

Summary
N6-methyladenosine (m6A) is the most abundant modified base in eukaryotic mRNA and has been linked to diverse effects on mRNA fate. Current m6A mapping approaches localize m6A residues to 100–200 nt-long regions of transcripts. The precise position of m6A in mRNAs cannot be identified on a transcriptome-wide level because there are no chemical methods to distinguish between m6A and adenosine. Here, we describe a method for using anti-m6A antibodies to induce specific mutational signatures at m6A residues after ultraviolet light-induced antibody-RNA crosslinking and reverse transcription. Then, we describe how to use these mutational signatures to map m6A residues at nucleotide resolution. Taken together, our protocol allows for high-throughput detection of individual m6A residues throughout the transcriptome.
Keywords: RNA, N6-methyladenosine, crosslinking, high-throughput sequencing sequencing
1. Introduction
N6-methyladenosine (m6A) is the most prevalent modified base in mRNA [1–5] and is found in several thousand transcripts, typically near the stop codon, but also in the coding sequence, 3′UTR, and 5′UTR of mRNAs [1–3].
The current m6A mapping approach, methyl-RNA immunoprecipitation and sequencing (MeRIP-Seq, also called m6A-Seq) [2,3], has allowed researchers to map regions of RNA methylation. MeRIP-Seq involves immunoprecipitation of ~100 nt-long RNA fragments using m6A-specific antibodies, followed by high-throughput sequencing of the immunoprecipitated fragments. m6A-containing fragments then generate overlapping sequencing reads that produce a “peak” whose summit reflects an underlying m6A residue [2]. However, the current mapping approach does not identify specific m6A residues.
Identifying m6A residues is challenging. Adenosine methylation is predominantly restricted to adenosines in a DRA*CH sequence context (D=A, G or U; R = purine; A* = methylatable A; H=A, C or U) [6]; however, not all DRACH motifs are methylated in vivo [1]. Exact positions of m6A residues can be bioinformatically predicted from MeRIP-Seq peaks by searching for the presence of a subset of DRACH motifs near the point of highest read coverage [7]. However, this approach is complicated because m6A often appears in clusters, which can result in large peaks spanning several m6A residues [2]. Additionally, multiple DRACH motifs can be present underneath a peak, making it difficult to predict the specific methylated adenosine.
Likewise, there is no chemical method that results in selective modification and detection of m6A residues. The nearly identical chemical properties of A and m6A have prevented the development of a chemical method to distinguish these nucleotides. Additionally, unlike other base modifications, m6A does not introduce errors during reverse transcription that would allow direct mapping of its position [8]. Thus, a major goal is to develop a method that provides a specific chemical signature that indicates the precise location of m6A residues in the transcriptome.
Here, we demonstrate how to use anti-m6A antibodies to induce specific mutational signatures that enable precise identification of m6A residues in RNA (Figure 1). In this approach, anti-m6A antibodies are crosslinked to RNA using UV light to create antibody-RNA crosslinks. Reverse transcription of crosslinked RNA then results in a highly specific pattern of mutations or truncations in the cDNA. These mutational signatures are then computationally identified to reveal precise positions of m6A residues. Using these signatures we map m6A residues throughout the transcriptome at single-nucleotide resolution.
Figure 1.
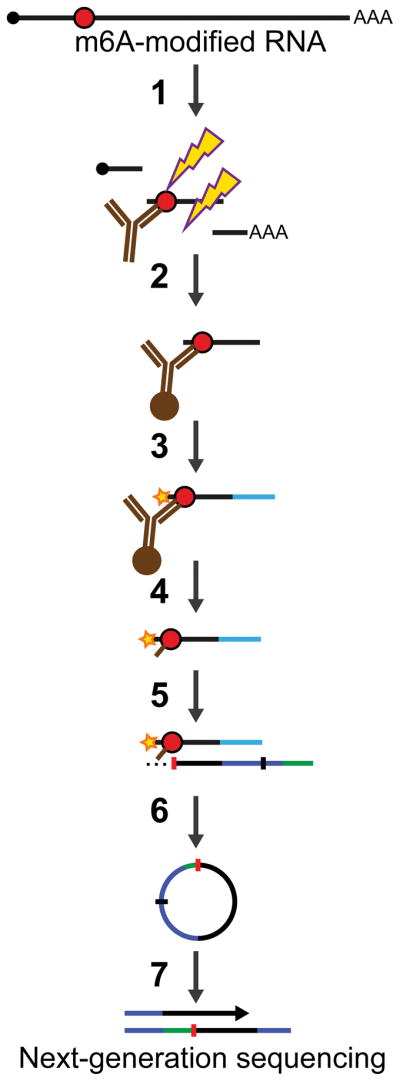
Schematic of the miCLIP protocol. Cellular RNA containing m6A (red circles) is fragmented, incubated with an anti-m6A antibody and UV-crosslinked (1). Then, antibody-RNA complexes are recovered by protein A/G-affinity purification (2). Next, a 3′-adapter is ligated and the 5′-end is radiolabeled (3). RNA-protein complexes are then purified (4). RNA fragments are then reverse transcribed, generating mutations or truncations in the resulting cDNA (5). The cDNA is then circularized (6), re-linearized and amplified by PCR (7).
2. Materials
All buffers and solutions should be made in ultrapure, nuclease- and nucleic-acid free water, and purified using vacuum filtration units (see below).
2.1. Buffers and solutions
Binding/low-salt buffer: 50 mM Tris HCl pH 7.4, 150 mM NaCl, 0.5 % Nonidet P-40 (NP-40).
High-salt buffer: 50 mM Tris HCl pH 7.4, 1 M NaCl, 1 % NP-40, 0.1 % sodium dodecyl sulfate (SDS).
