols approved by the Research Animal Resources Center of the University of Wisconsin-Madison.
Histology and Immunohistology
Pregnant females were sacrificed at 8.5 dpc, and uteruses collected. Embryos were dissected from uteruses, formalin fixed and paraffin embedded. Immunohistochemical staining details are supplied in Supplemental experimental Procedures.
Plasmid Construction and Recombinant Proteins
Plasmids and oligonucleotide primers used in this study are described in Supplemental Experimental Procedures.
N-terminal FLAG-tagged Recombinant WBSCR16, for in vitro pull down experiments and in vitro GEF/GTPase assays, was produced from human WBSCR16 sequences codon optimized, synthetically generated and subcloned into pFastBacI by GeneArt. Protein was produced in Sf9 insect cells and purified on an anti-FLAG M2 (Sigma) affinity column. Recombinant human proteins purchased for the GEF/GTPase assay and pull down experiments are described in Supplemental Experimental Procedures.
Cell Culture and Imaging
Mfn1−/−, Mfn2−/−, Mfn1/2−/−, and Opa1−/− MEFs11, 19 and Hela cells were obtained from ATCC. In live-cell experiments, cells were plated on collagen coated 14 mm glass bottom dishes (MatTek, USA). After incubation with 100 nM MitoTracker (Cell Signal), cells were imaged in live cell imaging solution (ThermoFisher) on the 37 °C preheated stage of a Nikon A1RS confocal microscope. Cells were imaged with an oil immersion PlanApo 60X objective and appropriate laser lines for each fluorophore were used.
Cell Proliferation and Apoptosis Assays
Proliferation was measured by standard MTT assay. Floating cells in medium and trypsinized adherent cells were harvested, combined, and fixed in 70% ethanol. Cells were then treated 1 h with RNase A (200 μg/ml) prior to propidium iodide (Invitrogen, 50 μg/ml) staining. DNA content was detected by flow cytometry and percent apoptotic cells with sub-G1 DNA content was determined.
In addition to the above, floating cells were harvested and combined with adherent cells for extraction with SDS-loading buffer. Samples were then boiled and subjected to SDS-PAGE and immunoblotting with antibody (1:1000, Cell Signaling) for cleaved caspase 3.
Assays of transfected cells were performed 36 h post transfection. Additional details are in Supplemental Experimental Procedures.
Cell Cycle Analysis of WBSCR16-ablated HeLa Cells
HeLa cells, transfected with empty vector or with vector containing WBSCR16-specific sequences for CRISPR-Cas9 knockout were stained 36 h later with Hoechst 33342 (2 mg/ml and 30 min incubation at 37°C), sorted by FACS into G1-, S-, and G2/M-phase cells, then released into culture medium. Cells were then harvested at different time points, fixed with 70% ethanol, and incubated 30 min with 200 μg/ml RNase A and 50 mg/ml propidium iodide at 37°C, for DNA profiling (Fig. S5). FACS was via FACSCalibur flow cytometer and data were analyzed with FlowJo software.
Cell lysis and Immunoprecipitation Assays
Cells were lysed with RIPA buffer containing EDTA-free protease inhibitors (Roche) and 25 units alkaline phosphatase (NEB) per ml cell lysate. After centrifugation, supernatants were incubated with anti-MFN1, -MFN2 (Origene), -OPA1 (cell signaling) or -WBSCR16 (Abcam) antibodies coupled to Aminolink Resin (Pierce Direct IP kit, Thermo Scientific). Resin/antibody-bound proteins were subjected to SDS-PAGE and immunoblotting. Additional details are in Supplemental Experimental Procedures, as is detailed description of in vitro pull down assays of recombinant proteins.
In vitro GEF Exchange and GTPase Activity Assays
GEF exchange and GTPase activity assays were carried out with the RhoGEF Exchange Assay Biochem Kit (BK100, Cytoskeleton), and GTPase/ATPase ELIPA Biochem Kit (BK051, Cytoskeleton), respectively, using manufacturer’s protocols, and with purified recombinant proteins, described above. Full details are in Supplemental Experimental Procedures.
