

Abstract
Natural products discovered using agnostic approaches, unlike rationally designed or HTS obtained leads, offer the ability to reveal new biological pathways, and hence, serve as an important vehicle to unveil new avenues in drug discovery. The ritterazine-cephalostatin family of natural products displays robust and potent anti-tumor activities, with sub-nanomolar growth inhibition against multiple cell lines and potent activity in xenograft models. Herein, we use comparative cellular and molecular biological methods to uncover the ritterazine/cephalostatin mode of cytotoxic action (MOA) in human tumor cells. Our findings indicate that while ritterostatin GN1N, a cephalostatin-ritterazine hybrid, binds to multiple HSP70s, its cellular trafficking confines activity to the endoplasmic reticulum (ER) based HSP70 isoform, GRP78. This targeting results in activation of the unfolding protein response (UPR) and subsequent apoptotic cell death.
Keywords: GRP78, HSP70, ritterazine, cephalostatin, drug discovery, natural products
Graphical Abstract
A combination of cellular microscopy and affinity methods are used to elucidate the targeting of the ritterazine-cephalostatin hybrid, ritterostatin GN1N, to GRP78/BIP within tumor cells. This discovery validates ER stress as the mode and GRP78 as the acting target in the mechanism of action of ritterostatin GN1N. It also further supports targeting the ER based GRP78 as means to terminate tumor cell growth.
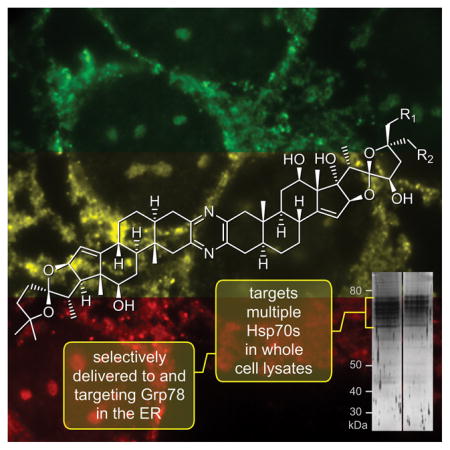
The ritterazine and cephalostatin trisdecacyclic pyrazines comprise a family of over 45 congeners obtained from marine tunicates and tube worms (Fig. 1).[1] In the 1980s, reports emerged describing the first members of these unique dimeric bis-steroidal pyrazine spiroketals with potent anti-tumor activity.[2,3] Soon thereafter, synthetic campaigns were launched, successfully validating and refining the structural assignments,[4] as well as providing materials to assess structure activity relationships (SARs).[5] These synthetic efforts were key in providing initial validation of the unique cytostatic activity of this class of molecules[6] and demonstrating that the ritterazines and cephalostatins share a common MOA.[7] This MOA was further validated through preparation of cephalostatin-ritterazine hybrid molecules, such as ritterostatin GN1N (3, Fig. 1).[8]
Figure 1.

Structure of cephalostatin 1 (1), ritterazine B (2), ritterostatin GN1N (3), 25-epi-ritterostatin GN1N (4) and the corresponding IAF probes 5–6.
While 1–4 are potent leads, a detailed understanding of the mode and mechanism leading to apoptotic death in tumor cells remained unexplained.[6] In 2011, a team led by the Shair laboratory reported that cephalostatin 1 (1, Fig. 1), ritterazine B (2, Fig. 1), OSW-1, and schweinfurthin A target oxysterol binding protein (OSBP). In addition, 1 and OSW-1 also target OSPB’s closest paralog, OSBP-related protein 4L (ORP4L).[9] While an important find, the fact that there is no direct correlation between OSBP or ORP4L to the activation of endoplasmic reticulum (ER) stress and apoptosis described by Vollmar to be a consequence of the cephalostatins,[10] suggested that perhaps other targets were yet to be discovered. A suggestion that is further supported by the fact that natural ligation of OSBP/ORP4L by intrinsic sterols such as 25-hydroxycholesterol does not correlate with cytotoxic responses in cells.[9]
Our search for these targets began by applying a combination of cellular imaging and affinity based methods. We first prepared immunoaffinity fluorescent (IAF) probes 5 and 6 from ritterostatin GN1N (3) and 25-epi-ritterostatin GN1N (4), respectively.[11] Our plan was to use both of the C25 isomers as SAR work suggested that this position was not critical for biological activity. Moreover, we anticipated that comparative studies with both isomers would further reduce concerns over potential activity losses due to labeling. Guided by previous results and NCI-60 data, we selected HCT116 (colon adenocarcinoma), SF268 (astrocytoma), and U2OS (bone carcinoma) human tumor cells for our MOA studies.
