

Abstract
Purpose
This trial evaluated the safety, pharmacokinetics, pharmacodynamics, and efficacy of selinexor (KPT-330), a novel, oral small-molecule inhibitor of exportin 1 (XPO1/CRM1), and determined the recommended phase II dose.
Patients and Methods
In total, 189 patients with advanced solid tumors received selinexor (3 to 85 mg/m2) in 21- or 28-day cycles. Pre- and post-treatment levels of XPO1 mRNA in patient-derived leukocytes were determined by reverse transcriptase quantitative polymerase chain reaction, and tumor biopsies were examined by immunohistochemistry for changes in markers consistent with XPO1 inhibition. Antitumor response was assessed according Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 guidelines.
Results
The most common treatment-related adverse events included fatigue (70%), nausea (70%), anorexia (66%), and vomiting (49%), which were generally grade 1 or 2. Most commonly reported grade 3 or 4 toxicities were thrombocytopenia (16%), fatigue (15%), and hyponatremia (13%). Clinically significant major organ or cumulative toxicities were rare. The maximum-tolerated dose was defined at 65 mg/m2 using a twice-a-week (days 1 and 3) dosing schedule. The recommended phase II dose of 35 mg/m2 given twice a week was chosen based on better patient tolerability and no demonstrable improvement in radiologic response or disease stabilization compared with higher doses. Pharmacokinetics were dose proportional, with no evidence of drug accumulation. Dose-dependent elevations in XPO1 mRNA in leukocytes were demonstrated up to a dose level of 28 mg/m2 before plateauing, and paired tumor biopsies showed nuclear accumulation of key tumor-suppressor proteins, reduction of cell proliferation, and induction of apoptosis. Among 157 patients evaluable for response, one complete and six partial responses were observed (n = 7, 4%), with 27 patients (17%) achieving stable disease for ≥ 4 months.
Conclusion
Selinexor is a novel and safe therapeutic with broad antitumor activity. Further interrogation into this class of therapy is warranted.
INTRODUCTION
The genomic surveillance function of most tumor-suppressor proteins (TSPs) is dependent on their localization within the cell nucleus.1 In cancer, increased nuclear export results in the inactivation of key TSPs.2 Exportin 1 (XPO1, CRM1) is a karyopherin that mediates the nuclear export of more than 200 mammalian cargo proteins, including the following TSPs: p53, p73, BRCA, IκB, p21, and FOXO transcription factors.3-5 XPO1 is overexpressed in most cancer types and is correlated with poor prognosis in acute myeloid leukemia, mantle-cell lymphoma, glioma, and pancreatic, cervical, and ovarian cancers.6-13 In addition, XPO1 activity is involved in the activation of oncogenic pathways, at least in part through enhanced nuclear export of eukaryotic initiation factor 4e (eIF4e), the sole transporter of guanine-capped mRNAs, including those for Myc, cyclin D1, and MDM2.13 Increased nuclear export of these oncogenic transcripts to the cytosol promotes synthesis of cognate oncoproteins, driving cell survival and proliferation. Therefore, inhibition of XPO1 is expected to activate TSPs by restricting them to the nucleus and prevent the translation of several oncoprotein transcripts by preventing their mRNA export to the cytoplasm. Thus, XPO1 inhibition is a novel and promising anticancer strategy with broad applicability across tumor types.
Selinexor (KPT-330) is a first-in-class, orally bioavailable, highly specific, slowly reversible, covalent inhibitor of XPO1. Preclinical studies demonstrated dose-dependent cytotoxicity in multiple cell lines and potent in vivo antitumor activity in xenograft and orthotopic murine models.2,5,14-24 Comparatively, the use of a sister compound, verdinexor, in canine species resulted in partial responses in lymphomas with limited toxicities.25 Here, we assess the safety, pharmacokinetics, pharmacodynamics, and preliminary antitumor efficacy of selinexor in patients with advanced solid tumors and propose a recommended phase II dose (RP2D).
PATIENTS AND METHODS
Patient Selection
Eligible patients had advanced solid malignancy unresponsive to available therapies or for which no standard therapy exists, an Eastern Cooperative Oncology Group performance status of 0 of 1, adequate organ function, and absolute neutrophil counts of ≥ 1,500/μL and platelet counts of ≥ 100,000/μL. Key exclusion criteria included prior therapy within 21 days or major surgery within 28 days of study enrollment, unstable cardiac function, uncontrolled active infection or symptoms, and significantly diseased or obstructed GI tract. In the dose expansion cohort, patients of any solid tumor type were enrolled, but selected numbers of patients with melanoma and colon, gynecologic, prostate, brain, and squamous cancers were included. All patients had documented disease progression at study entry. Full eligibility criteria are provided in the Data Supplement.
Study Design and Oversight
This study used a modified 3 + 3 design in the escalation phase to evaluate selinexor when administered orally in 21- or 28-day cycles, with subsequent expansion cohorts (same eligibility criteria). The primary objectives of the study were to assess safety and identify the maximum-tolerated dose (MTD) and RP2D for selinexor. The secondary objectives were to assess pharmacokinetics, pharmacodynamics, and efficacy. The starting dose of selinexor (3 mg/m2) was determined based on extrapolation from rat and monkey toxicology studies. A dose escalation of 100% was used from cohorts 1 to 2 and 2 to 3, and a dose escalation of 30% to 40% was used for cohorts ≥ 3. One patient was enrolled at the first dose level, whereas three to six patients were recruited for the second and subsequent dose levels in escalation phase. Dose escalation continued until at least two patients among a cohort of three to six patients experienced a dose-limiting toxicity (DLT; defined Data Supplement) during the first treatment cycle. The study was approved by the independent ethics committee for each site and was conducted in accordance with the Declaration of Helsinki. All patients provided informed written consent. The study was sponsored by Karyopharm Therapeutics (Newton, MA) and registered at ClinicalTrials.gov (identifier: NCT01607905).