Assays of Total GTPase Activity of Isolated Mitochondria
4 × 107 cells were harvested, washed with phosphate-free buffer A (20 mM Tris-HCl, 150 mM NaCl, pH 7.4), and mitochondria were isolated with a Mitochondria Isolation Kit (ThermoFisher). Isolated mitochondria were suspended in EBc buffer (10 mM Tris-MOPS, 125 mM KCl, 5 mM MgCl2, pH 7.4). After 30 min, mitochondria were centrifuged (7,000 g, 10 min), resuspended in EBc buffer, and 50 μl cell suspension was then aliquoted per tube. Tubes were supplemented with 50 μl EBc buffer containing 100 μM GTP, incubated at room temperature, and supernatant was harvested after 10, 20, and 30 min, or after 1, 2 and 4 h. Free Pi concentrations were analyzed via the GTPase/ATPase ELIPA Biochem Kit (BK051).
Subfractionation of Mouse Liver Mitochondria
Mouse liver mitochondria were isolated as described (Frezza et al., 2007), and subfractionated by a modification of the procedure of Hovius et al. (Hovius et al., 1990). Briefly, mitochondrial pellets were suspended in 10 mM KH2PO4 (pH 7.4), incubated 15 min, diluted 2-fold with buffer B (32%(w/v) sucrose, 30% glycerol (v/v), 10 mM MgCl2 in 10 mM KH2PO4, pH 7.4), followed by sonication and then centrifugation. Supernatant was loaded onto a discontinuous sucrose gradient and 4 fractions were collected: Top supernatant (intermembrane space, IS); 25.3/37.7% interface membrane protein (outer membrane, OM); and 37.7/51.3% interface membrane protein (inner membrane, IM) fractions were diluted 3-fold in buffer A (250 mM mannitol, 0.5 mM EGTA, 5 mM Hepes, pH 7.4). The bottom pellet (inner membrane plus matrix, IMM) was resuspended in buffer A, or in buffer B, diluted 2-fold with 10 mM KH2PO4 (pH 7.4), for further sonication. Additional sonication was followed by centrifugation, and the supernatant was then loaded onto a 2 ml discontinuous sucrose gradient and centrifuged, as above. Supernatant from this gradient contained soluble matrix proteins (MX), while the pellet contained the “2nd IM” fraction of inner membrane purified away from soluble matrix proteins. Proteins of each fraction were collected 10 min at 16,000 g at 4 °C, resuspended in SDS loading buffer, and immunoblotted.
To determine the orientation of WBSCR16 bound to the inner membrane, mitoplasts were prepared and subjected to immunoprecipitations before and after lysis, as described (Tokarska-Schlattner et al., 2008). Briefly, purified mitochondria were suspended in isotonic buffer A, containing 0.1% (w/v) bovine serum albumin and protease inhibitors on ice for 15 min with 0.3 mg digitonin/mg protein. After washing with buffer A, mitoplasts were resuspended in buffer A with antibodies coupled to Aminolink Resin (Thermo Fisher). Antibodies were then recovered, rinsed with RIPA buffer (150 mM NaCl, 50 mM Tris-HCl, pH7.5, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 1 mM phenylmethylsulfonyl fluoride and protease inhibitors), and suspended in Laemmli buffer. After 3 washes in buffer A to remove unbound antibodies, intact mitoplasts were suspended in RIPA buffer containing Aminolink-coupled antibodies, and lysed mitoplasts were incubated 1 h at 4 °C. Antibodies linked to resin were then recovered, rinsed with RIPA, and suspended in Laemmli buffer. Full subfractionation and mitoplast study details are in Supplemental Experimental Procedures.
Proximity Ligation Assay (PLA)
The proximity ligation assay was performed essentially as described by Gauthier et al (Gauthier et al., 2015). Briefly, after culturing 24 h on coverslips, cells were fixed 15 min with 4% paraformaldehyde at room temperature, and then permeabilized 20 min in 4°C methanol. After washing twice with PBS, coverslips were blocked 1 h with 1% BSA in PBS at room temperature, and then incubated overnight at 4°C with mouse anti-WBSCR16 primary antibodies (Sigma), together with rabbit primary antibodies to MFN1, MFN2 (Origene), or OPA1 (Cell Signaling). In Situ PLA Probes Anti-Mouse MINUS (DUO82004) and Anti-Rabbit PLUS (DUO82002) and additional reagents were from the Duolink In situ-Fluorescence kit (Sigma), and were employed using manufacture’s guidelines. Coverslips were mounted and Fluorescence was visualized with a Nikon A1RS confocal microscope with an oil immersion PlanApo 60X objective.