PNK wash buffer: 20 mM Tris HCl pH 7.4, 10 mM MgCl2, 0.2 % Tween-20.
5X PNK pH 6.5 buffer: 350 mM Tris HCl pH 6.5, 50 mM MgCl2, 5 mM dithiothreitol (DTT).
Proteinase K (PK) buffer: 100 mM Tris HCl pH 7.4, 50 mM NaCl, 10 mM ethylenediaminetetraacetic acid (EDTA).
Proteinase K urea (PKU) buffer: 100 mM Tris HCl pH 7.4, 50 mM NaCl, 10 mM EDTA, 7 M urea.
RNase-free sodium acetate: 3 M, pH 5.5.
RNase-free TE buffer: 10 mM Tris-HCl pH 7.5, 1mM EDTA pH 8.0.
NuPage MES SDS Running Buffer: 20X (Novex), diluted to 1X in water.
Bis-Tris Transfer Buffer: 5X (Invitrogen), diluted to 1X in water.
TBE Running Buffer: 5X (Invitrogen), diluted to 1X in water.
2.2. Reagents
Cellular RNA (Note 1 )
Anti-m6A antibodies: 1 mg/mL (Note 2 )
Crushed ice
Ethanol, pure
Methanol, pure
RNase-free DNase I: 1 U/μL.
RNA fragmentation reagent, including stop solution: 10X (Thermo Fisher).
Ribonuclease inhibitor: 40U/μL.
Protein A/G magnetic beads
T4 polynucleotide kinase supplied with buffer: 1000 U/μL.
T4 RNA ligase I supplied with buffer: 1000 U/μL.
Polyethylene glycol (PEG) 400
[γ-32P]ATP: 3000 Ci/mmol (Perkin Elmer) (Note 3 )
Lithium dodecyl sulfate (LDS) sample buffer: 4X (Thermo Fisher).
Dithiothreitol: 1 M in water.
Prestained protein standard
Proteinase K: 10 mg/mL
Acidic phenol:chloroform:isoamyl alcohol, pH 6.5. (Note 4 )
GlycoBlue coprecipitant:15 mg/mL (Thermo Fisher)
Superscript III kit, supplied with buffer and 10 mM dNTPs (Thermo Fisher)
Deoxynucleotide triphosphates: 10 mM in water.
Low molecular weight DNA ladder: 25 to 800 nucleotides
TBE sample buffer: 5X (Thermo Fisher)
TBE-urea sample buffer: 2X (Thermo Fisher)
SYBR gold: 10,000X (Note 5 )
CircLigase II kit, supplied with buffer and 50 mM MnCl2 (Epicentre)
FastDigest BamHI (800 reactions), supplied with buffer (Thermo Fisher)
Accuprime Supermix I (Thermo Fisher)
Agencourt Ampure XP magnetic beads (Beckman Coulter)
2.3. Oligonucleotides
All oligonucleotides are from Integrated DNA Technologies (IDT) with standard purification unless otherwise indicated.
L3 linker (HPLC-purified): /5rApp/AGATCGGAAGAGCGGTTCAG/3ddC/, 20 μM stock
Cut oligo (PAGE-purified): GTTCAGGATCCACGACGCTCTTC/3ddC/, 10 μM stock
P5 Solexa PCR primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT, 10 μM stock
P3 Solexa PCR primer: CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT, 10 μM stock
-
Reverse transcription primers: 0.5 μM stocks of each
RT1: /5phos/NnaaccNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT2: /5phos/NNacaaNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT3: /5phos/NNattgNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT4: /5phos/NNaggtNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT5: /5phos/NNcgccNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT6: /5phos/NNccggNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT7: /5phos/NNctaaNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT8: /5phos/NNcattNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT9: /5phos/NNgccaNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT10: /5phos/NNgaccNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT11: /5phos/NNggttNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT12: /5phos/NNgtggNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT13: /5phos/NNtccgNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT14: /5phos/NNtgccNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT15: /5phos/NNtattNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
RT16: /5phos/NNttaaNNNAGATCGGAAGAGCGTCGTGgatcCTGAACCGC
2.4 Supplies
Vacuum filter units for purifying buffers and solutions
Tissue culture clusters, 12 wells per cluster
Plastic wrap
Microcentrifuge tubes of various sizes: 1.5 mL, 0.5 mL 0.2 mL
Pipettors and filtered tips in various sizes: 1000 μL, 200 μL, 20 μL, 10 μL.
4–12 % bis-tris protein gels (Thermo Fisher)
Nitrocellulose membrane, 0.45 μm pore size
Carestream BioMax MS films, 8×10 inches (Kodak)
Carestream BioMax intensifying screen, 8×10 inches (Kodak)
Luminescent stickers
Phase-lock gel heavy tubes (5 PRIME)
6 % TBE-urea gels (Thermo Fisher)
19G syringe needles or gel breaker tubes
CoStar Spin-X columns, 0.22 μm pore size (Corning)
6 % TBE gels (Thermo Fisher)
2.5 Equipment
Cold room and freezers (−20°C and −80°C)
End-over-end mixer for rotating microcentrifuge tubes
Vortexer
Magnetic rack for microcentrifuge tubes
Shallow tray (~5 cm high) for crosslinking samples
Stratalinker 2400 (Stratagene)
Electrophoretic gel chamber
Electrophoretic transfer unit
Geiger counter
Autoradiography cassette
Temperature-adjustable microcentrifuge mixer
Refrigerated microcentrifuge capable of spins >/=20,000 RCF
UV transilluminator or imager for visualizing SYBR gold-stained gels
Thermocycler
Access to a facility capable of Illumina high-throughput sequencing
2.6 Software
Flexbar 2.5 (available at: https://github.com/seqan/flexbar.git)
pyCRAC 1.1.3 (available at: https://bitbucket.org/sgrann/pycrac)
Novoalign 3.02.12 (available at: http://www.novocraft.com/products/novoalign)
CIMS and CITS packages (available at: http://zhanglab.c2b2.columbia.edu/index.php/CIMS_Documentation)
Bedtools 2.25.0 (available at: https://github.com/arq5x/bedtools2.git)
Samtools 1.2 (available at: http://samtools.sourceforge.net/)
Perl, python, and basic unix command line tools
File of the reference genome in .fasta format (various reference genomes are available at: http://hgdownload.cse.ucsc.edu/downloads.html)
Bedtools-compatible genome index file in .fasta.fai format (produced with samtools index command)
3. Methods
For any steps requiring the use or addition of water, use ultrapure, nuclease-, and nucleic-acid free water.