Safety and Tolerability Assessment
All patients who received at least one dose of selinexor were evaluated for safety. Standard safety assessments were conducted at baseline and during the study, with adverse events (AEs) graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03.26
Pharmacokinetic Assessment
In cycle 1, serial blood samples were obtained before dose and at 0.5, 1, 2, 4, 8, 24, and 48 hours after dose on day 1 and before dose and 0, 1, 2, 4, and 8 hours after dose on day 15 or 17. Plasma concentrations of selinexor were measured at Tandem Laboratories (Durham, NC) using a validated liquid chromatography–tandem mass spectrometry method (Sciex API 4000; Sciex, Framingham, MA). The lower limit of quantification for selinexor was 1.0 ng/mL. Pharmacokinetic parameters were calculated by standard noncompartmental analysis using PK Solutions Software (Summit Research Services, Montrose, CO).
Pharmacodynamics
XPO1 mRNA level in leukocytes was assessed by reverse transcriptase quantitative polymerase chain reaction before dose and up to 48 hours after selinexor treatment. In addition, immunohistochemistry was performed on paired tumor biopsies (before dose and after approximately 4 weeks on treatment) to investigate the effects of selinexor on tumor cell morphology (hematoxylin and eosin; Richard-Allan Scientific, San Diego, CA), apoptosis (Apoptag, S71003; Millipore, Billerica, MA; and cleaved caspase 3, #9661; Cell Signaling Technology, Beverly, MA), proliferation (Ki-67, 275R-18; Cell Marque, Rocklin, CA), and TSP expression and localization (p53, SC-126; Santa Cruz Biotechnology, Dallas, TX; and FOXO3A, AB47409; Abcam, Cambridge, United Kingdom).
Tumor Assessment
Tumor size was assessed at baseline and every 8 weeks during the treatment period. Tumor response was determined using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1.27
Statistics
The paired t test was used to compare XPO1 expression in leukocytes at baseline and after treatment. χ2 tests were used to assess correlations between dose level and frequency of AEs and response to selinexor. Data were considered significant at P < .05.
RESULTS
Patient Characteristics
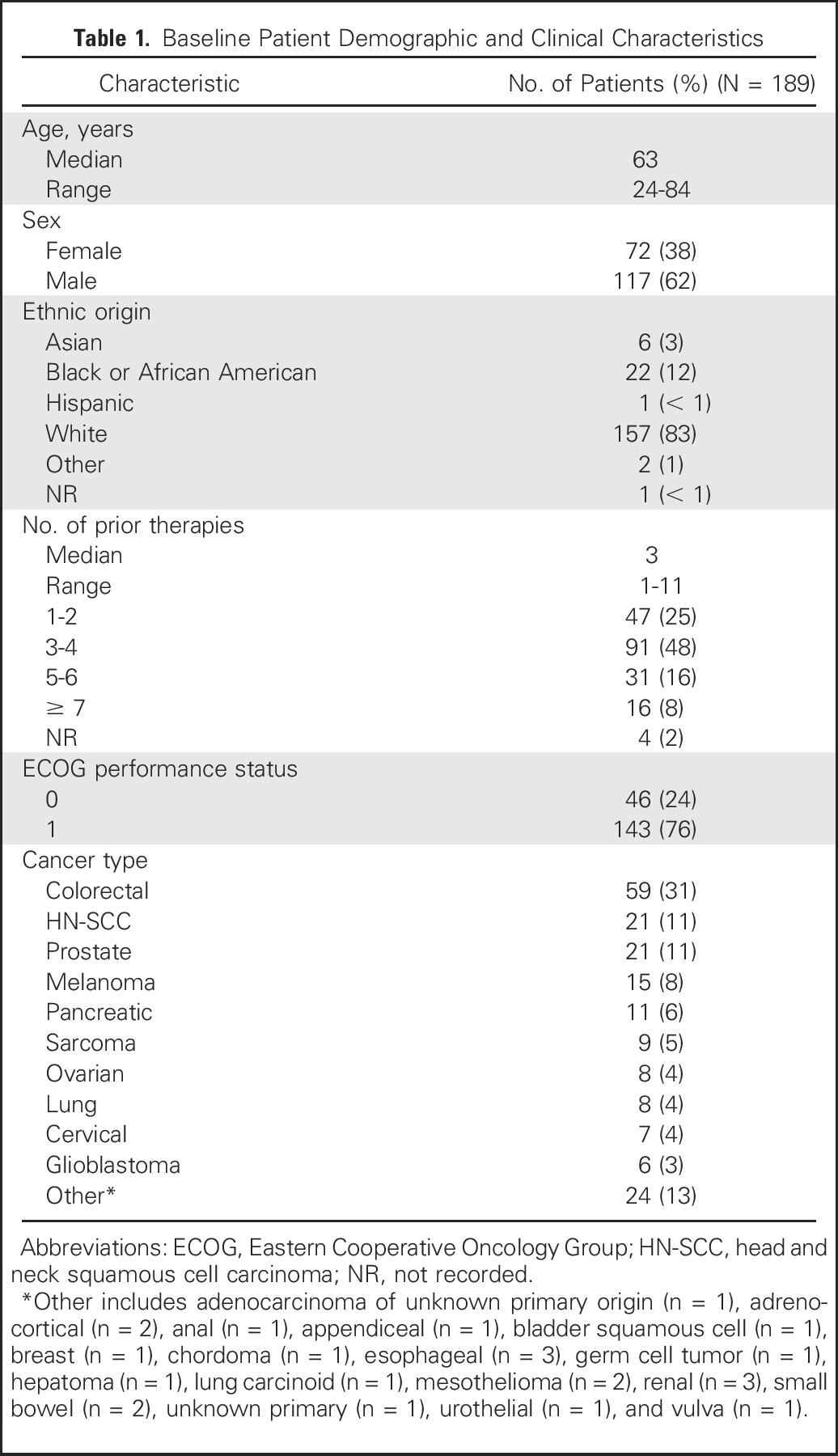
A total of 189 patients were enrolled and treated between June 2012 and October 2015. The study population was predominantly male (62%) and heavily pretreated (73% of patients had three or more previous lines of therapies), and all patients had disease progression at study entry. Baseline characteristics are listed in Table 1.
