

Abstract
Purpose
There is growing evidence that inflammation is a central and reversible mechanism through which obesity promotes cancer risk and progression.
Methods
We review recent findings regarding obesity-associated alterations in the microenvironment and the local and systemic mechanisms through which these changes support tumor growth.
Results
Locally, hyperadiposity is associated with altered adipose tissue function, adipocyte death, and chronic low-grade inflammation. Most individuals who are obese harbor inflamed adipose tissue, which resembles chronically injured tissue, with immune cell infiltration and remodeling. Within this distinctly altered local environment, several pathophysiologic changes are found that may promote breast and other cancers. Consistently, adipose tissue inflammation is associated with a worse prognosis in patients with breast and tongue cancers. Systemically, the metabolic syndrome, including dyslipidemia and insulin resistance, occurs in the setting of adipose inflammation and operates in concert with local mechanisms to sustain the inflamed microenvironment and promote tumor growth. Importantly, adipose inflammation and its protumor consequences can be found in some individuals who are not considered to be obese or overweight by body mass index.
Conclusion
The tumor-promoting effects of obesity occur at the local level via adipose inflammation and associated alterations in the microenvironment, as well as systemically via circulating metabolic and inflammatory mediators associated with adipose inflammation. Accurately characterizing the obese state and identifying patients at increased risk for cancer development and progression will likely require more precise assessments than body mass index alone. Biomarkers of adipose tissue inflammation would help to identify high-risk populations. Moreover, adipose inflammation is a reversible process and represents a novel therapeutic target that warrants further study to break the obesity-cancer link.
INTRODUCTION
The importance of the tumor microenvironment in the development, growth, and progression of cancer is now well recognized. Studies of the diverse constituents of the microenvironment, such as immune cells, and their complex interactions are leading to the identification of novel therapeutic targets. Composed of an intricate network of heterogeneous cell types, the tumor microenvironment includes infiltrating immune cells, such as lymphocytes (ie, T and B cells), mast cells, and antigen-presenting cells (ie, macrophages and dendritic cells), as well as granulocytes, cancer-associated fibroblasts, endothelial cells, extracellular matrix (ECM), and other stromal components. Disruption of the normal functions of this complex network can cause epithelial dysfunction and ultimately carcinogenesis, as well as tumor promotion.
Chronic inflammation is a classic and prevalent example of ongoing perturbation within the microenvironment.1-3 Moreover, epidemiologic data have established an association between chronic inflammation and the development and progression of several cancers, including gastric, esophageal, colorectal, liver, pancreatic, bladder, and lung.1,3 Infectious agents that cause chronic inflammation and an increased risk of cancer include Helicobacter pylori, Schistosoma haematobium, and hepatitis B and C viruses.4-6 Noninfectious inflammatory stimuli, such as tobacco smoking and alcohol consumption, have also been identified.7,8 In addition, obesity, which is rapidly increasing in global prevalence, is now a recognized cause of chronic subclinical inflammation.9-12 Multiple systemic and local pathways mediate the pleiotropic effects of obesity, which include cancer promotion.10,13 Our understanding of the mechanisms linking obesity to cancer has rapidly evolved within the past decade, and the microenvironment is a critical component.
Obesity is currently defined by an elevated body mass index (BMI), typically as a consequence of excess adipose tissue. Adipose tissue is a dynamic endocrine organ that is responsible for energy homeostasis. Unchecked hyperadiposity commonly leads to metabolic disorders, altered production of steroid hormones, and chronic subclinical inflammation.10,14,15 These pathophysiologic effects have been associated with tumor development and progression.10,15,16 It is particularly interesting to note that several obesity-associated cancers, such as those of the breast and visceral organs, arise either within or adjacent to adipose depots. This suggests that altered adipose biology, commonly found in the setting of elevated BMI, locally promotes several types of cancer. A challenge to using BMI to identify at-risk individuals, however, is that adipose tissue dysfunction and its consequences do not exclusively occur in patients who are currently defined as obese. For example, insulin resistance and inflammation have been reported in individuals who have a normal BMI.17-19 Conversely, a minority of individuals with elevated BMI are metabolically healthy.20-22 Accordingly, the current definition of obesity on the basis of BMI has recently been called into question,23,24 and BMI is inadequate for the identification of patients with adipose inflammation. If anthropometric phenotype does not always accurately reflect overall health or, specifically, the inflammatory status of adipose tissue, then identifying underlying alterations in pathophysiology may be more useful in defining the obesity-cancer relationship. Characterizing the obese state by systemic and tissue-specific measures could provide more reliable identification of high-risk populations and a mechanistically informed approach for the development of effective prevention and treatment strategies that target cancers arising in the setting of adipose inflammation. Here, we discuss the mechanisms through which adipose tissue inflammation fosters a tumor-supportive microenvironment through local and systemic effects.