First, we confirmed that probes 5 and 6 retained potent activity in the MTT assay with IC50 values of 84±3 nM and 79±4 nM, respectively, against HCT116 cells.[11] Using methods established for imaging (an improved means to validate the phenotypic response),[11] we were able to confirm that in SF268 cells both probes 5 and 6 were trafficked to the endoplasmic reticulum (ER) within 30 min, where they remained for up to 24 h (limit of evaluation) even after removing the probes from the media by washing (Fig. 2).
Figure 2.

Subcellular localization of probes 5 and 6 in SF268 cells. Confocal microscopy confirms that a) 5 and b) 6 localize within the ER of SF268 cells. Cells were incubated with 2.1 μM 5 or 2.1 μM 6 for 24 h then washed with buffer prior to imaging. Imaging was conducted on live cells and screened over concentrations of 0.5–50 μM for up to 24 h. The subcellular localization was consistent over this concentration range. c)–f) Counterstaining with a green fluorescent mitochondrial stain, 10 μM rhodamine 123, further argued for the assignment of the ER as the site of localization. d) A close-up of c) showing mitochondrial stain (green) and localization from 5 (blue) within the ER. Comparable localization was observed in HCT116 cells.[9] e) Isolated green and f) blue fluorescent channels observed in the image in d). Images were collected at λex 405 nm and λex 408–508 nm for blue and λex 514 nm and λex 518–555 nm for green. Bars denote 10 μm.
We then turned to the affinity properties of the IAF approach, and immunoprecipitated (IP) whole cell lysates from both HCT116 and SF268 cells with 5 and 6 using Affi-gel 10 conjugated with an mAb against the IAF tag (XRI-TF35).[12] As shown in Fig. 3a, common sets of ~6 bands were identified between 70–80 kDa. As expected from SAR studies, both probes 5 and 6 returned the same set of proteins in both cell lines, with only the relative levels differing between the cell lysates. Furthermore, we were able to determine that addition of ritterostatin GN1N (3) blocked the IP of these proteins in a dose-dependent manner (Fig. 3b) providing additional support for their target specific role.
Figure 3.

Immunoaffinity analyses. a) Immunoprecipitation (IP) of HCT116 (1 mg/mL total protein) and SF268 cell lysates (1 mg/mL total protein) in PBS pH 7.2 containing 5 or 6 returned 6+ protein bands that could be detected in a 12% SDS PAGE gel after silver staining. IP was conducted using 40 μL of an Affi-gel 10 resin bearing 3.5 mg/mL of the anti-IAF mAb (XRI-TF35, Xenobe Research Institute). Lanes L1–L2 depict IP products from an HCT116 cell lysate from probes 5 and 6, respectively. L3 depicts the parent HCT116 whole cell lysate. Lanes L4–L5 depict IP products from SF268 cell lysate from probes 5 and 6, respectively. b) Chasing studies. Treatment of cell lysate with 3 prior to lysis and IP with 5 for 1 h at 4 °C reduced the level of protein returned. Analysis was conducted on a 4–12% Bis-Tris gradient gel after silver staining. Lane L6 denotes control untreated IP and L7–L8 indicate reduced levels of returned protein upon pre-treatment with 3. c) Protein sequence depicting the detected peptides (red) for the top band obtained in lane L1. Exemplary analyses of the other are provided in Supporting Fig. S1. d) Western blot analysis depicting the validation of GRP78, using a GRP78 specific mAb (3177, Cell Signaling) on the IP fractions in lanes L1 (HCT116) and L4 (SF268). e) Western blot analysis using a general HSP70 mAb (JG1, Thermo Fisher Scientific) on the IP fractions in lanes L1 (HCT116) and L4 (SF268).