Table 1.
Baseline Patient Demographic and Clinical Characteristics
Safety
Overall, the study drug was investigated across 20 dose levels (3 to 85 mg/m2). A variety of treatment schedules were investigated, as incremental knowledge of the toxicity profile for selinexor emerged, summarized in Table 2. At the start of the trial, a dose was administered three times a week, every other day, on weeks 1 and 3 and twice a week on weeks 2 to 4, with more than 48 hours between doses, for a total of 10 target doses per cycle (schedule 1). After the 12 mg/m2 cohort, to improve the tolerability of selinexor at higher dose levels, a run-in week was introduced, where selinexor was administered at 12 mg/m2, three times per week every other day, before the start of target dose (schedule 2). However, because there was no apparent difference in toxicity profiles observed at doses ≥ 16.8 mg/m2, the run-in week was abandoned. To improve tolerability and patient compliance, the treatment schedule proceeded with twice-a-week doses in a 3- or 4-week cycle (schedules 3, 4, 5, and 8) and once-a-week dosing (schedule 7). Because in vitro analysis suggested that the main catabolic pathway of selinexor is through glutathionylation, selinexor was administered with a standard dose of acetaminophen (1,000 mg) twice a week to reduce glutathione levels and examine the effect on selinexor pharmacokinetics and tolerability (schedule 6).
Table 2.
Dose Levels and Schedule Used and Occurrence of DLT
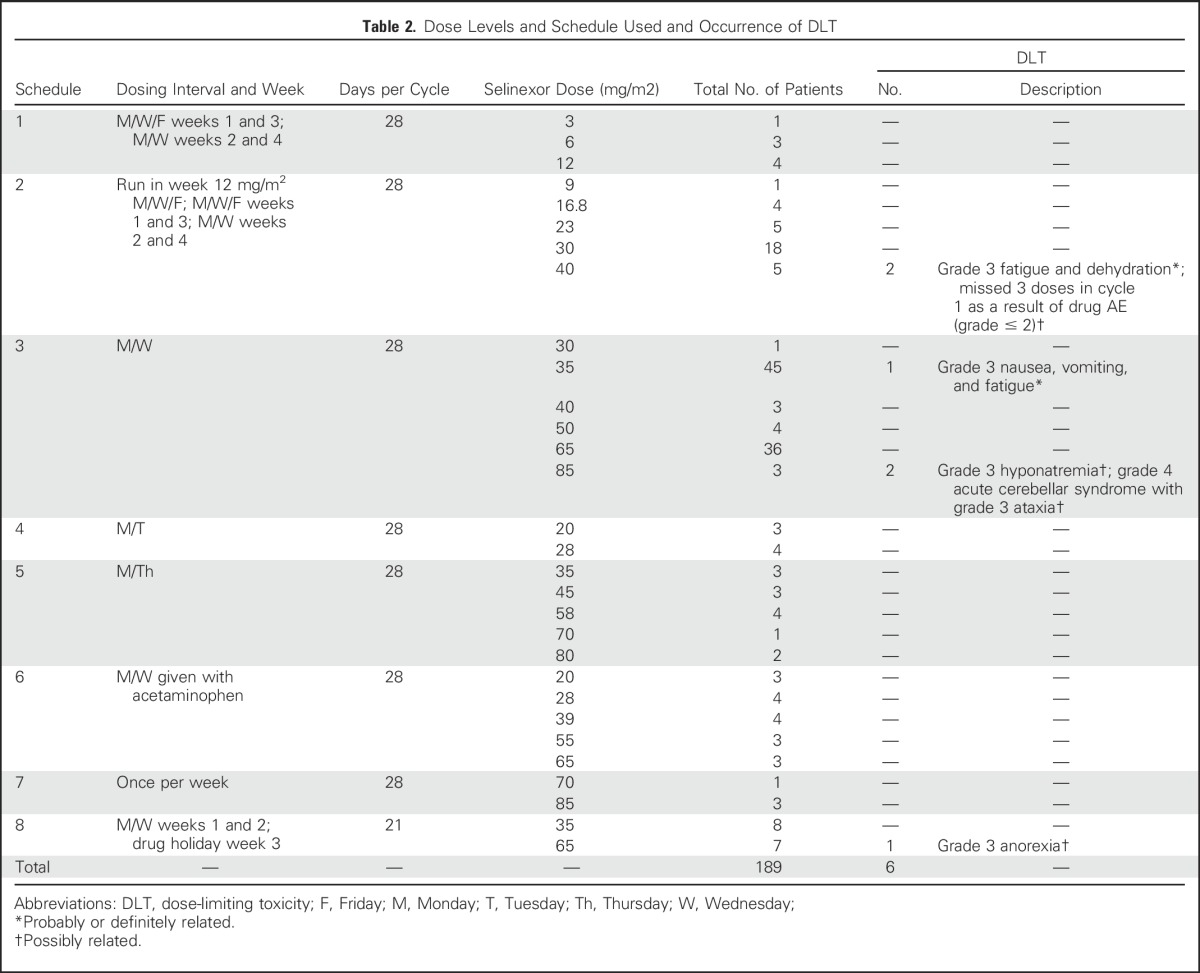
In 189 patients evaluable for safety, there were six DLTs (Table 2). Two patients experienced DLTs at 40 mg/m2 on schedule 2 (10 doses per cycle); the first patient had grade 3 fatigue and dehydration, whereas the second patient had three missed doses in cycle 1 as a result of an AE ≥ grade 2, establishing an MTD of 30 mg/m2 for schedules 1 and 2. Therefore, the frequency of selinexor administration was reduced to twice a week (eight doses per cycle), and selinexor dose was incrementally escalated from 30 to 85 mg/m2. On schedule 3 (eight doses per cycle), one DLT of grade 3 nausea, vomiting, and fatigue occurred at 35 mg/m2. Two patients at the 85 mg/m2 dose experienced DLTs; one patient had grade 3 hyponatremia resistant to standard intravenous fluid correction, and the other patient had reversible grade 4 cerebellar syndrome with associated ataxia and dysarthria. The patient with grade 4 cerebellar syndrome developed symptoms after three doses of trial drug. Selinexor was discontinued, and the patient recovered significantly to near baseline over 6 weeks. No other cerebellar events related to selinexor were reported in this study or in the concurrent studies in advanced hematologic tumors or sarcomas (food-effect and formulation study). On the basis of these results, the MTD was declared at 65 mg/m2 for schedule 3. In schedules 4 to 8, one DLT was observed (grade 3 anorexia). All DLTs resolved when drug administration was stopped, and patients returned to baseline or near baseline. To enable determination of a dose level best tolerated for chronic dosing while maintaining antitumor activity, patients were enrolled into twice-a-week dose-expansion cohorts at 35 mg/m2 (approximately 60-mg fixed dose) and 65 mg/m2 (approximately 100-mg fixed dose).