3.1. Fragment RNA
(Note 6.)
Start with 5–20 μg of DNase-treated, polyA+-selected (optional) RNA suspended in 20 μL of water in a sterile, nuclease-free microcentrifuge tube.
Add 2 μL of RNA fragmentation reagent to the RNA, and mix gently with a pipettor. Incubate the mixture in a heat block at 75°C for exactly 12 min.
Immediately after fragmentation has completed, place the tube on ice, add 2.2 μL of the stop solution to the mixture, and mix gently with the pipettor. (Note 7.)
3.2. Crosslink RNA to anti-m6A antibody
Bring up the volume of the sample to 500 μL with an appropriate volume of the binding/low-salt buffer and mix gently with the pipettor.
Add 10 μL of anti-m6A antibody and 2 μL of ribonuclease inhibitor to the sample and mix gently with the pipettor. (Note 8.)
Rotate the sample tube at 4°C (e.g. in a cold room) for 1.5 to 2 h.
Prior to crosslinking, pre-chill a sterile 12-well tissue culture plate on ice. (Note 9.)
After RNA-antibody binding has completed, transfer the sample to a single well of the 12-well tissue culture plate. Place the plate on crushed ice in a shallow (~5 cm) tray.
Remove the lid from the plate, and crosslink twice in a Stratalinker 2400 using 254 nm light and 150 mJ/cm2. Between each irradiation, gently agitate the tray to mix the sample.
Transfer the crosslinked sample to a new microcentrifuge tube, and place on ice. (Note 10.)
3.3. Immunoprecipitate the RNA-antibody complexes
Resuspend protein A/G magnetic beads by gentle vortexing. Aliquot 50 μL of the magnetic beads to a new microcentrifuge tube. Wash the beads twice in 500 μL of binding/low-salt buffer. Resuspend the washed beads in 100 μL of binding/low-salt buffer.
Add the bead slurry to the crosslinked sample and mix gently with the pipettor.
Rotate the sample tube in a cold room for 1.5 h to overnight. (Note 11.)
Wash the magnetic beads twice in 900 μL of high-salt buffer. (Note 12.)
Wash the magnetic beads twice in 900 μL of binding/low-salt buffer.
Wash the magnetic beads twice in 500 μL of PNK wash buffer.
3.4. Dephosphorylate 3′ ends of the RNA fragments
-
Resuspend the magnetic beads in 20 μL of dephosphorylation master mix:
15 μL water
4 μL 5X PNK pH 6.5 buffer
0.5 μL T4 polynucleotide kinase
0.5 μL ribonuclease inhibitor
Incubate the sample in a thermal mixer at 37°C for 20 min, shaking constantly at 1,100 RPM.
Wash the beads once in 500 μL of PNK wash buffer.
Wash the beads once in 500 μL of high-salt buffer.
Wash the beads twice in 500 μL of PNK wash buffer.
3.5. Ligate linker to 3′ ends of RNA fragments
-
Resuspend the magnetic beads in 20 μL of ligation master mix:
11 μL water
2 μL 10X T4 RNA ligase buffer
1 μL T4 RNA ligase I
0.5 μL ribonuclease inhibitor
1.5 μL L3 linker
4 μL PEG 400
Incubate the sample in a thermal mixer at 16°C overnight, shaking intermittently at 1,100 RPM (30 s shaking followed by 4 min and 30 s without shaking).
Wash the beads once in 500 μL of PNK wash buffer.
Wash the beads twice in 500 μL of high-salt buffer. For each wash, rotate the sample tube at 4°C for 5 min.
Wash the beads twice in 500 μL of PNK wash buffer.
3.6. Radiolabel the 5′ ends of RNA fragments
(Note 13.)
-
Resuspend the magnetic beads in 40 μL of radiolabeling master mix:
33 μL water
4 μL 10X T4 polynucleotide kinase buffer
1 μL [γ-32P]ATP
2 μL T4 polynucleotide kinase
Incubate the sample in a thermal mixer at 37°C for 20 min, shaking constantly at 1,100 RPM.
Wash beads twice in 500 μL of binding/low-salt buffer.
Wash beads twice in 500 μL of PNK wash buffer.
3.7. Purify RNA-antibody complexes using SDS-PAGE and membrane transfer
-
Resuspend the magnetic beads in 20 μL of sample buffer:
14 μL water
5 μL 4X LDS sample buffer
1 μL 1M DTT
Incubate the sample in a thermal mixer at 70°C for 15 min, shaking constantly at 1,100 RPM.
Place the sample tube in a magnetic rack and allow the magnetic beads to pellet completely for 2 min. (Note 14.)
Load the supernatant on a 4–12% bis-tris protein gel according to the manufacturer’s instructions using MES buffer. In a separate lane, load 10 μL of a prestained protein standard.