Next, we applied trypsin digestion and LC-MS/MS analysis to identify the proteins. To our surprise, each of the proteins was identified as an HSP70 isoform, with GRP78 (HSPA5) being confirmed as the most prevalent band (Fig. 3c). Using western blotting, we were able to validate that the dominant band was indeed GRP78[13] (Fig. 3d). Further analysis using a non-isoform selective HSP70 mAb indicated that the other bands were indeed other HSP70 isoforms, but the individual identities were not confirmed (Fig. 3e).
With these targets identified, we prepared recombinant GRP78, HSPA8 (the constitutive HSP70), and HSPA1A (the major inducible HSP70) for further analysis.[14] Using isothermal calorimetry (ITC), we were able to demonstrate binding of 3 to GRP78 with a Kd value of 190±38 nM and stoichiometric ratio of n=1.07±0.11 (Fig. 4). Interestingly, the binding was found to be endothermic, which likely reflects the hydrophobic nature of this interaction. Comparable binding was also found for cephalostatin 1 (1) with a Kd value of 679±29 nM and stoichiometric ratio n=1.07±0.06 (Fig. S2). We also detected binding of 3 to HSPA1A with a Kd value of 625 nM and n=0.9 (Supporting Fig. S3) and 3 to HSPA8 with a Kd value of 1.63 μM and n=1.1 (Supporting Fig. S4).
Figure 4.

ITC confirmed GRP78 binding. This plot was obtained by addition of 200 μM 3 into 30 μM GRP78. Data were fit to an independent binding model using NanoAnalyze (TA Instruments).
As GRP78 is a di-domain protein, we were interested in exploring which domain was targeted by 3. We prepared both the isolated N- and C-terminal domains and submitted these proteins to ITC analyses. From these studies, we determined the binding site of 3 was in the C-terminal domain (Kd = 3.12±1.45 μM and stoichiometric ratio n=1.01±0.10, Supporting Fig. S5). No binding was observed for the N-terminal domain (Supporting Fig. S6). Finally, through biochemical investigations, we did not observe inhibition of GRP78 ATPase activity (Supporting Fig. S7) suggesting that 3 did not target the N-terminal ATPase site or induce allosteric effects.
While the binding and IP data indicated a lack of HSP70 isoform specificity, the fact that probes 5 and 6 localized predominantly in the ER (Fig. 2) was suggestive that ER-resident GRP78 was selectively targeted in cells. To explore this possibility, we turned to a large cell line, U2OS cells, which allowed improved tracking of the uptake of 5 and 6. As depicted in Fig. 5, we were able to observe rapid formation of vesicular clusters containing 5 (green, Fig. 5a). Counterstaining for caveolin[15] indicated that these clusters of vesicles were endophagosomes (red, Fig. 5a), suggesting that 5 entered the cell by endocytosis, as indicated by the direct overlap of green and red fluorescence in Fig. 5a. After 75 min, probe 5 had left these endophagosomes and moved into the ER, as illustrated by the lack of overlap in Fig. 5b.
Figure 5.

Epifluorescence imaging of the uptake of probe 5. U2OS cells were incubated with 50 μM 5 in DMEM buffer for 10 min. After treatment, the cells were fixed and stained with 1:200 dilution anti-CAVEOLIN mAb (D46G3, Cell Signaling) and 80 μM Alexa647-conjugated anti-IAF mAb (XRI-TF35, Xenobe Research Institute). Localization of CAVEOLIN (red) was completed by staining with a Cy3B-labeled secondary antibody and imaging with λex 535–585 nm and λem 600–660 nm. The localization of IAF tag (green) was conducted by imaging with λex 590–650 nm and λem 663–738 nm. b) Comparable image collected after incubation with 50 μM 5 for 75 min indicating complete localization in the ER. Bars denote 10 μm.