Treatment-related AEs that were at least possibly related to selinexor occurring in ≥ 10% of patients are listed in Table 3. The most frequently reported AEs were fatigue (70%), nausea (70%), anorexia (66%), and vomiting (49%), which were generally grade 1 or 2 and manageable with supportive care agents, such as standard antiemetics, corticosteroids, or megestrol acetate (anorexia), and/or olanzapine for anorexia and nausea; in latter cohorts, these agents were used as primary prophylaxis. The commonly reported grade 3 or 4 AEs were thrombocytopenia (16%), fatigue (15%), hyponatremia (13%), anemia (9%), neutropenia (8%), and anorexia (6%). A summary of all toxicities, regardless of attribution, can be found in the Data Supplement.
Table 3.
Treatment-Related Adverse Events
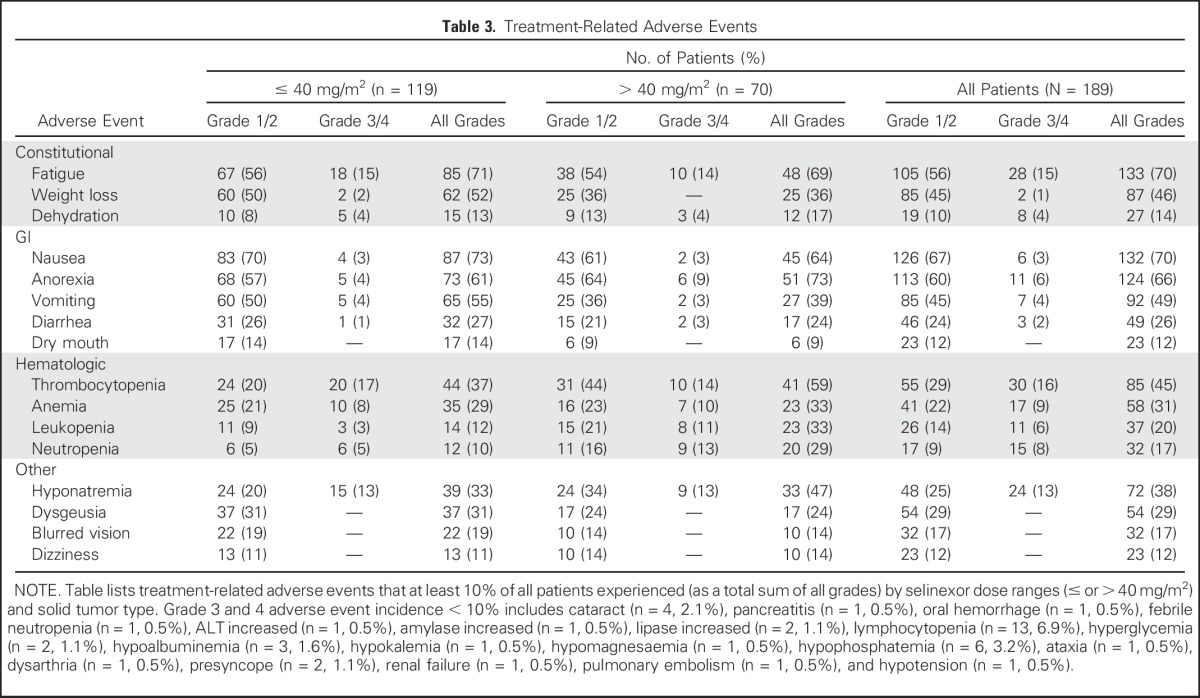
On the basis of drug toxicity, 32 patients (17%) underwent temporary treatment discontinuation, and 52 patients (28%) had dose reductions as a result of toxicities and/or intolerance, with 12 patients (6%) undergoing two or more dose reductions. A total of 57 patients (30%) withdrew consent from the study, 34 (18%) of whom withdrew consent as a result of known treatment-related toxicities, whereas the rest withdrew as a result of patient discretion or early disease progression. Consent withdrawal as a result of AEs was clearly related to dose level, as described in the following paragraph.