INFLAMMATION AND THE MICROENVIRONMENT
The tumor microenvironment closely resembles that of a healing wound.2 Tissue injury and the ensuing inflammation promote enhanced cellular proliferation via influx of immune cells, production of proinflammatory mediators and growth factors, tissue remodeling, and angiogenesis.3 The initiating events in response to tissue injury include platelet activation and aggregation, and stimulation of the coagulation cascade. In addition to achieving hemostasis, these initiating events also lead to the production and secretion of several proteins that stimulate a local inflammatory response. For example, platelet-derived growth factor, transforming growth factor-β, and several complement factors stimulate neutrophil chemotaxis.3 Once engaged, neutrophils continue the cascade by producing cytokines and chemotactic factors that recruit and activate effector cells. Specifically, factors such as platelet-derived growth factor, transforming growth factor-β, monocyte chemoattractant protein-1 (MCP-1), interleukin (IL)-1β, tumor necrosis factor-α (TNF-α), and others guide circulating mononuclear phagocytes to the site of injury.3 Once present, these progenitor cells differentiate into mature macrophages, which assume the main role of cytokine and growth factor production. These macrophage products have profound effects on the local microenvironment, including stimulation of angiogenesis and modulation of the ECM.
Chronic tissue injury, such as adipose tissue inflammation, can stimulate the same wound healing mechanisms and generate a proneoplastic microenvironment.25 Once established, malignant cells may co-opt the inflammatory mechanisms responsible for tissue repair and instead promote tumor growth and invasion. Obesity is a common cause of chronic inflammation, both systemically and at the tissue level.10 Locally, white adipose tissue (WAT) in patients who are obese is infiltrated by immune cells, including macrophages and lymphocytes. In this manner, the obese fat pad resembles chronically injured tissue and can be a rich source of proinflammatory mediators, potentially fostering tumor growth. Local inflammatory mechanisms that promote tumor development and growth will be discussed in further detail.
WAT INFLAMMATION
Adipose tissue inflammation may be a key process by which obesity promotes cancer (Fig 1).10 As adipose tissue outgrows its blood supply, leading to hypoxia, adipocyte stress and death may occur.15 This is paralleled by the increased production of MCP-1 and several other cytokines that are traditionally involved in wound healing.26 In addition to recruitment, MCP-1 may also stimulate proliferation of macrophages within adipose tissue.27 Macrophages form an envelope around dead or dying adipocytes in a configuration termed crown-like structures (CLS), a histologic biomarker of inflammation.28 The macrophages that form CLS engage in phagocytosis of a dead or dying adipocyte and become lipid loaded, forming foam cells.29 Free fatty acids (FFAs) released from the entrapped adipocyte and from other sources can activate toll-like receptor (TLR) 4 on the macrophage plasma membrane, leading to increased nuclear factor kappa B-dependent expression of proinflammatory genes, including TNF-α, IL-1β, and cyclooxygenase-2 (COX-2).30 Lipolysis and release of FFAs are further stimulated by TNF-α and other cytokines, thereby sustaining WAT inflammation. Consistent with this potential mechanism, elevated levels of proinflammatory mediators are found in association with CLS in visceral fat. Enlarged adipocytes are another potential source of proinflammatory mediators.
Fig 1.

Local and circulating effects of white adipose tissue inflammation can promote tumor growth. Several circulating and local alterations that promote tumor growth occur in the setting of white adipose tissue inflammation manifested as CLS. CYP19A1 encodes aromatase, which catalyzes the conversion of androgen to estrogen. CLS, crown-like structure; CRP, C-reactive protein; IL-6, interleukin-6; MCP-1, monocyte chemoattractant protein-1.
The presence of WAT inflammation manifested as CLS was first reported in visceral and subcutaneous fat depots in association with obesity and insulin resistance.9 Similar lesions were identified in the mammary glands of obese mice, and WAT inflammation has also been reported to occur in the human breast.31-33 Detected by the presence of CLS of the breast (Fig 2), breast WAT inflammation occurs in the majority of individuals who are obese and overweight.19 The inflammatory process associated with CLS of the breast includes activation of nuclear factor-kappa B, a transcription factor linked to both inflammation and the development and progression of tumors.31,32 Recently, we reported that WAT inflammation in the breast at the time of treatment mastectomy in women with early-stage breast cancer is associated with shortened distant recurrence-free survival in those who develop metastatic disease.34 Another recent study of patients with breast cancer demonstrated that WAT inflammation of the breast, as defined by the density of CLS, was associated with reduced overall survival.35 In this regard, the observation that WAT inflammation may also be found in one third of individuals with a normal BMI is a concern. Further, breast WAT inflammation is associated with an unfavorable clinical course in patients with breast cancer independent of BMI.19,34 In patients with early-stage tongue cancer, the presence of CLS in tongue fat is associated with reduced disease-specific and overall survival.36 Hence, adipose inflammation may well stimulate the progression of multiple tumor types. These initial observations provide the first evidence that WAT inflammation affects disease course. Larger-scale studies investigating the impact of WAT inflammation on cancer-specific and overall survival in independent cohorts are needed. Nonetheless, these findings are consistent with preclinical and mechanistic observations linking WAT inflammation to tumor growth and thereby highlight the importance of adipose tissue in the tumor microenvironment. Collectively, these findings help to establish the rationale for interventions aimed at improving adipose tissue health as a novel opportunity to reduce the risk of cancer and improve outcomes.