Run the gel at 4–8°C in a cold room at 200 V for 35 min. (Note 15.)
After the gel has finished running, remove the wells and the running front of the gel and discard in radioactive waste.
Set up transfer of the RNA-antibody complexes to a nitrocellulose membrane in an electrophoretic transfer unit according to the manufacturer’s instructions.
Transfer the RNA-protein complexes to the membrane in a cold room at 400 mA for 30 min. (Note 16.)
Wrap the membrane in plastic wrap and expose to a Kodak BioMax MS film in an autoradiography cassette with an intensifying screen at −80°C. (Note 17 and 18.)
3.8. Elute RNA-antibody complexes
Align the developed film to the membrane and mark regions of the membrane containing the RNA-antibody complexes (Figure 2).
Using a sterile razor blade or small surgical scissors, cut the marked region of the membrane into small fragments (~1×2 mm) and place the fragments into a new microcentrifuge tube.
Quick-spin the membrane fragments and add 200 μL of PK buffer to the sample tube. The membrane pieces must be submerged.
Add 10 μL proteinase K to the sample and mix gently with the pipettor.
Incubate the sample in a thermal mixer at 37°C for 25 min, shaking constantly at 1,100 RPM.
Add 200 μL PKU buffer to the sample and mix gently with the pipettor.
Incubate the sample in a thermal mixer at 37°C for an additional 25 min, shaking constantly at 1,100 RPM.
Pre-spin a phase-lock gel heavy tube for 2 min at 20,000 RCF in a microcentrifuge.
Quick-spin the membrane fragments and transfer the supernatant to the phase-lock gel heavy tube. Add 400 μL of acidic phenol:chloroform:isoamyl alcohol to the tube.
Incubate the sample in a thermal mixer at 30°C for 5 min, shaking constantly at 1,100 RPM.
Centrifuge the sample tube at 20,000 RCF for 5 min.
Transfer the supernatant to a new microcentrifuge tube, and properly discard the phase-lock gel heavy tube. (Note 19.)
-
Add the following to the supernatant in order, mixing gently and thoroughly at each step:
1 μL GlycoBlue coprecipitant
40 μL 3 M sodium acetate, pH 5.5
1000 μL 100% ethanol
Precipitate the RNA fragments at −20°C overnight.
Figure 2.

Sample miCLIP autoradiogram. Here, poly(A)+ RNA was crosslinked to an anti-m6A antibody. The crosslinked RNA-protein complexes were then reduced and purified with SDS-PAGE and nitrocellulose membrane transfer. Crosslinked RNA fragments are found as smears extending from the light and heavy chains of the reduced antibody, at 25 kD and 50 kD, respectively.
3.9. Reverse transcribe RNA fragments
Spin down precipitated RNA fragments in a refrigerated centrifuge at 20,000 RCF for 20 min. Discard the supernatant.
Add 800 μL of 75% ethanol to the sample tube. Spin the sample tube in a refrigerated centrifuge at 20,000 RCF for 5 min. Discard the supernatant.
Invert the sample tube onto a clean tissue paper and allow the RNA pellet to air dry for 5 to 10 min.
-
Resuspend the RNA pellet in 7 μL of denaturing master mix:
5 μL water
1 μL RT primer (Note 20 )
1 μL dNTPs
-
Transfer the resuspended RNA to a new 0.2 mL PCR tube. Incubate the sample in a thermocycler using the following settings:
70°C for 5 min
Pause at 25°C
-
Add reverse transcription master mix to the sample:
7 μL water
4 μL 5X First strand buffer
1 μL DTT
0.5 μL ribonuclease inhibitor
0.5 μL SuperScript III
-
Incubate the sample in the thermocycler using the following settings:
25°C for 5 min
42°C for 20 min
50°C for 40 min
80°C for 5 min
Pause at 4°C
After reverse transcription has completed, transfer the sample to a microcentrifuge tube.
-
Add the following to the sample in order, mixing gently and thoroughly at each step:
350 μL TE buffer
1 μL GlycoBlue coprecipitant
40 μL 3 M sodium acetate, pH 5.5
1000 μL 100% ethanol
Precipitate the cDNA at −20°C overnight or at −80°C for 2 h.
3.10. Purify the cDNA fragments
Spin down precipitated cDNA in a refrigerated centrifuge at 20,000 RCF for 20 min. Discard the supernatant.
Add 800 μL of 75% ethanol to the sample tube. Spin the sample tube in a refrigerated centrifuge at 20,000 RCF for 5 min. Discard the supernatant.
Invert the sample tube onto a clean tissue paper and allow the cDNA pellet to air dry for 5 to 10 min.
Resuspend the cDNA pellet in 6 μL of water.
Add 6 μL of 2X TBE-urea sample buffer to the cDNA and mix gently with the pipettor.
Incubate the sample at 65°C for 5 min to denature the cDNA. Immediately place the tube on ice when finished.
Load the sample on a 6% TBE-urea gel according to the manufacturer’s instructions. In a separate lane, load 0.5 μL of a low molecular weight DNA ladder.
Run the gel at 200 V for 35 min, or until the dark blue dye of the sample buffer has migrated approximately three-quarters of the way through the gel.
Using a sterile razor blade, remove the lane of the gel carrying the ladder and stain this portion of the gel using a 1:10,000 dilution of SYBR gold in gel running buffer for 5 to 10 min.
Place the gel and the stained ladder lane on clean plastic wrap on a UV transilluminator. Using the stained ladder and dye markers as a guide, cut three bands with a sterile razor blade: 120–200 nt (high), 85–120 nt (medium), and 70–85 nt (low). (Note 21.)