We then compared the localization of 5 with HSP70 isoforms. As shown in Fig. 6, probe 5 matched the localization of GRP78 providing a less resolved but nearly identical match. While 5 bound to HSPA1A in vitro and in cell lysates, it did not co-localize in cells (Supporting Fig. S8) suggesting that the endocytic trafficking observed in Fig. 5 served to selectively deliver these materials to the ER-localized GRP78. While new for this class of molecules, toxins such as ricin are known to enter the cell by hijacking endocytosis and subsequently operating on ER specific pathways.[16]
Figure 6.

GRP78 colocalization studies. Epifluorescence imaging of probe 5. a) An image collected after incubation of U2OS cells with 50 μM probe 5 for 75 min. After treatment the cells were fixed and staining with 1:20 dilution of the anti-GRP78 mAb (3177S, Cell Signaling) and 80 μM Alexa647-conjugated anti-IAF mAb (XRI-TF35, Xenobe Research Institute). Localization of GRP78 (red) was completed by staining with a Cy3B-labeled secondary antibody and imaging with λex 535–585 nm and λem 600–660 nm. The localization of IAF tag (green) was conducted by imaging with λex 590–650 nm and λem 663–738 nm b) Expansion of the image (white box) in a). Bars denote 10 μm.
Next we wanted to see if the cell death activity of the parent ritterostatin GN1N (3) correlated with levels of GRP78 in cells. Generally, the levels of GRP78 inversely correlate with cytotoxicity. As shown in Fig. 7, increased expression of GRP78 was observed in HCT116, SK-MEL-147 and MRC-5 (non-tumor) cell lines. Both SK-MEL-147 and MRC-5 cells demonstrated higher IC50 values, while HCT116 was very sensitive to 3. The corresponding response was evident, but not as profound as the levels of GRP78 expression observed. This was indicative that additional actions such as cell-line specific uptake, a differential reliance on downstream events, like the unfolded protein response (UPR) within the different cell lines, or differential access to feedback loops within each cell line[17] may also complicate the modulatory activity of 3.
Figure 7.

GRP78 expression correlates with cytotoxicity data. IC50 values (blue) for ritterostatin GN1N (3) in select cell lines after 72 h of treatment with the corresponding levels of GRP78 (black) observed in the cell lines as determined by Western blot analysis using antibodies against GRP78 (3177, Cell Signaling) and GAPDH (5174, Cell Signaling) as a loading control. IC50 values were collected using the MTT assay (see Supporting Information).
Finally, it was anticipated that blocking GRP78 function should lead to ER stress, increasing the unfolded protein response (UPR).[18] To test the effects of 3 on UPR, we used a western blot analysis of the IRE1α and PERK arms of the UPR. HCT116 cells were treated with four concentrations of 3. To measure the IRE1α arm, we evaluated XBP1 splicing (Fig. 8). Although the increase was modest relative to the positive control, a dose-dependent increase in XBP1s was observed. To measure the PERK arm, we looked at GADD34, ATF4, and phosphorylation of eIF2α. Each of these increased in a dose dependent manner, indicating activation of the PERK arm, which seemed to be more susceptible to modulation by 3 than the IRE1α arm (Fig. 8). In addition, one of the consequences of the UPR is an increase in the levels of chaperones such as GRP78 and we observed a dose-dependent increase in GRP78 levels. This collective demonstrates a dose-dependent activation of the UPR. Uncorrected, UPR leads to ER associated protein degradation (ERAD) and ultimately apoptosis,[19] and hence provides a direct connection between bioactive lead, ritterostatin GN1N (3), its target (GRP78) and downstream ER stress and apoptotic cell death as noted by Vollmar.[10]
Figure 8.