Fig 2.

Breast white adipose tissue inflammation is detected by the presence of crown-like structures of the breast (CLS-B). (A) Hematoxylin and eosin section of breast tissue showing CLS-B (arrow; ×200 magnification) and (B) anti-CD68 immunostaining showing CLS-B (arrow; ×200 magnification).
Enhanced Hormone Signaling
In addition to directly stimulating tumor growth, cytokines generated in inflamed WAT, such as IL-6, are also known to activate the androgen receptor and promote prostate cancer cell survival and proliferation.37 Similarly, several of the proinflammatory mediators associated with WAT inflammation, including TNF-α, IL-1β, and COX-2–derived prostaglandin E2, can induce aromatase, the rate-limiting enzyme for estrogen biosynthesis.38,39 WAT inflammation is also associated with elevated circulating levels of leptin, another known inducer of aromatase (Fig 1).34,40 Breast WAT inflammation is paralleled by increased tissue levels of aromatase.32,38 It is therefore plausible that locally produced estrogens, as a consequence of WAT inflammation and related systemic effects, may represent a key driver of postmenopausal hormone receptor-positive breast cancer.10 Despite near-complete cessation of ovarian estradiol biosynthesis and dramatically diminished levels of circulating estrogens after menopause, the incidence of estrogen-dependent breast cancer increases with age and is the most common breast tumor subtype in postmenopausal women. Independent of BMI, postmenopausal women have a higher incidence of breast WAT inflammation than do their premenopausal counterparts.19 Possibly, enhanced estrogen production in inflamed breast WAT will help to explain the paradoxical increase in the incidence of estrogen-dependent tumors in postmenopausal women.
Endoplasmic Reticulum Stress
Individuals who are obese have elevated levels of FFAs, which can induce endoplasmic reticulum (EndoR) stress in adipocytes.41 The EndoR is a key organelle involved in protein folding. It has been suggested that FFA-mediated reactive oxygen species oxidize proteins and increase unfolded proteins in the EndoR. In the setting of EndoR stress, unfolded proteins accumulate resulting, in turn, in an adaptive unfolded protein response. Three major transducers of the unfolded protein response have been identified, including protein kinase RNA-like EndoR kinase, activating transcription factor 6, and inositol-requiring enzyme 1-α. If the unfolded protein response proves insufficient to mitigate EndoR stress, the cell undergoes apoptosis, which can elicit an inflammatory response. Notably, EndoR stress-related WAT inflammation has been implicated in metabolic disorders, including glucose intolerance.41,42 In light of these findings, future studies to determine the role of adipocyte EndoR stress in obesity-associated cancers are warranted.
ECM Alterations
Recently, obesity-associated WAT inflammation was shown to correlate with mechanical changes in the ECM that promote tumor growth.43 Specifically, myofibroblast enrichment and increased density and stiffness are observed in mammary gland ECM of obese versus lean mice. Moreover, these profibrotic features in the ECM are found in the breasts of women who are obese and occur in association with WAT inflammation. Altered mechanotransduction as a result of a stiffer ECM may promote breast tumorigenesis.43 The observation that ECM remodeling is also associated with WAT inflammation demonstrates a convergence of pathways that are dysregulated in obesity and locally promote tumor growth.
Cancer-Associated Adipocytes
Direct interactions between peritumoral adipocytes and cancer cells have also been reported to contribute to tumor growth. In the breast, tumor invasion promotes morphologic changes in adipocytes at the tumor-stromal interface. These cancer-associated adipocytes (CAAs) acquire a fibroblast-like phenotype, which promotes further tumor invasion via secretion of numerous proteases and cytokines, including IL-6.44,45 Additionally, CAAs constitute and contribute to the accumulation of a dense collagenous stroma, further increasing ECM stiffness. Secretion of IL-6 and IL-8 by adipocytes in the omentum, the most common site of ovarian cancer metastasis, has been shown to promote homing, migration, and invasion of ovarian cancer cells.46 Furthermore, release of FFAs due to increased lipolysis in omental adipocytes has been implicated as an energy source for growing ovarian metastases. In prostate cancer, secretion of chemokines by periprostatic adipocytes has been reported to stimulate prostate cancer cell migration.47 Thus, adipocyte–tumor cell cross-talk, characterized by reciprocal changes in tumor cells and CAAs, is an important process within the microenvironment that can promote aggressive tumor behavior.