Study of Neurons from heterozygous mutant Wbscr16 Mice
After rinsing with cold high glucose DMEM (HGDMEM), neonatal hippocampus was dissociated with papain (2.5 mg/ml, 30 min at 37°C), with suspensions passed through a 100 μm cell strainer. Centrifuged (200 g for 5 min) cells were then resuspended in HGDMEM, 10% fetal bovine serum, followed by plating on poly-D-lysine-coated cover slips. Cells were switched to neurobasal B27 medium after 5 h of plating, and after 48 h were treated 48 h with 1 μM Ara-c. Cells were then maintained in neurobasal B27 medium. Mitochondrial membrane potential was measured with potentiometric fluorescence dye TMRM, as described (Joshi and Bakowska, 2011). For fragmentation assays, cells were labeled 30 min with 250 nM MitoTracker Green and time-lapse imaging was performed to observe mitochondrial morphology before/after 50 μM glutamate exposure. Mitochondrial aspect ratio (length/width) was calculated by averaging thousands of mitochondria from multiple images, in an unbiased way (using overall, rather than region-specific fields), using NIH imageJ software. After color thresholding, MitoTracker Green images were binarized and subjected to particle analysis to obtain aspect ratios.
Statistics
Analyses were performed with two-tailed Student’s t test, or a chi-square test where noted, with differences considered significant at P < 0.05. Data are presented as mean ± S.D., except for figure 7, in which data are presented as mean ± S.E. Linkage analysis was performed using two-point LOD scores, with scores above 3.0 considered significant evidence of linkage.
Supplementary Material
Cells were stained with MitoTracker-Green. One frame was acquired every 15 sec in a 10 min span. Selected fusion/fission events are highlighted with red arrows.
Cells were stained with MitoTracker-Green. One frame was acquired every 15 sec in a 10 min span. Selected fusion/fission events are highlighted with red arrows.
Acknowledgments
We thank Karen Downs, Bill Bement and David Pagliarini for helpful conversations, Kevin Sonnemann for help in producing recombinant WBSCR16, and Eshwar Udho for excellent technical assistance. This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Disease (NIAMS) grant R01AR047746 to D.S.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes Supplemental Experimental Procedures, Figures S1–S5, Table S1, and Movies S1 and S2.
AUTHOR CONTRIBUTIONS
G.H., D.M., A.M.M., C-L.Z. and D.C.J. performed and helped conceptualize experiments. S.Y.C. helped conceptualize and supervise some experiments. D.S.G. conceptualized the study and some experiments and wrote the paper.
References
- Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, Mootha VK. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab. 2016 doi: 10.1016/j.cmet.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai H, Tamada Y, Maruyama O, Nakai K, Miyano S. Extensive feature detection of N-terminal protein sorting signals. Bioinformatics. 2002;18:298–305. doi: 10.1093/bioinformatics/18.2.298. [DOI] [PubMed] [Google Scholar]
- Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, Arnaune-Pelloquin L, Davezac N, Mils V, Miquel MC, et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol Dis. 2016;90:3–19. doi: 10.1016/j.nbd.2015.10.011. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Emerging functions of mammalian mitochondrial fusion and fission. Hum Mol Genet. 2005;14:R283–289. doi: 10.1093/hmg/ddi270. Spec No 2. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, Zeviani M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015;21:845–854. doi: 10.1016/j.cmet.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- DeVay RM, Dominguez-Ramirez L, Lackner LL, Hoppins S, Stahlberg H, Nunnari J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J Cell Biol. 2009;186:793–803. doi: 10.1083/jcb.200906098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Henriques M, Anton F. Mechanistic perspective of mitochondrial fusion: tubulation vs. fragmentation. Biochim Biophys Acta. 2013;1833:162–175. doi: 10.1016/j.bbamcr.2012.07.016. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K. MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics. 2015;14:1113–1126. doi: 10.1074/mcp.M114.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco C, Micale L, Augello B, Teresa Pellico M, Menghini D, Alfieri P, Cristina Digilio M, Mandriani B, Carella M, Palumbo O, et al. Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur J Hum Genet. 2014;22:64–70. doi: 10.1038/ejhg.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier T, Claude-Taupin A, Delage-Mourroux R, Boyer-Guittaut M, Hervouet E. Proximity Ligation In situ Assay is a Powerful Tool to Monitor Specific ATG Protein Interactions following Autophagy Induction. PLoS One. 2015;10:e0128701. doi: 10.1371/journal.pone.0128701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. 2004;279:18792–18798. doi: 10.1074/jbc.M400920200. [DOI] [PubMed] [Google Scholar]