Transfer each of the gel bands into a gel breaker tube placed into a new microcentrifuge tube. (Note 22.)
Centrifuge each sample at 20,000 RCF for 2 min to crush the gel slices. Discard the gel breaker tubes.
Add 400 μL of 1X TE buffer to the crushed gel. Freeze the sample in a −80°C freezer for 5 min. Then, incubate the sample in a thermal mixer at 4°C overnight, shaking constantly at 1,100 RPM.
The following morning, transfer the sample to a CoStar SpinX column and centrifuge at 20,000 RCF for 2 min. Transfer the eluate to a phase-lock gel heavy tube.
Add 400 μL of acidic phenol:chloroform:isoamyl alcohol to the tube.
Incubate the samples in a thermal mixer at 30°C for 5 min, shaking constantly at 1,100 RPM.
Centrifuge the sample tube at 20,000 RCF for 5 min.
Transfer the supernatant to a new microcentrifuge tube, and properly discard the phase-lock gel heavy tube.
-
Add the following to the sample in order, mixing gently and thoroughly at each step:
1 μL GlycoBlue coprecipitant
40 μL 3 M sodium acetate, pH 5.5
1000 μL 100% ethanol
Precipitate the cDNA at −20°C overnight or at −80°C for 2 h.
3.11. Circularize and cut the cDNA fragments
Spin down precipitated cDNA in a refrigerated centrifuge at 20,000 RCF for 20 min. Discard the supernatant.
Add 800 μL of 75% ethanol to the sample tube. Spin the sample tube in a refrigerated centrifuge at 20,000 RCF for 5 min. Discard the supernatant.
Invert the sample tube onto a clean tissue paper and allow the cDNA pellet to air dry for 5 to 10 min.
-
Resuspend the cDNA pellet in 8 μL of circularization master mix:
6.5 μL water
0.8 μL CircLigase buffer
0.4 μL MnCl2
0.3 μL CircLigase II
-
Transfer the sample to a 0.2 mL PCR tube and incubate in a thermocycler using the following settings:
60°C for 60 min
Pause at 4°C
-
Add 30 μL of oligo annealing master mix to the sample and mix gently:
26 μL water
3 μL FastDigest buffer
1 μL Cut oligo
-
Anneal the cut oligo to the circularized cDNA using the following settings:
95°C for 2 min
Decrease temperature by 1°C every 20 sec until 25°C is reached
Pause at 25°C
-
Add 2 μL FastDigest BamHI to the sample and linearize the cDNA using the following settings:
37°C for 30 min
80°C for 5 min
-
Transfer the cDNA to a new microcentrifuge tube. Add the following in order, mixing gently at each step:
350 μL TE
1 μL GlycoBlue coprecipitant
40 μL 3 M sodium acetate, pH 5.5
1000 μL 100% ethanol
Precipitate the cDNA at −20°C overnight or at −80°C for 2 h.
3.12. PCR-amplify the cDNA fragments
Spin down precipitated cDNA in a refrigerated centrifuge. Centrifuge at 20,000 RCF for 20 min. Discard the supernatant.
Add 800 μL of 75% ethanol to the sample tube. Spin the sample tube in a refrigerated centrifuge at 20,000 RCF for 5 min. Discard the supernatant.
Invert the sample tube onto a clean tissue paper and allow the cDNA pellet to air dry for 5 to 10 min.
Resuspend the cDNA pellet in 21 μL of water.
-
Prepare three test PCR reactions for each sample, consisting of:
3.75 μL water
1 μL cDNA
0.25 μL P5 and P3 Solexa PCR primer mix
5 μL Accuprime I supermix
-
Incubate the reactions in a thermocycler using the following settings:
94°C for 2 min
Then, 15, 20, or 25 cycles of:
94°C for 15 sec
65°C for 30 sec
68°C for 30 sec
Pause at 4°C
After the PCR reactions have completed, place the reactions on ice and add 2.5 μL of 5X TBE sample buffer to each.
Load the samples on a 6% TBE gel according to the manufacturer’s instructions. In a separate lane, load 0.5 μL of a low molecular weight DNA ladder.
To visualize the PCR products, stain the gel using a 1:10,000 dilution of SYBR gold in gel running buffer for 5 to 10 min. Visualize the stained gel on a UV transilluminator or imager (Figure 3).
Determine the appropriate number of PCR cycles for the preparative PCR reaction. (Note 23.)
-
Assemble the preparative PCR reaction, consisting of:
11 μL water
8 μL cDNA
1 μL of P5 and P3 Solexa primer mix
20 μL Accuprime I Supermix
Amplify the cDNA in a thermocycler for the appropriate number of cycles using the settings described in step 12.6 (Figure 4).
Purify the PCR products using Agencourt Ampure XP magnetic beads using the manufacturer’s instructions. Elute the purified PCR products in 30 μL to 50 μL of water. (Note 24.)
Submit half of the purified library for high-throughput sequencing. (Note 25.)
Figure 3.

Test PCR reactions. This step helps determine the number of cycles used in the preparative PCR reaction. Here, a miCLIP library of medium cDNA size was amplified using PCR for 15, 20, or 25 cycles, and visualized with gel electrophoresis and SYBR gold. While 15 cycles leads to under-amplification, 20 or 25 cycles lead to overamplified products.
Figure 4.

Preparative PCR reactions. Here, miCLIP libraries of high, medium, and low cDNA sizes were PCR-amplified for the number of cycles determined to be appropriate in the test PCR reactions. The intensity of the SYBR gold signal here represents an appropriate amount of amplification prior to sequencing of libraries.