WAT and the Microbiome
Apart from activation by FFAs, TLR4 is prototypically stimulated via binding of lipopolysaccharide, a constituent of the cell wall of gram-negative bacteria, which activates the innate immune response. Preclinical data demonstrate that intake of diets high in saturated fat can alter intestinal microbiota, leading to TLR activation, WAT inflammation, and insulin resistance.48 Additionally, commensal intestinal microorganisms can drive systemic IL-6 production via TLR5 and promote tumor growth.49 Depleting gut microbiota, for example, with antibiotic treatment, can reverse the effects of a high-fat diet in obese mice by inducing a phenotypic switch from white to brown adipose tissue, which is thought to have antiobesity effects.50,51 It has been suggested that in humans, high fat consumption promotes a TLR4-mediated inflammatory response as a consequence of altered gut permeability and perturbations of intestinal microbiota.52 The potential relationships between the microbiome and cancer represent an emerging and promising field,53 and WAT may be a key mediator.
EXTENDED IMPACT OF THE INFLAMED MICROENVIRONMENT
The effects of adipose tissue dysfunction extend well beyond the tumor microenvironment and affect systemic processes that may synergistically fuel local tumor growth while also promoting metastasis. Importantly, WAT inflammation occurs synchronously in multiple fat depots, indicating that WAT inflammation at one site (ie, the breast) is a sentinel for inflammation in other regional as well as distant adipose depots.19 Consistently, WAT inflammation correlates with increased circulating levels of C-reactive protein (CRP) and IL-6.34 These findings are supported by reports of elevated levels of CRP and IL-6 found in the blood of individuals who are obese (Fig 1).54,55 Furthermore, the observation that breast WAT inflammation portends an inferior clinical course for patients with breast cancer is consistent with earlier reports demonstrating that TNF-α, IL-1β, and IL-6 promote tumor growth in mouse models of obesity and that elevated levels of IL-6 and CRP are found in women who are obese and have been associated with the development and progression of breast tumors.55-58 In addition, increased prostaglandin E2 metabolite in the urine is found in women who are obese and is associated with an increased risk of postmenopausal breast cancer.59,60 This could be a consequence of increased COX-2 levels in inflamed fat. Thus, smoldering WAT inflammation and low-grade systemic inflammation are inexorably coupled, and each perpetuates the other. In this manner, the local and systemic environments are together programmed to promote tumor growth and metastasis in individuals with poor adipose tissue health.
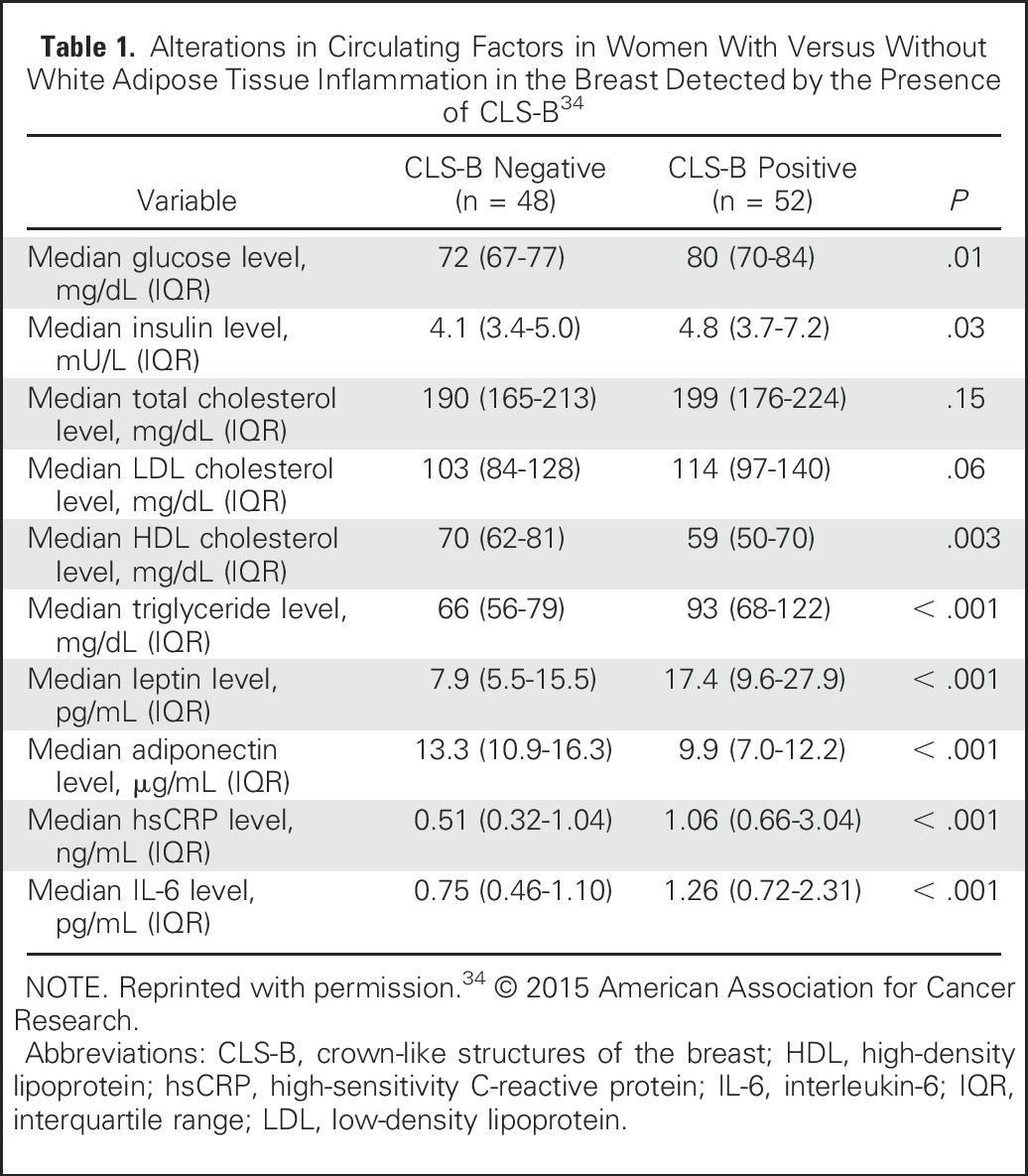