UPR response. Western blot data from HCT116 cells treated with 0.5–10 μM of 3 for 12 h, collected, lysed and blotted using the ascribed mAb as given by XBP1-s (12782S, Cell Signaling), GRP78 (3183S, Cell Signaling), pEIF2α (9721S, Cell Signaling), EIF2α (9722S, Cell Signaling), ATF-4 (11815S, Cell Signaling), 1:1000 GAPDH (sc-32233, Santa Cruz Biotechnology), and 1:3000 GADD 34 (sc-825, Santa Cruz Biotechnology). GAPDH was used as a loading control, 5 μM MG132[20] as a positive control (+), and DMSO alone as a negative control (–).
Overall this study provides a detailed understanding of how this potent family of bis-steroidal pyrazines induce ER stress and result in tumor cell death. Using a combination of fluorescence and affinity methods, we now identify the targeting of GRP78, the ER resident HSP70, by ritterostatin GN1N (3). We were able to rapidly correlate molecular specificity with temporal cellular trafficking. Interestingly, while 3 demonstrated comparable binding to different HSP70 isoforms, its function was directed to the ER resident GRP78 by endocytic trafficking. Ultimately, we found that this event resulted in induction of the UPR, a finding that was directly in line with that observed in prior studies of cephalostatin-induced apoptosis.[8] Given the bis-steroidal motif within these families of natural products, it is quite likely that they engage with multiple receptors within a cell. Critically, these studies identify an important point within a MOA program, namely, the need to unite cellular and molecular data into a single study. Here, both molecular affinity data and cellular imaging data were used to guide each other. This feedback allowed us not only to fully characterize the activated targets, but also to rapidly validate their function in terms of the overall phenotypic response to a natural product. In this study, we have found an additional target for this family of natural products, one that is directly in-line with its apoptotic activity within tumor cells. Studies are now underway to evaluate if the observed HSP70 (GRP78) activation is the primary mechanism of apoptotic induction.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institutes of Health (NIH) T32 Training Grant GM008804 (A.J.A.), Brazilian National Council for Scientific and Technological Development CNPq (L.V.C.L), National Institutes of Health (CA 60548) for financial (P.F.), NIH New Innovator Award 1-DP2-EB020400 (H.C.), RTEF Career Development Award (H.C.), Ellison Medical Foundation New Scholar in Aging Award (H.C.), the UA Start-up funds (E.C.) and the Xenobe Research Institute (J.J.L.). We also wish to thank Nobuhiro Fusetani (Hokkaido University), John Beutler (National Cancer Institute), Bob Pettit (Arizona State University) for insightful discussions and on going encouragement.
Footnotes
This article is dedicated to Nobuhiro Fusetani and George R. Pettit
Supporting information for this article is available on the WWW under http://
Contributor Information
Andrew J. Ambrose, Department of Pharmacology and Toxicology, College of Pharmacy, University of Arizona, 1703 East Mabel Street, P. O. Box 210207, Tuscon, AZ 85721 (USA)
Evelyne A. Santos, Departamento de Fisiologia e Farmacologia, Universidade Federal do Ceará, Fortaleza, CE, 60.430-270 (Brazil)
Prof. Dr. Paula C. Jimenez, Departamento de Fisiologia e Farmacologia, Universidade Federal do Ceará, Fortaleza, CE, 60.430-270 (Brazil). Instituto do Mar, Universidade Federal de São Paulo, Santos, SP, 11.070-100 (Brazil)
Dr. Danilo D. Rocha, Departamento de Fisiologia e Farmacologia, Universidade Federal do Ceará, Fortaleza, CE, 60.430-270 (Brazil)
Dr. Diego V. Wilke, Departamento de Fisiologia e Farmacologia, Universidade Federal do Ceará, Fortaleza, CE, 60.430-270 (Brazil)
Dr. Paolo Beuzer, Waitt Advanced Biophotonics Center, The Salk Institute for Biological Sciences, 10010 North Torrey Pines Rd., La Jolla, CA 92037 (USA)
Josh Axelrod, Waitt Advanced Biophotonics Center, The Salk Institute for Biological Sciences, 10010 North Torrey Pines Rd., La Jolla, CA 92037 (USA).