A number of end points related to tolerability were found to be dose dependent by comparing effects of ≤ 40 mg/m2 (median, 35 mg/m2; n = 70) and greater than 40 mg/m2 (median, 65 mg/m2; n = 59) of selinexor administered on a twice-a-week schedule (eight doses per 28-day cycle). The percentage of patients on twice-a-week dosing who withdrew consent on greater than 40 mg/m2 (43%; 30 of 70 patients) was 1.9 times greater than the percentage of those who withdrew consent on ≤ 40 mg/m2 (23%; 27 of 119 patients). In addition, of the 16 scheduled doses over the first two cycles, patients on greater than 40 mg/m2 took a median of 14 doses, and 27% took 15 to 16 doses, whereas on ≤ 40 mg/m2, patients took a median of 15 doses, with 61% taking 15 to 16 doses. Weight loss was greater in the high- versus low-dose group after two cycles, with average losses of 5.6% and 3.4%, respectively. Furthermore, the percentage of patients who made it through two cycles without either grade 2 anorexia or fatigue was significantly higher in the low-dose group (Data Supplement). The most common reasons given for drug withdrawal were fatigue, nausea, and vomiting.
Pharmacokinetics
Pharmacokinetic parameters of selinexor are listed in Table 4 and demonstrated a median time to peak serum concentration of 2 to 4 hours with a terminal half-life of approximately 6 to 7 hours. There was no evidence of drug accumulation or changes in clearance across all dose levels. Analysis of plasma and urine samples from patients treated with selinexor revealed glucuronidation as the primary route of drug metabolism. In addition, analysis of drug levels based on milligram per meter squared dosing suggests minimal variability as a result of body surface area, such that fixed doses of selinexor show comparable correlation with pharmacokinetic parameters.
Table 4.
Selinexor Pharmacokinetics as a Function of Dose

Pharmacodynamics
XPO1 mRNA levels were measured in leukocytes isolated from patient blood after the first dose of selinexor and after the fifth or sixth dose (days 15 to 17). The level of XPO1 mRNA increased, by virtue of a positive loop feedback, by 2 hours after selinexor administration, with peak induction by 4 to 8 hours after dosing of greater than five-fold versus before dosing. The induction declined to greater than three-fold at 48 hours but remained at that level through day 15 or 17, before the fifth or sixth dose (Data Supplement). XPO1 transcription increased dose proportionally from 3 to 28 mg/m2, reaching an induction plateau of 5.3- ± 2.9-fold at doses ≥ 28 mg/m2 (Data Supplement). Therefore, the half-life of selinexor pharmacodynamic activity, based on circulating leukocytes, is at least 48 hours and consistent with twice-a-week dosing.
Tumor biopsies from accessible lesions of nine patients (cervical cancer, two colorectal cancers [CRCs], Ewing sarcoma, head and neck squamous cell carcinoma, lung cancer, melanoma, mesothelioma, and prostate cancer) were examined before and approximately 4 weeks after initiation of selinexor. Hematoxylin and eosin staining showed an overall reduction in malignancy in all responders (Figs 1A and 1B). Immunohistochemical analysis using ApopTag, cleaved caspase 3, and Ki-67 antibodies showed increased apoptosis and decreased proliferation after selinexor treatment (Figs 1C to 1H). In addition, nuclear localization of key TSPs and XPO1 cargos p53 and FOXO3A were observed after selinexor treatment (Figs 1I to 1L).
Fig 1.

Pharmacodynamics of selinexor treatment: immunohistochemistry (IHC) of paired tumor biopsies. Patient tumor biopsies were analyzed by IHC for morphologic and biochemical changes in response to selinexor treatment. (A and B) Hematoxylin and eosin (H&E) staining of tumor samples taken from a patient with colorectal cancer (CRC) at baseline and 5 weeks after treatment with selinexor 23 mg/m2. IHC staining (brown) for apoptosis markers (C and D) Apoptag (Millipore, Billerica, MA) and (E and F) cleaved caspase 3 in a patient with CRC at baseline and after treatment with selinexor 12 mg/m2. (G and H) Decreased staining intensity for the proliferation marker Ki-67 in a patient with melanoma treated with selinexor 16.8 mg/m2. Nuclear accumulation of XPO1 cargos (I and J) p53 and (K and L) FOXO3A in a patient with lung adenosquamous cell carcinoma treated with selinexor 35 mg/m2. Images are × 10 magnification with smaller inserts magnified to × 40.
Antitumor Activity
Among the 157 patients who were evaluable for efficacy, one patient had a complete response (melanoma), and six other patients (melanoma, CRC, ovarian cancer, prostate cancer, thymoma, and cervical cancer) had radiographic partial responses, for an objective response rate of 4%. In addition, 67 patients (43%) had stable disease, including 27 patients (17%) with durable disease control (≥ 4 months), and 83 patients (53%) had progressive disease as their best response. Thirty-two patients (17%) were nonevaluable, mainly as a result of consent withdrawal before the first radiologic assessment. There was no relationship between dose range and response when patients were evaluated by dose groups (≤ or > 40 mg/m2). A waterfall plot depicting target lesion change is depicted in Figure 2, and best response is summarized in the Data Supplement.
Fig 2.

Waterfall plot of change in target lesion volume after selinexor treatment. Best response in target lesions for 129 evaluable patients. Quantitative target lesion assessment was not available for the remaining 28 evaluable patients. These included 22 patients with progressive disease based solely on clinical symptoms and six patients with stable disease (SD), two of whom were patients with prostate cancer who had bone scans and remained on study for 325 and 72 days before showing disease progression. One patient with ovarian cancer had SD and remained on study for 365 days despite not having quantifiable baseline scans. The remaining three patients (two with prostate cancer and one with nasopharyngeal cancer) remained on study for 117, 71, and 44 days, respectively, without evidence of disease progression before withdrawing consent.