In addition to systemic inflammation, WAT inflammation is associated with the metabolic syndrome.34 The metabolic syndrome comprises a number of clinical disorders, such as obesity, insulin resistance, and dyslipidemia, and is associated with worse outcomes for patients with breast cancer.61-65 Breast WAT inflammation may constitute a local mechanism contributing to the association between the metabolic syndrome systemically and adverse breast cancer outcomes. Elevated levels of insulin and glucose are found in individuals with WAT inflammation.34 Insulin can stimulate the synthesis of insulin-like growth factor-1 (IGF-1), and both have potent mitogenic effects on tumor cells. For example, insulin and IGF-1 can activate the PI3K/Akt/mTOR and Ras/Raf/MAPK pathways, thereby stimulating tumor growth.10 Additionally, specific gene expression signatures enriched for IGF-1 signaling have been observed in breast tumors from patients who are obese.66 The mechanisms by which tumor gene expression is altered by obesity-related processes, including insulin resistance and WAT inflammation, are currently under study. Furthermore, transcriptomic alterations in nontumorous tissue may also occur in association with obesity-related processes, such as WAT inflammation, and thereby reprogram the microenvironment to support tumor growth and invasion. Taken together, WAT inflammation is associated with several systemic alterations, including the metabolic syndrome and elevated levels of circulating proinflammatory mediators. For example, changes in circulating blood factors in a cohort of 100 women who underwent mastectomy for breast cancer treatment or prevention stratified by the presence or absence of WAT inflammation in the breast are listed in Table 1.34
Table 1.
Alterations in Circulating Factors in Women With Versus Without White Adipose Tissue Inflammation in the Breast Detected by the Presence of CLS-B34
CLINICAL-TRANSLATIONAL APPLICATION
The totality of evidence relating adipose inflammation to a protumor state both within the microenvironment and in peripheral circulation supports the development of interventions that reduce adipose inflammation as a new strategy to prevent and treat cancer. Weight reduction via diet and/or exercise can restore many pathways that are dysregulated in the obese state and is of significant interest for cancer prevention and treatment. In a proof-of-principal study, significant calorie restriction for 28 days resulted in improvements in inflammatory gene expression signatures within subcutaneous WAT.67 Additionally, bariatric surgery is associated with reduced macrophage infiltration into subcutaneous WAT.68 These findings might help to explain the reduced cancer incidence observed in individuals who are obese who undergo gastric bypass surgery.69,70 Medications that stimulate weight loss could, in theory, be beneficial in the treatment of patients with cancer who are overweight and obese. Finally, several commonly used medications, including nonsteroidal anti-inflammatory drugs, aspirin, metformin, and statins, could be beneficial in treating the local or systemic consequences of WAT inflammation. However, to date, no study has determined whether the efficacy of these agents in reducing cancer risk or progression varies on the basis of the presence or absence of WAT inflammation. Given its many associated molecular aberrations, WAT inflammation represents a convergence of pathways that are deregulated in the obese state, which is not consistently identified by BMI. Accordingly, adipose inflammation is likely to be a highly informative end point and possibly a critical inclusion parameter in studies testing weight reduction and/or medications that target obesity-related cancers.