3.13. Preprocess sequencing reads and align them to the reference genome
-
Trim 3′ adaptor sequences using flexbar [9]: (Note 26.)
flexbar -r forward_reads.fastq -p reverse_reads.fastq -f i1.8 - a flexbar_adapters.fasta --pre-trim-phred 30 -s -t exp_prefix
-
Demultiplex reads using the barcodes in the RT primers used in the experiment using the command below. We routinely use the pyCRAC suite [11] because it preserves the random portion of the barcode when demultiplexing paired-end reads. This step will also move the first three nucleotides of the random barcode into the header of each read. (Note 27.)
pyBarcodeFilter.py –f exp_prefix_1.fastq –r exp_prefix_2.fastq – b pyCRAC_barcodes.txt
-
Move the remainder of the random barcode--the two nucleotides following the experimental barcode--to the read headers: (Note 28.)
awk -F "##" '{sub(/.../,"##"$2, $2); getline($3); $4 = substr($3,1,2); $5 = substr($3,3); print $1 $2 $4"\n"$5}' exp_prefix_1_NNNGGTT_cond1.fastq > exp_prefix_1_NNNGGTTNN_cond1.fastq -
Collapse PCR duplicates based on the read sequence using pyCRAC [10]:
pyFastqDuplicateRemover.py -f exp_prefix_1_NNNGGTTNN_cond1.fastq -r exp_prefix_2_NNNGGTT_cond1.fastq -o dedup_reads
-
Transform the header in the forward read file to be compatible with downstream CIMS analysis:
awk -F '[_/]' '/^>/{print $1"_"$2"_"$3"/"$4"#"$3"#"$2; getline($9); print $9}' dedup_reads_1.fasta > dedup_reads_1.cims.fasta -
Use the header information of the forward reads to create a CIMS-compatible header for the reverse reads, and reverse-complement these reads using the command below. This strategy reduces the noise of mutations introduced by sequencing errors. We have developed a perl script for this purpose that can be found at https://github.com/jaffreylab/miCLIP_MiMB_2016. (Note 29.)
perl match_barcodes_and_reverse_complement.pl dedup_reads_1.cims.fasta dedup_reads_2.fasta
-
Concatenate the forward reads and modified reverse reads using the command below. Note that orphan reads generated in step 13.1 can be concatenated into this file.
cat dedup_reads_1.cims.fasta dedup_reads_2.cims.rc.fasta > reads.cims.cat.fasta
-
Use Novoalign to align the concatenated reads to an index of the reference genome: (Note 30.)
novoalign -t 85 -d genome_index.nix -f reads.cims.cat.fasta -F FA -l 16 -s 1 -o Native -r None -a > reads.cims.cat.novoalign
3.14. Call of m6A sites with the CIMS pipeline
-
Parse novoalign output to generate a bed file of read coordinates and a separate file of mutation coordinates:
perl novoalign2bed.pl -v --mismatch-file reads.cims.mutation.txt reads.cims.cat.novoalign reads.cims.tag.bed
-
Collapse PCR duplicates based on read coordinates and barcode identities: (Note 31.)
perl tag2collapse.pl -v --random-barcode -EM 30 --seq-error- model alignment --weight-in-name --keep-max-score --keep-tag- name reads.cims.tag.bed reads.cims.tag.uniq.bed
-
Cluster overlapping CLIP tags:
perl tag2cluster.pl -v -s -maxgap "−1" reads.cims.tag.uniq.bed reads.cims.tag.uniq.cluster.bed
-
Use the CIMS algorithm to call mutation sites:
python joinWrapper.py reads.cims.mutation.txt reads.cims.tag.uniq.bed 4 4 N reads.cims.tag.uniq.mutation.txt
-
Filter for C→T transitions and generate a .bed file of their coordinates:
awk '{if($6=="+" && $8=="C" && $9==">" && $10=="T" || $6=="-" && $8=="G" && $9==">" && $10=="A") {print $0}}' reads.cims.tag.uniq.mutation.txt | cut -f 1–6 > reads.cims.tag.uniq.C2T.bed -
Evaluate the reproducibility of mutations by permutation:
perl CIMS.pl -v -n 5 -p -c ./$1_cache_C2T --keep-cache reads.cims.tag.uniq.bed reads.cims.tag.uniq.C2T.bed reads.cims.tag.uniq.C2T.CIMS.txt
-
Create a sorted bed file of C→T transitions: (Note 32.)
awk -v prefix="rep1" '{if($9<=1){print $1"\t"$2"\t"$3"\t"prefix"_"$4"\t"$5"\t"$6}}' reads.cims.tag.uniq.C2T.CIMS.txt | sort -k 9,9n -k 8,8nr -k 7,7n > reads.cims.tag.uniq.C2T.CIMS.p1.bed -
Call and annotate m6A sites using the script below, available in the supplementary information (Note 33 ). This generates a .bed file centered on coordinates of putative m6A residues (i.e. at position −1 of the C→T transition), and extracts their sequence environment. To identify putative m6A residues, this script filters C→T transitions based on the number of transitions (m ≥ 2) and the transition frequency (1% ≥ m/k ≤ 50%) at any given coordinate. Additionally, the script generates statistics describing the ratio of called adenosines, and those called within the m6A consensus motif relative to the total number of filtered transitions. This information allows for a quick assessment of the success of the experiment.
kmer.annotate.cims.sh reads.cims.tag.uniq.C2T.CIMS.p1.bed genome.fasta
3.15. Calling m6A sites with CITS
We recommend using only forward reads for calling truncations. If only CITS analysis is desired, pre-processing of reverse reads in steps 13.5 and 13.6 can be omitted. After alignment of the forward reads, proceed to steps 14.2, 14.3, and 14.4 to generate files necessary for CITS analysis. Finally, proceed to CITS analysis as described below:
-
Identify coordinates of truncation sites:
perl bedExt.pl -n up −l "−1" -r "−1" -v reads.cims.tag.uniq.bed reads.cims.tag.uniq.trunc.bed
-
Shuffle truncation events to evaluate their significance using the command below. The cluster file used here is derived from step 14.3. (Note 34.)