Patients with CRC were the largest population represented (59 patients, 52 evaluable patients). Tumor biopsies from 43 patients identified 29 with KRAS mutation. The patient with CRC who achieved a partial response had a KRAS mutation. A greater percentage of patients with stable disease (44%) was observed in the KRAS-mutant group versus the wild-type RAS group; however, the difference from wild-type (21%) was not enough to suggest mutational status as predictive (Data Supplement).
DISCUSSION
The recognition that tumor cells use XPO1 to functionally inactivate many TSPs led to the design of selective inhibitor of nuclear export compounds. Here, we report that the selective inhibitor of nuclear export compound selinexor has an acceptable toxicity profile, induces expected pharmacodynamic changes in both normal blood leukocytes and in tumor tissue, and can lead to tumor shrinkage across a broad array of chemotherapy-refractory neoplastic disorders. The most commonly reported toxicities were low-grade GI (vomiting, nausea, anorexia) and constitutional (fatigue, weight loss) AEs. Supportive treatment, with aggressive use of antiemetics and nutritional supplements, increased tolerability to selinexor, as demonstrated by patients remaining on selinexor for greater than 1 year. DLTs were all reversible and included hyponatremia, anorexia, nausea, vomiting, fatigue, dehydration, and one case of cerebellar syndrome. Unusual toxicities such as hyponatremia and blurred vision may be a class effect phenomenon, but they seem to be self-limiting and reversible in most patients. Dose interruptions and dose reductions occurred most commonly as a result of GI AEs. Patients were noted to be less tolerant to higher dose levels regardless of dosing schedule.
The RP2D of selinexor of 35 mg/m2 (approximately 60-mg fixed dose) twice a week was established based on greater tolerability in patients receiving ≤ 40 mg/m2 (median, 35 mg/m2) compared with greater than 40 mg/m2. This is evidenced by fewer missed doses, less weight loss, and decreased prevalence of grade 2 anorexia and fatigue, which are all relevant for chronic dosing. The superior tolerability of 35 mg/m2 is coupled with efficacy that is comparable to the higher doses evaluated, and therefore, justifies our decision for the RP2D. However, one still has to be mindful that challenges with toxicities in the setting of chronic dosing schedules exist, because 19% of patients still withdrew treatment at doses less than 40 mg/m2, which highlights the need for a proactive stance on supportive treatment when using this agent.
Target engagement, reflected by changes in XPO1-related biomarkers as well as nuclear accumulation of TSPs, supports the novel mode of action for this drug. Until now, targeting XPO1-mediated nuclear transport in patients has never been successfully carried out, because clinical trials with leptomycin B, an irreversible inhibitor of XPO1, were abandoned as a result of toxicities.28
In this study, treatment with selinexor resulted in one complete response, six partial responses, and prolonged disease stabilization (≥ 4 months) in 27 patients (17%) over a number of tumor types. Seven patients remained on selinexor for more than 1 year, and in two patients, treatment with selinexor continued and was well tolerated for almost 2 years (668 and 679 days; soft tissue sarcoma and thymoma), suggesting that long-term treatment is feasible. Interestingly, selinexor treatment was associated with stable disease in 44% of CRC tumors with KRAS mutations, which is interesting because these patients tend to have fewer systemic treatment options. Efforts to identify protein, mRNA, and/or miRNA biomarkers that predict response to selinexor are ongoing.
Given the demonstrated activity as monotherapy, clinical combinations of selinexor with other cytotoxic and/or targeted agents are ongoing. The majority of current anticancer agents require induction of apoptosis with at least transient activation of TSPs such as p53, p73, BRCA1, IκB, p21, and/or the forkhead box transcription factors (eg, FOXO1, FOXO3a, and FOXO4).13 The elevated levels of XPO1 observed across essentially all cancers mediate the efficient nuclear exclusion of these key TSPs, thereby attenuating the anticancer effects of available agents. By inhibiting XPO1, selinexor forces the nuclear retention of TSPs and restores their function, and these effects are potentially synergistic with existing therapies. Additionally, given that the metabolism of selinexor is independent of the cytochrome P450 pathways, the possible combinations with this agent are vast. Preclinical evaluation of selinexor in combination therapies, with platins, taxanes, irinotecan, temozolomide, gemcitabine, vemurafenib, and ABT737, demonstrated enhanced efficacy in mouse xenograft models.29-34 In addition, selinexor acted synergistically with radiation therapy in non–small-cell lung cancer to enhance cell death, at least in part through the inhibition of DNA damage repair enzymes.35 Together, these data highlight the potential for selinexor in combination with commonly used therapeutic regimens and warrant further investigation of selinexor in clinical trials, some of which are currently ongoing.
In conclusion, selinexor has an acceptable safety profile with evidence of clinical benefit in advanced metastatic solid tumors. Given the promising safety and efficacy profile of selinexor, multiple trials in are under way.
ACKNOWLEDGMENT
We thank Lillian Siu, MD (Princess Margaret Cancer Centre, Toronto, Ontario, Canada), Philippe Bedard, MD (Princess Margaret Cancer Centre), and Hemchandra Mahaseth, MD (Karmanos Cancer Centre, Detroit, MI), for their help in patient recruitment and care.
Footnotes
Supported by Karyopharm Therapeutics.