The disruption of numerous tissue-specific and systemic physiologic processes in the obese state generates and sustains a transformed local environment primed to support tumor development and growth. Adipose tissue is a key component of the local environment, and increasing evidence points to the critical role of adipose health as an indicator of multiple system-wide and local processes. By transforming the local landscape through the mechanisms reviewed here, WAT inflammation seems to be a key process by which obesity promotes carcinogenesis and tumor progression. Numerous systemic changes that are implicated in cancer growth, including insulin resistance, elevated levels of proinflammatory mediators, and altered levels of adipokines, correlate with WAT inflammation. These observations suggest that therapeutic targeting of WAT inflammation may be more effective than other strategies that aim to reduce or limit the burden of obesity-associated cancers. Given that WAT inflammation also occurs in a subset of patients with a normal BMI, assessing WAT inflammatory status (as opposed to BMI) may represent an even more appropriate method of defining the pathophysiologic consequences of the obese state. The development of noninvasive methods to assay adipose health, including blood-based biomarker signatures and/or radiographic techniques, is therefore a critical research goal. A clinically feasible biomarker of adipose inflammation would allow for broader study of the relationships between the obese state and a variety of cancers, and would potentially provide a surrogate marker of efficacy for interventions aimed at alleviating the obesity-cancer burden.
Footnotes
Supported by the Breast Cancer Research Foundation, the Botwinick-Wolfensohn Foundation (in memory of Mr. and Mrs. Benjamin Botwinick), and Memorial Sloan Kettering Cancer Center Support Grant/Core Grant No. P30 CA008748.
AUTHOR CONTRIBUTIONS
Conception and design: Neil M. Iyengar, Ayca Gucalp, Clifford A. Hudis
Collection and assembly of data: Neil M. Iyengar, Ayca Gucalp, Clifford A. Hudis
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Neil M. Iyengar
No relationship to disclose
Ayca Gucalp
Research Funding: Pfizer, Innocrin Pharmaceuticals
Andrew J. Dannenberg
Consulting or Advisory Role: SynDevRx, Euclises
Clifford A. Hudis
Consulting or Advisory Role: Pfizer, Genentech, Novartis, Eli Lilly
Other Relationship: Breast Cancer Research Foundation
REFERENCES
- 1.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 3.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ernst PB, Gold BD. The disease spectrum of Helicobacter pylori: The immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol. 2000;54:615–640. doi: 10.1146/annurev.micro.54.1.615. [DOI] [PubMed] [Google Scholar]
- 5.zur Hausen H. Papillomaviruses and cancer: From basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 6.Tsukuma H, Hiyama T, Tanaka S, et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med. 1993;328:1797–1801. doi: 10.1056/NEJM199306243282501. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi H, Ogata H, Nishigaki R, et al. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garro AJ, Lieber CS. Alcohol and cancer. Annu Rev Pharmacol Toxicol. 1990;30:219–249. doi: 10.1146/annurev.pa.30.040190.001251. [DOI] [PubMed] [Google Scholar]
- 9.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 10.Iyengar NM, Hudis CA, Dannenberg AJ. Obesity and cancer: Local and systemic mechanisms. Annu Rev Med. 2015;66:297–309. doi: 10.1146/annurev-med-050913-022228. [DOI] [PubMed] [Google Scholar]
- 11.Flegal KM, Carroll MD, Ogden CL, et al. Prevalence and trends in obesity among US adults, 1999-2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 12. Levi J, Segal LM, St. Laurent R, et al: F as in fat: How obesity threatens America’s future 2012. http://healthyamericans.org/assets/files/TFAH2012FasInFatFnlRv.pdf. [Google Scholar]
- 13.Sinicrope FA, Dannenberg AJ. Obesity and breast cancer prognosis: Weight of the evidence. J Clin Oncol. 2011;29:4–7. doi: 10.1200/JCO.2010.32.1752. [DOI] [PubMed] [Google Scholar]
- 14.Howe LR, Subbaramaiah K, Hudis CA, et al. Molecular pathways: Adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res. 2013;19:6074–6083. doi: 10.1158/1078-0432.CCR-12-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Kruijsdijk RC, van der Wall E, Visseren FL. Obesity and cancer: The role of dysfunctional adipose tissue. Cancer Epidemiol Biomarkers Prev. 2009;18:2569–2578. doi: 10.1158/1055-9965.EPI-09-0372. [DOI] [PubMed] [Google Scholar]
- 17.Deepa M, Papita M, Nazir A, et al. Lean people with dysglycemia have a worse metabolic profile than centrally obese people without dysglycemia. Diabetes Technol Ther. 2014;16:91–96. doi: 10.1089/dia.2013.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S, Chen Y, Liu X, et al. Insulin resistance and metabolic syndrome in normal-weight individuals. Endocrine. 2014;46:496–504. doi: 10.1007/s12020-013-0079-8. [DOI] [PubMed] [Google Scholar]