perl tag2peak.pl -c ./reads.cims_cache_trunc -ss -v -gap 25 -p 0.05 reads.cims.tag.uniq.cluster.bed reads.cims.tag.uniq.trunc.bed reads.cims.tag.uniq.CITS.p005.bed
-
Annotate m6A sites with their sequence environment and generate a summary of called DRACH sites using the script below, available in the supplementary information:
kmer_annotate_cits.sh reads.cims.tag.uniq.CITS.p005
Acknowledgments
We thank members of the Jaffrey lab for their helpful comments and support. This work was supported by NIH grants NIDA DA037150 (S.R.J.), T32 CA062948 (A.O.-G.), and a German Research Foundation (DFG) fellowship (B.L.).
Footnotes
We usually prepare sample RNA by harvesting total RNA from cultured cells using TRIzol Reagent (Thermo Fisher) according to the manufacturer’s instructions. Then the RNA is treated with DNase I (see Materials) to remove DNA contamination. Finally, if desired, we enrich for messenger RNA by selecting polyadenylated RNAs using oligo d(T) magnetic beads. We recommend that the total amount of RNA should be 10 μg or higher for each sample subjected to miCLIP.
We have successfully used rabbit polyclonal antibodies from Synaptic Systems and Abcam. Other anti-m6A antibodies may also be tested in miCLIP.
32P is a high-energy beta emitter that poses a dose hazard. Perform all work using this isotope behind Plexiglas shielding of 3/8-inch thickness or greater, and wear protective equipment, including gloves, wrist guards, a lab coat, and eye protection. Additionally, radioisotopes must be used and disposed of in accordance with federal and institutional regulations and guidelines. 3. Consult your institution’s radiation safety specialist for institution-specific radiation safety requirements.
Phenol is corrosive to the skin and mucous membranes, and should only be handled in a chemical hood with proper protective equipment, including gloves, a lab coat, and eye protection.
SYBR Gold binds DNA and is therefore a potential mutagenic agent. Wear protective equipment, including gloves, a lab coat, and eye protection when handling SYBR Gold.
This step will generate RNA fragments of 30–130 nt in length which are compatible with downstream radiolabeling and linker ligation procedures. In order to preserve the greatest number of fragments for sequencing, we avoid generating too many fragments under 30 nt or over 130 nt in length. Fragments which are too short or too long are not appropriate for downstream sequencing and will be lost during size selection of cDNA.
If desired, a small portion of the fragmented RNA (2 μL) can be saved at this point. This is important if RNA-seq analysis of the same sample is desired. These RNA fragments are compatible with a variety of small RNA library preparation protocols and commercial kits. (such as those from Illumina or NEB).
For samples with lower quantities of input RNA (5–10 μg), it may be possible to generate a miCLIP library. To do so, we recommend using a greater amount of antibody at this step in order to help capture the greatest possible number of methylated RNA fragments. For example, for 5 μg of input RNA, we recommend using 25 μL of antibody.
We recommend using the 12-well tissue culture cluster because the diameter of each well allows for the sample to have optimum depth and surface area for efficient crosslinking, and allows for crosslinking of multiple samples at once. Alternatively, a similarly-sized sterile round petri dish may be used.
At this point, crosslinked samples may be frozen at −80°C until ready for immunoprecipitation. The RNA-antibody complexes will remain stable for months. However, we do not recommend thawing and refreezing the samples.
After immunoprecipitation has completed, place the sample tube in a magnetic rack to pellet the magnetic beads, and save the supernatant (non-bound fraction) in a new microcentrifuge tube if desired. This is useful if downstream analysis of the non-bound fraction is desired.
For steps involving the washing of magnetic beads, remove the tube from the magnet, resuspend the beads in the new wash buffer and mix gently by pipetting. Then, return the tube to the magnet, allow the beads to pellet and remove the wash buffer.
The following steps involve use of a radioisotope. All steps involving use of radioactive materials require use of protective equipment, including gloves, wrist guards, a lab coat, and eye protection, and must be carried out behind Plexiglas shielding of 3/8-inch thickness or greater. Radioactive materials must be used and disposed of in strict accordance with federal and institutional regulations and guidelines.
At this point, the supernatant can be transferred to a new microcentrifuge tube and frozen at −80C until ready for SDS-PAGE. However, we recommend that SDS-PAGE be carried out within several days to prevent loss of radioactive signal.
The gel may be run for longer times to better resolve the antibody (150 kD) and its respective reduced fragments (50 kD and 25 kD). We recommend that the prestained protein standard be used as a guide for determining whether the gel should be run for longer than 35 minutes.
After transfer has completed, verify the presence of radiolabeled RNA-protein complexes on the membrane and gel using a Geiger counter.
We recommend that the gel be sealed in plastic wrapping (such as a Kapak SealPAK pouch, Ampac Flexibles 400–24) and exposed to the film along with the membrane to validate that transfer was complete. We usually find that some radioactive signal remains in the gel from loading of residual magnetic beads into the wells of the gel. However, the resolved RNA-antibody complexes are always nearly completely transferred to the membrane.
Exposure times will vary depending on the amount of input RNA, antibody binding and immunoprecipitation efficiency, and activity of the [γ-32P]ATP at the time of labeling. We recommend to develop the film after 30–60 min to determine whether the membrane should be re-exposed to a new film for a longer period (up to 2–3 days).