Presented at the 49th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 31-June 3, 2013, and the 50th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 30-June 3, 2014.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
See accompanying article on page 4180
AUTHOR CONTRIBUTIONS
Conception and design: Albiruni R. Abdul Razak, Morten Mau-Soerensen, John F. Gerecitano, Thaddeus J. Unger, Jean R. Saint-Martin, Robert Carlson, Mansoor R. Mirza, Michael Kauffman, Sharon Shacham, Amit Mahipal
Financial support: Sharon Shacham
Administrative support: Albiruni R. Abdul Razak, Thaddeus J. Unger, Robert Carlson, Michael Kauffman, Sharon Shacham
Provision of study materials or patients: Morten Mau-Soerensen, Nashat Y. Gabrail, John F. Gerecitano, Thaddeus J. Unger, Jean R. Saint-Martin, Robert Carlson, Yosef Landesman, Michael Kauffman, Sharon Shacham
Collection and assembly of data: Albiruni R. Abdul Razak, Anthony F. Shields, Thaddeus J. Unger, Jean R. Saint-Martin, Robert Carlson, Dilara McCauley, Sharon Shacham, Amit Mahipal
Data analysis and interpretation: Albiruni R. Abdul Razak, Morten Mau-Soerensen, Nashat Y. Gabrail, John F. Gerecitano, Anthony F. Shields, Thaddeus J. Unger, Jean R. Saint-Martin, Robert Carlson, Yosef Landesman, Dilara McCauley, Tami Rashal, Ulrik Lassen, Richard Kim, Lee-Anne Stayner, Michael Kauffman, Sharon Shacham, Amit Mahipal
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
First-in-Class, First-in-Human Phase I Study of Selinexor, a Selective Inhibitor of Nuclear Export, in Patients With Advanced Solid Tumors
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Albiruni R. Abdul Razak
Research Funding: Karyopharm Therapeutics
Morten Mau-Soerensen
Honoraria: Karyopharm Therapeutics
Research Funding: Karyopharm Therapeutics
Nashat Y. Gabrail
Research Funding Karyopharm Therapeutics, Taiho Pharmaceutical, Bayer, Bristol-Myers Squibb, OncoMed, Novartis, Boehringer Ingelheim
John F. Gerecitano
Research Funding: Karyopharm Therapeutics
Anthony F. Shields
Consulting or Advisory Role: GE Healthcare, Endocyte, Bayer, Bristol-Myers Squibb, Siemens Healthcare Diagnostics, Merck, Merrimack, Taiho Pharmaceutical, Perceptive Informatics
Speakers' Bureau: GE Healthcare
Research Funding: Karyopharm Therapeutics, Merrimack, Taiho Pharmaceutical, Bayer, Bristol-Myers Squibb, Novartis, Boehringer Ingelheim, OncoMed
Travel, Accommodations, Expenses: GE Healthcare, Merrimack, Endocyte
Thaddeus J. Unger
Employment: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Jean R. Saint-Martin
Employment: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Patents, Royalties, Other Intellectual Property: Karyopharm Therapeutics
Robert Carlson
Employment: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Yosef Landesman
Employment: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Patents, Royalties, Other Intellectual Property: Karyopharm Therapeutics
Dilara McCauley
Employment: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Patents, Royalties, Other Intellectual Property: Karyopharm Therapeutics
Travel, Accommodations, Expenses: Karyopharm Therapeutics
Tami Rashal
Employment: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Patents, Royalties, Other Intellectual Property: Karyopharm Therapeutics
Ulrik Lassen
No relationship to disclose
Richard Kim
Honoraria: Bristol-Myers Squibb
Consulting or Advisory Role: Bristol-Myers Squibb, Eli Lilly, Bayer, Genentech
Speakers' Bureau: Eli Lilly, Sanofi
Research Funding: Bayer, Novartis
Lee-Anne Stayner
No relationship to disclose
Mansoor R. Mirza
Leadership: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Consulting or Advisory Role: Karyopharm Therapeutics
Travel, Accommodations, Expenses: Karyopharm Therapeutics
Michael Kauffman
Employment: Karyopharm Therapeutics, Karyopharm Therapeutics (I)
Leadership: Karyopharm Therapeutics, Karyopharm Therapeutics (I)
Stock or Other Ownership: Karyopharm Therapeutics, Karyopharm Therapeutics (I)
Patents, Royalties, Other Intellectual Property: Karyopharm Therapeutics, Karyopharm Therapeutics (I)
Sharon Shacham
Employment: Karyopharm Therapeutics
Leadership: Karyopharm Therapeutics
Stock or Other Ownership: Karyopharm Therapeutics
Amit Mahipal
Research Funding: Karyopharm Therapeutics
REFERENCES
- 1.Zaidi SK, Young DW, Javed A, et al. Nuclear microenvironments in biological control and cancer. Nat Rev Cancer. 2007;7:454–463. doi: 10.1038/nrc2149. [DOI] [PubMed] [Google Scholar]
- 2.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83:1021–1032. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook A, Bono F, Jinek M, et al. Structural biology of nucleocytoplasmic transport. Annu Rev Biochem. 2007;76:647–671. doi: 10.1146/annurev.biochem.76.052705.161529. [DOI] [PubMed] [Google Scholar]
- 4.Kau TR, Way JC, Silver PA. Nuclear transport and cancer: from mechanism to intervention. Nat Rev Cancer. 2004;4:106–117. doi: 10.1038/nrc1274. [DOI] [PubMed] [Google Scholar]
- 5.Turner JG, Marchion DC, Dawson JL, et al. Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res. 2009;69:6899–6905. doi: 10.1158/0008-5472.CAN-09-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang WY, Yue L, Qiu WS, et al. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med. 2009;32:E315. [PubMed] [Google Scholar]
- 7.Lesinski GB, Yang J, Bill MA, et al. Effect of small inhibitors of nuclear export (SINE) on growth inhibition and apoptosis of human melanoma cells. J Clin Oncol 30, 2012 (suppl; abstr e13549)
- 8.Noske A, Weichert W, Niesporek S, et al. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008;112:1733–1743. doi: 10.1002/cncr.23354. [DOI] [PubMed] [Google Scholar]