Dr. Ananda Kumar Kanduluru, Department of Chemistry, Purdue University, West Lafayette, IN 47907 (USA).
Prof. Philip L. Fuchs, Department of Chemistry, Purdue University, West Lafayette, IN 47907 (USA)
Prof. Hu Cang, Waitt Advanced Biophotonics Center, The Salk Institute for Biological Sciences, 10010 North Torrey Pines Rd., La Jolla, CA 92037 (USA)
Prof. Letícia V. Costa-Lotufo, Departamento de Fisiologia e Farmacologia, Universidade Federal do Ceará, Fortaleza, CE, 60.430-270 (Brazil). Departamento de Farmacologia, Universidade de São Paulo, São Paulo, SP, 05508-900 (Brazil)
Prof. Eli Chapman, Department of Pharmacology and Toxicology, College of Pharmacy, University of Arizona, 1703 East Mabel Street, P. O. Box 210207, Tuscon, AZ 85721 (USA)
Dr. James J. La Clair, Xenobe Research Institute, P. O. Box 3052, San Diego, CA 92163-1052 (USA)
References
- 1.a) Menna M. Curr Top Med Chem. 2014;14:207. doi: 10.2174/1568026613666131213155813. [DOI] [PubMed] [Google Scholar]; b) Iglesias-Arteaga MA, Morzycki JW. Alkaloids Chem Biol. 2013;72:153. doi: 10.1016/b978-0-12-407774-4.00002-9. [DOI] [PubMed] [Google Scholar]; c) Moser BR. J Nat Prod. 2008;71:487. doi: 10.1021/np070536z. [DOI] [PubMed] [Google Scholar]
- 2.For early manuscripts on the ritterazines, see: Fukuzawa S, Matsunaga S, Fusetani N. J Org Chem. 1994;59:6164. doi: 10.1021/jo970091r.Fukuzawa S, Matsunaga S, Fusetani N. J Org Chem. 1995;60:608. doi: 10.1021/jo970091r.Fukuzawa S, Matsunaga S, Fusetani N. Tetrahedron. 1995;51:6707.
- 3.For early manuscripts on the isolation the cephalostatins, see: Pettit GR, Inoue M, Kamano Y, Herald DL, Arm C, Dufresne C, Christie ND, Schmidt JM, Doubek DL, Krupa TS. J Am Chem Soc. 1988;110:2006.Petit GR, Inoue M, Kamano Y, Dufresne C, Christie ND, Niven L, Herald DL. J Chem Soc, Chem Comm. 1988:865.
- 4.a) Lee S, LaCour TG, Fuchs PL. Chem Rev. 2009;109:2275. doi: 10.1021/cr800365m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kou Y, Cheun Y, Koag MC, Lee S. Steroids. 2013;78:304. doi: 10.1016/j.steroids.2012.10.021. [DOI] [PubMed] [Google Scholar]; c) Shi Y, Jia L, Xiao Q, Lan Q, Tang X, Wang D, Li M, Ji Y, Zhou T, Tian W. Chem Asian J. 2011;6:786. doi: 10.1002/asia.201000882. [DOI] [PubMed] [Google Scholar]; d) Fortner KC, Kato D, Tanaka Y, Shair MD. J Am Chem Soc. 2010;132:275. doi: 10.1021/ja906996c. [DOI] [PubMed] [Google Scholar]; e) Taber DF, Joerger JM. J Org Chem. 2008;73:4155. doi: 10.1021/jo800454w. [DOI] [PubMed] [Google Scholar]; f) Phillips ST, Shair MD. J Am Chem Soc. 2007;129:6589. doi: 10.1021/ja0705487. [DOI] [PubMed] [Google Scholar]; g) Lee S, Fuchs PL. Org Lett. 2002;4:317. doi: 10.1021/ol016572l. [DOI] [PubMed] [Google Scholar]; h) Kim S, Sutton SC, Guo C, LaCour TG, Fuchs PL. J Am Chem Soc. 1999;121:2056. [Google Scholar]; i) Jeong JU, Guo C, Fuchs PL. J Am Chem Soc. 1999;121:2071. [Google Scholar]; j) LaCour TG, Guo C, Bhandaru S, Fuchs PL, Boyd MR. J Am Chem Soc. 1998;120:692. [Google Scholar]; k) Jeong JU, Sutton SC, Kim S, Fuchs PL. J Am Chem Soc. 1995;117:10157. [Google Scholar]