Avoid touching or disrupting the gel interphase at this step and subsequent steps involving the use of the phase-lock gel tubes.
Use an RT primer with a unique experimental barcode for each sample.
We recommend placing a clean paper under the portion of the plastic wrap underlying lanes of the gel that contain the cDNA products. This way, the UV illumination will allow visualization of the ladder without illuminating the cDNA products, potentially introducing mutations. Avoid cutting gel slices that are higher than 200 nt or lower than 70 nt to prevent inclusion of cDNA inserts that are too long or unligated L3 linker sequence. We strongly recommend that for each sample (i.e. each individual gel lane), the three gel slices (high, medium, and low) be collected separately.
As an alternative to using gel breaker tubes, you may use 0.5 mL tubes by piercing their bottom with a 19–20 G syringe needle. Because other CLIP-based protocols do not use gel elution kits or dissolving agents for cDNA extraction, we do not recommend using such kits for this protocol.
We recommend to determine the optimum number of PCR cycles for each sample using the outcome of the three test PCR reactions. In general, the optimum number of PCR cycles is the number required to faintly see the PCR products on the gel. However, because the amount of cDNA template used for the preparative PCR is greater than the amount used for the test PCR, we recommend that the number of cycles used for the preparative PCR be one less than the optimum number determined in the test PCR.
The unique barcodes used during reverse transcription allow for pooling of samples without loss of sample identity. Thus, given that the proper number of preparative PCR cycles was chosen individually for each sample, we pool completed preparative PCR reactions from multiple samples prior to purification. To elute the purified PCR products, we use 30 μL to 50 μL of water depending on the number of samples pooled for the purification.
We recommend sequencing miCLIP libraries in paired-end mode with a read length of 50 base pairs. For libraries of up to 6 unique samples, we find that sequencing the samples on a single lane of a high-throughput flowcell is sufficient for adequate sequencing coverage. However, for a greater number of unique samples, or if higher sequencing depth is desired, we recommend sequencing the libraries on up to 3 lanes.
If this step results in a substantial number of orphan reads, which are missing a mate due to quality filters but are written to a unique file, we recommend running subsequent steps for the orphan reads as well as paired reads. The orphan reads can be concatenated into the processed read file before alignment during subsequent steps as described.
The “barcodes.txt” file should contain one line for each barcode. The barcode is written in the format NNNXXXX, where NNN is the first part of the iCLIP random barcode and XXXX is the experiment barcode (e.g. barcode 1 is NNNGGTT—note that the experimental portion of the barcode is the reverse complement of the RT primer). Like flexbar, the pyBarcodesFilter.py script will also generate files of orphan reads. If this constitutes a substantial amount of reads, we recommend to process them in parallel with paired reads and include them in the downstream analysis by concatenating them at a downstream step as described.
The “getline” function used here requires that this command is run with the “mawk” implementation of awk on some systems. If this is not the standard implementation used by your system (e.g. on Debian Wheezy, which uses gawk), you should explicitly write “mawk” instead of “awk” in this command.
This script reverse-complements the reverse reads and their barcodes to use them as a sequencing replicate of the forward reads, thereby filtering for sequencing errors. Note that if your experiment uses an even number of random barcode nucleotides (e.g. when not using standard iCLIP barcodes), reads with palindromic barcodes will be flagged as duplicates and removed in step two of the CIMS analysis.
The option “-c 36” starts Novoalign with 36 processing threads. Note that multithreading is only available for a licensed version of Novoalign; otherwise, only a single thread can be used. When multithreading, more or less processing threads may be used depending on the computational power of your machine.
At this step, it is essential that the random barcode in the header of the reverse reads was reverse complemented (step 13.5). Otherwise, read pairs will be collapsed as PCR duplicates.
The “if($9<=1)” statement defines a threshold for the false-discovery-rate determined by the CIMS analysis. Here, we allow an FDR of 1 because we apply downstream filtering to acquired C→T transitions. Also, the “prefix=” portion of this command allows for adding an experimental identifier to the fourth column of the resulting .bed file. If sites from multiple experiments are to be concatenated later, we recommend using a unique identifier for each experiment.
This script makes extensive use of the bedtools suite [11]. The second argument of the command refers to a .fasta file of the reference genome that has an accompanying index file in the same directory (e.g. genome.fasta and an accompanying genome.fasta.fai). Note that this script uses genomic coordinates to extract the sequence context of m6A residues. Thus, some m6A residues may be misannotated if located at an exon boundary.
We use an initial p-value cutoff of 0.05 for filtering truncation sites. Depending on the enrichment of DRACH sites that is evaluated in the next step, the stringency of this filter can be adjusted accordingly.
References
- 1.Linder B, Grozhik A, Olarerin-George A, et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Meth. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer K, Saletore Y, Zumbo P, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 4.Perry R, Kelley D, Friderici K, et al. The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′ terminus. Cell. 1975;4:387–394. doi: 10.1016/0092-8674(75)90159-2. [DOI] [PubMed] [Google Scholar]
- 5.Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71:3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schibler U, Kelley D, Perry R. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J Mol Biol. 1977;115:695–714. doi: 10.1016/0022-2836(77)90110-3. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz S, Mumbach M, Jovanovic M, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:294–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimoto Y, Konig J, Hussain S, et al. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol. 2012;13:R67. doi: 10.1186/gb-2012-13-8-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dodt M, Roehr J, Ahmed R, et al. FLEXBAR—flexible barcode and adapter processing for next-generation sequencing platforms. Biology. 2012;1:895–905. doi: 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webb S, Hector R, Kudla G, et al. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biol. 2014;15:R8. doi: 10.1186/gb-2014-15-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quinlan A, Hall I. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]