- Gu JM, Crawley JT, Ferrell G, Zhang F, Li W, Esmon NL, Esmon CT. Disruption of the endothelial cell protein C receptor gene in mice causes placental thrombosis and early embryonic lethality. J Biol Chem. 2002;277:43335–43343. doi: 10.1074/jbc.M207538200. [DOI] [PubMed] [Google Scholar]
- Hadjebi O, Casas-Terradellas E, Garcia-Gonzalo FR, Rosa JL. The RCC1 superfamily: from genes, to function, to disease. Biochim Biophys Acta. 2008;1783:1467–1479. doi: 10.1016/j.bbamcr.2008.03.015. [DOI] [PubMed] [Google Scholar]
- Hovius R, Lambrechts H, Nicolay K, de Kruijff B. Improved methods to isolate and subfractionate rat liver mitochondria. Lipid composition of the inner and outer membrane. Biochim Biophys Acta. 1990;1021:217–226. doi: 10.1016/0005-2736(90)90036-n. [DOI] [PubMed] [Google Scholar]
- Ivanusic D, Eschricht M, Denner J. Investigation of membrane protein-protein interactions using correlative FRET-PLA. Biotechniques. 2014;57:188–191. 193–188. doi: 10.2144/000114215. [DOI] [PubMed] [Google Scholar]
- Jarvius M, Paulsson J, Weibrecht I, Leuchowius KJ, Andersson AC, Wahlby C, Gullberg M, Botling J, Sjoblom T, Markova B, et al. In situ detection of phosphorylated platelet-derived growth factor receptor beta using a generalized proximity ligation method. Mol Cell Proteomics. 2007;6:1500–1509. doi: 10.1074/mcp.M700166-MCP200. [DOI] [PubMed] [Google Scholar]
- Joshi DC, Bakowska JC. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J Vis Exp. 2011 doi: 10.3791/2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Ichas F, Holmuhamedov EL, Camacho P, Lechleiter JD. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 1995;377:438–441. doi: 10.1038/377438a0. [DOI] [PubMed] [Google Scholar]
- Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev. 2009;89:799–845. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- MacVicar T, Langer T. OPA1 processing in cell death and disease - the long and short of it. J Cell Sci. 2016;129:2297–2306. doi: 10.1242/jcs.159186. [DOI] [PubMed] [Google Scholar]
- Merla G, Ucla C, Guipponi M, Reymond A. Identification of additional transcripts in the Williams-Beuren syndrome critical region. Hum Genet. 2002;110:429–438. doi: 10.1007/s00439-002-0710-x. [DOI] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papini D, Langemeyer L, Abad MA, Kerr A, Samejima I, Eyers PA, Jeyaprakash AA, Higgins JM, Barr FA, Earnshaw WC. TD-60 links RalA GTPase function to the CPC in mitosis. Nat Commun. 2015;6:7678. doi: 10.1038/ncomms8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L, Scorrano L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu Rev Physiol. 2016;78:505–531. doi: 10.1146/annurev-physiol-021115-105011. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Andrews SB. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010;277:3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- Schubert C. The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci. 2009;66:1178–1197. doi: 10.1007/s00018-008-8401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits P, Smeitink J, van den Heuvel L. Mitochondrial translation and beyond: processes implicated in combined oxidative phosphorylation deficiencies. J Biomed Biotechnol. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiglitz BM, Kreider JM, Frankenburg EP, Pappano WN, Hoffman GG, Meganck JA, Liang X, Hook M, Birk DE, Goldstein SA, et al. Procollagen C proteinase enhancer 1 genes are important determinants of the mechanical properties and geometry of bone and the ultrastructure of connective tissues. Mol Cell Biol. 2006;26:238–249. doi: 10.1128/MCB.26.1.238-249.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarska-Schlattner M, Boissan M, Munier A, Borot C, Mailleau C, Speer O, Schlattner U, Lacombe ML. The nucleoside diphosphate kinase D (NM23-H4) binds the inner mitochondrial membrane with high affinity to cardiolipin and couples nucleotide transfer with respiration. J Biol Chem. 2008;283:26198–26207. doi: 10.1074/jbc.M803132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cells were stained with MitoTracker-Green. One frame was acquired every 15 sec in a 10 min span. Selected fusion/fission events are highlighted with red arrows.
Cells were stained with MitoTracker-Green. One frame was acquired every 15 sec in a 10 min span. Selected fusion/fission events are highlighted with red arrows.