- 19.Iyengar NM, Morris PG, Zhou XK, et al. Menopause is a determinant of breast adipose inflammation. Cancer Prev Res (Phila) 2015;8:349–358. doi: 10.1158/1940-6207.CAPR-14-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu XJ, Gauthier MS, Hess DT, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53:792–801. doi: 10.1194/jlr.P022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denis GV, Obin MS. ‘Metabolically healthy obesity’: Origins and implications. Mol Aspects Med. 2013;34:59–70. doi: 10.1016/j.mam.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klöting N, Fasshauer M, Dietrich A, et al. Insulin-sensitive obesity. Am J Physiol Endocrinol Metab. 2010;299:E506–E515. doi: 10.1152/ajpendo.00586.2009. [DOI] [PubMed] [Google Scholar]
- 23.Sahakyan KR, Somers VK, Rodriguez-Escudero JP, et al. Normal-weight central obesity: Implications for total and cardiovascular mortality. Ann Intern Med. 2015;163:827–835. doi: 10.7326/M14-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poirier P. The many paradoxes of our modern world: Is there really an obesity paradox or is it only a matter of adiposity assessment? Ann Intern Med. 2015;163:880–881. doi: 10.7326/M15-2435. [DOI] [PubMed] [Google Scholar]
- 25.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 26. Monteiro R, Azevedo I: Chronic inflammation in obesity and the metabolic syndrome. Mediators Inflamm 10.1155/2010/289645 [epub ahead of print on July 14, 2010] [DOI] [PMC free article] [PubMed]
- 27.Amano SU, Cohen JL, Vangala P, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 2014;19:162–171. doi: 10.1016/j.cmet.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cinti S, Mitchell G, Barbatelli G, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Shapiro H, Pecht T, Shaco-Levy R, et al. Adipose tissue foam cells are present in human obesity. J Clin Endocrinol Metab. 2013;98:1173–1181. doi: 10.1210/jc.2012-2745. [DOI] [PubMed] [Google Scholar]
- 30.Lee JY, Sohn KH, Rhee SH, et al. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 31.Subbaramaiah K, Howe LR, Bhardwaj P, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res (Phila) 2011;4:329–346. doi: 10.1158/1940-6207.CAPR-10-0381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Morris PG, Hudis CA, Giri D, et al. Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev Res (Phila) 2011;4:1021–1029. doi: 10.1158/1940-6207.CAPR-11-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun X, Casbas-Hernandez P, Bigelow C, et al. Normal breast tissue of obese women is enriched for macrophage markers and macrophage-associated gene expression. Breast Cancer Res Treat. 2012;131:1003–1012. doi: 10.1007/s10549-011-1789-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyengar NM, Zhou XK, Gucalp A, et al. Systemic correlates of white adipose tissue inflammation in early-stage breast cancer. Clin Cancer Res. 2016;22:2283–2289. doi: 10.1158/1078-0432.CCR-15-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koru-Sengul T, Santander AM, Miao F, et al. Breast cancers from black women exhibit higher numbers of immunosuppressive macrophages with proliferative activity and of crown-like structures associated with lower survival compared to non-black Latinas and Caucasians. Breast Cancer Res Treat. 2016;158:113–126. doi: 10.1007/s10549-016-3847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iyengar NM, Ghossein RA, Morris LG, et al. White adipose tissue inflammation and cancer-specific survival in patients with squamous cell carcinoma of the oral tongue. Cancer. doi: 10.1002/cncr.30251. 10.1002/cncr.30251 [epub ahead of print on August 10, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. J Carcinog. 2011;10:20. doi: 10.4103/1477-3163.83937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subbaramaiah K, Morris PG, Zhou XK, et al. Increased levels of COX-2 and prostaglandin E2 contribute to elevated aromatase expression in inflamed breast tissue of obese women. Cancer Discov. 2012;2:356–365. doi: 10.1158/2159-8290.CD-11-0241. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Irahara N, Miyoshi Y, Taguchi T, et al. Quantitative analysis of aromatase mRNA expression derived from various promoters (I.4, I.3, PII and I.7) and its association with expression of TNF-alpha, IL-6 and COX-2 mRNAs in human breast cancer. Int J Cancer. 2006;118:1915–1921. doi: 10.1002/ijc.21562. [DOI] [PubMed] [Google Scholar]
- 40.Brown KA, McInnes KJ, Hunger NI, et al. Subcellular localization of cyclic AMP-responsive element binding protein-regulated transcription coactivator 2 provides a link between obesity and breast cancer in postmenopausal women. Cancer Res. 2009;69:5392–5399. doi: 10.1158/0008-5472.CAN-09-0108. [DOI] [PubMed] [Google Scholar]
- 41.Kawasaki N, Asada R, Saito A, et al. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. doi: 10.1038/srep00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang L, Calay ES, Fan J, et al. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. 2015;349:500–506. doi: 10.1126/science.aaa0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seo BR, Bhardwaj P, Choi S, et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. 2015;7:301ra130. doi: 10.1126/scitranslmed.3010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dirat B, Bochet L, Dabek M, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455–2465. doi: 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 45.Bochet L, Lehuédé C, Dauvillier S, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73:5657–5668. doi: 10.1158/0008-5472.CAN-13-0530. [DOI] [PubMed] [Google Scholar]