- 9.Shen A, Wang Y, Zhao Y, et al. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009;65:153–159. doi: 10.1227/01.NEU.0000348550.47441.4B. [DOI] [PubMed] [Google Scholar]
- 10.van der Watt PJ, Maske CP, Hendricks DT, et al. The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer. 2009;124:1829–1840. doi: 10.1002/ijc.24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Watt PJ, Leaner VD. The nuclear exporter, Crm1, is regulated by NFY and Sp1 in cancer cells and repressed by p53 in response to DNA damage. Biochim Biophys Acta. 2011;1809:316–326. doi: 10.1016/j.bbagrm.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 12.Yao Y, Dong Y, Lin F, et al. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep. 2009;21:229–235. [PubMed] [Google Scholar]
- 13.Tan DS, Bedard PL, Kuruvilla J, et al. Promising SINEs for embargoing nuclear-cytoplasmic export as an anticancer strategy. Cancer Discov. 2014;4:527–537. doi: 10.1158/2159-8290.CD-13-1005. [DOI] [PubMed] [Google Scholar]
- 14.Azmi AS, Kauffman M, McCauley D, et al. Novel small-molecule CRM-1 inhibitor for GI cancer therapy. J Clin Oncol 30, 2012 (suppl 4; abstr 245)
- 15.Chung HW, Salas Fragomeni RA, Shacham S, et al. Effect of inhibition of nuclear protein export using a novel CRM1 inhibitor on the apoptotic response to SN38 in preclinical colon cancer models. J Clin Oncol 30, 2012 (suppl 4; abstr 609)
- 16.Inoue H, Kauffman M, Shacham S, et al. Evaluation of selective inhibitors of nuclear export (SINE) CRM1 inhibitors for the treatment of renal cell carcinoma (RCC). J Clin Oncol 30, 2012 (suppl; abstr 4634)
- 17.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120:4621–4634. doi: 10.1182/blood-2012-05-429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCauley D, Landesman Y, Senapedis W, et al. Preclinical evaluation of selective inhibitors of nuclear export (SINE) in basal-like breast cancer (BLBC). J Clin Oncol 30, 2012 (suppl; abstr 1055)
- 19.Shacham S, Kauffman M, Sandanayaka V, et al. Preclinical development of small-molecule CRM1 inhibitors as novel therapy for the treatment of colorectal cancer (CRC). J Clin Oncol 29, 2011 (suppl 4; abstr 430)
- 20.Green AL, Ramkissoon SH, McCauley D, et al. Preclinical antitumor efficacy of selective exportin 1 inhibitors in glioblastoma. Neuro Oncol 17:697-707, 2015. [DOI] [PMC free article] [PubMed]
- 21.Chen Y, Kalir E, Camacho-Venegas C, et al. Increased overall survival in platinum-resistant ovarian cancer: paradigmatic use of novel SINE (selective inhibitor of nuclear export), which restores p53 nuclear localization and activation. Cancer Res. 2013;73:2163. (abstr) [Google Scholar]
- 22.Parikh K, Cang S, Sekhri A, et al. Selective inhibitors of nuclear export (SINE): A novel class of anti-cancer agents. J Hematol Oncol. 2014;7:78. doi: 10.1186/s13045-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahaseth H, Maity S, Gabrail N, et al. Selinexor (KPT-330), an oral, selective inhibitor of nuclear export (SINE) shows anti-prostate cancer (PrCa) activity preclinically and disease control in patients (pts) with chemotherapy refractory, castrate-resistant prostate cancer (CRPC). Ann Oncol 25, 2014 (suppl 4; abstr iv257)
- 24.Etchin J, Sun Q, Kentsis A, et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013;27:66–74. doi: 10.1038/leu.2012.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shacham S, Barnard S, Kisseberth W, et al. Results of a phase I dose escalation study of the novel, oral CRM1 selective inhibitor of nuclear export (SINE) KPT-335 in dogs with spontaneous non-Hodgkin’s lymphomas (NHL) Blood. 2012;120:161. (abstr) [Google Scholar]
- 26. doi: 10.1200/JOP.2015.006106. National Cancer Institute: Common Terminology Criteria for Adverse Events v4.03. http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf. [DOI] [PMC free article] [PubMed]
- 27.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 28.Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer. 1996;74:648–649. doi: 10.1038/bjc.1996.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martingnetti J, Razak A, Chen Y, et al. Preclinical and early clinical activity of the oral selective inhibitor of nuclear export (SINETM) exportin 1 (XPO1) antagonist selinexor (KPT-330) in patients with platinum resistant/refractory ovarian cancer (OvCa). Ann Oncol 25, 2014 (suppl 4; abstr iv310)
- 30.Kazim S, Malafa MP, Coppola D, et al. Selective nuclear export inhibitor KPT-330 enhances the antitumor activity of gemcitabine in human pancreatic cancer. 2015. Mol Cancer Ther 14:1570-1581, 2015. [DOI] [PMC free article] [PubMed]
- 31.Salas Fragomeni RA, Chung HW, Landesman Y, et al. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol Cancer Ther. 2013;12:1171–1179. doi: 10.1158/1535-7163.MCT-12-1171. [DOI] [PubMed] [Google Scholar]
- 32.Miyake TM, Pradeep S, Zand B, et al. Therapeutic targeting of CRM1 in ovarian cancer. Cancer Res. 2013;73:5541. [Google Scholar]
- 33.Chung HW, Salas Fragomeni RA, Shacham S, et al. Use of selective inhibition of nuclear export (SINE) using a CRM1/XPO1 antagonist to overcome resistance to CPT-11 in colon cancer in preclinical models. J Clin Oncol 31, 2013 (suppl 4; abstr 396)
- 34.Miyake T, Pradeep S, Wu SY, et al. XPO1/CRM1 inhibition causes antitumor effects by mitochondrial accumulation of eIF5A. Clin Cancer Res. 2015;21:3286–3297. doi: 10.1158/1078-0432.CCR-14-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rashal T, Elloul S, Crochiere M, et al. Selinexor (KPT-330) radio-sensitizes non-small cell lung cancer cells in vitro and in vivo. Cancer Res. 2015;75:4490. (abstr) [Google Scholar]