- 46.Nieman KM, Kenny HA, Penicka CV, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laurent V, Guérard A, Mazerolles C, et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat Commun. 2016;7:10230. doi: 10.1038/ncomms10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caesar R, Tremaroli V, Kovatcheva-Datchary P, et al. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 2015;22:658–668. doi: 10.1016/j.cmet.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rutkowski MR, Stephen TL, Svoronos N, et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell. 2015;27:27–40. doi: 10.1016/j.ccell.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suárez-Zamorano N, Fabbiano S, Chevalier C, et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat Med. 2015;21:1497–1501. doi: 10.1038/nm.3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen P, Spiegelman BM. Brown and beige fat: Molecular parts of a thermogenic machine. Diabetes. 2015;64:2346–2351. doi: 10.2337/db15-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pendyala S, Walker JM, Holt PR: A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 142:1100-1101.e2, 2012. [DOI] [PMC free article] [PubMed]
- 53.Garrett WS. Cancer and the microbiota. Science. 2015;348:80–86. doi: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Visser M, Bouter LM, McQuillan GM, et al. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131–2135. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 55.Hursting SD, Digiovanni J, Dannenberg AJ, et al. Obesity, energy balance, and cancer: New opportunities for prevention. Cancer Prev Res (Phila) 2012;5:1260–1272. doi: 10.1158/1940-6207.CAPR-12-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bachelot T, Ray-Coquard I, Menetrier-Caux C, et al. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br J Cancer. 2003;88:1721–1726. doi: 10.1038/sj.bjc.6600956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sicking I, Edlund K, Wesbuer E, et al. Prognostic influence of pre-operative C-reactive protein in node-negative breast cancer patients. PLoS One. 2014;9:e111306. doi: 10.1371/journal.pone.0111306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allin KH, Bojesen SE, Nordestgaard BG. Baseline C-reactive protein is associated with incident cancer and survival in patients with cancer. J Clin Oncol. 2009;27:2217–2224. doi: 10.1200/JCO.2008.19.8440. [DOI] [PubMed] [Google Scholar]
- 59.Morris PG, Zhou XK, Milne GL, et al. Increased levels of urinary PGE-M, a biomarker of inflammation, occur in association with obesity, aging, and lung metastases in patients with breast cancer. Cancer Prev Res (Phila) 2013;6:428–436. doi: 10.1158/1940-6207.CAPR-12-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim S, Taylor JA, Milne GL, et al. Association between urinary prostaglandin E2 metabolite and breast cancer risk: A prospective, case-cohort study of postmenopausal women. Cancer Prev Res (Phila) 2013;6:511–518. doi: 10.1158/1940-6207.CAPR-13-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goodwin PJ, Ennis M, Pritchard KI, et al. Fasting insulin and outcome in early-stage breast cancer: Results of a prospective cohort study. J Clin Oncol. 2002;20:42–51. doi: 10.1200/JCO.2002.20.1.42. [DOI] [PubMed] [Google Scholar]
- 62.Duggan C, Irwin ML, Xiao L, et al. Associations of insulin resistance and adiponectin with mortality in women with breast cancer. J Clin Oncol. 2011;29:32–39. doi: 10.1200/JCO.2009.26.4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berrino F, Villarini A, Traina A, et al. Metabolic syndrome and breast cancer prognosis. Breast Cancer Res Treat. 2014;147:159–165. doi: 10.1007/s10549-014-3076-6. [DOI] [PubMed] [Google Scholar]
- 64.Barone BB, Yeh HC, Snyder CF, et al. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: A systematic review and meta-analysis. JAMA. 2008;300:2754–2764. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Erickson K, Patterson RE, Flatt SW, et al. Clinically defined type 2 diabetes mellitus and prognosis in early-stage breast cancer. J Clin Oncol. 2011;29:54–60. doi: 10.1200/JCO.2010.29.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Creighton CJ, Sada YH, Zhang Y, et al. A gene transcription signature of obesity in breast cancer. Breast Cancer Res Treat. 2012;132:993–1000. doi: 10.1007/s10549-011-1595-y. [DOI] [PubMed] [Google Scholar]
- 67.Clément K, Viguerie N, Poitou C, et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J. 2004;18:1657–1669. doi: 10.1096/fj.04-2204com. [DOI] [PubMed] [Google Scholar]
- 68.Cancello R, Henegar C, Viguerie N, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54:2277–2286. doi: 10.2337/diabetes.54.8.2277. [DOI] [PubMed] [Google Scholar]
- 69.Adams TD, Stroup AM, Gress RE, et al. Cancer incidence and mortality after gastric bypass surgery. Obesity (Silver Spring) 2009;17:796–802. doi: 10.1038/oby.2008.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sjöström L. Review of the key results from the Swedish Obese Subjects (SOS) trial: A prospective controlled intervention study of bariatric surgery. J Intern Med. 2013;273:219–234. doi: 10.1111/joim.12012. [DOI] [PubMed] [Google Scholar]