- 5.a) Pettit GR, Xu JP, Chapuis JC, Melody N. J Nat Prod. 2015;78:1446. doi: 10.1021/acs.jnatprod.5b00129. [DOI] [PubMed] [Google Scholar]; b) Pettit GR, Moser BR, Herald DL, Knight JC, Chapuis JC, Zheng X. J Nat Prod. 2015;78:1067. doi: 10.1021/np501033u. [DOI] [PubMed] [Google Scholar]; c) Poza JJ, Rodríguez J, Jiménez C. Bioorg Med Chem. 2010;18:58. doi: 10.1016/j.bmc.2009.11.018. [DOI] [PubMed] [Google Scholar]; d) Taber D, Joerger JM. J Org Chem. 2008;73:4155. doi: 10.1021/jo800454w. [DOI] [PubMed] [Google Scholar]; e) Flessner T, Jautelat R, Scholz U, Winterfeldt E. Fortschr Chem Org Naturst. 2004;87:1. doi: 10.1007/978-3-7091-0581-8_1. [DOI] [PubMed] [Google Scholar]; f) LaCour TG, Guo C, Ma S, Jeong JU, Boyd MR, Matsunaga S, Fusetani N, Fuchs PL. Bioorg Med Chem Lett. 1999;9:2587. doi: 10.1016/s0960-894x(99)00430-8. [DOI] [PubMed] [Google Scholar]; g) Guo C, LaCour TG, Fuchs PL. Bioorg Med Chem Lett. 1999;9:419. doi: 10.1016/s0960-894x(98)00743-4. [DOI] [PubMed] [Google Scholar]
- 6.a) Komiya T, Fusetani N, Matsunaga S, Kubo A, Kaye FJ, Kelley MJ, Tamura K, Yoshida M, Fukuoka M, Nakagawa K. Cancer Chemother Pharmacol. 2003;51:202. doi: 10.1007/s00280-002-0558-8. [DOI] [PubMed] [Google Scholar]; b) Rudy A, López-Antón N, Dirsch VM, Vollmar AM, Rudy A. J Nat Prod. 2008;71:482. doi: 10.1021/np070534e. [DOI] [PubMed] [Google Scholar]; c) Müller IM, Dirsch VM, Rudy A, López-Antón N, Pettit GR, Vollmar AM. Mol Pharmacol. 2005;67:1684. doi: 10.1124/mol.104.004234. [DOI] [PubMed] [Google Scholar]; d) Dirsch VM, Müller IM, Eichhorst ST, Pettit GR, Kamano Y, Inoue M, Xu JP, Ichihara Y, Wanner G, Vollmar AM. Cancer Res. 2003;63:8869. [PubMed] [Google Scholar]
- 7.We refer to mechanism of action or mechanism of apoptotic action as the phenotypic pathway of apoptosis induced as best described in [6]. The mode of action (MOA) is described as the entire story including the cellular localization, molecular targets and resulting mechanism of apoptotic induction.
- 8.Kanduluru AK, Banerjee P, Beutler JA, Fuchs PL. J Org Chem. 2013;78:9085. doi: 10.1021/jo401171q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgett AW, Poulsen TB, Wangkanont K, Anderson DR, Kikuchi C, Shimada K, Okubo S, Fortner KC, Mimaki Y, Kuroda M, Murphy JP, Schwalb DJ, Petrella EC, Cornella-Taracido I, Schirle M, Tallarico JA, Shair MD. Nat Chem Biol. 2011;7:639. doi: 10.1038/nchembio.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.López-Antón N, Rudy A, Barth N, Schmitz ML, Pettit GR, Schulze-Osthoff K, Dirsch VM, Vollmar AM. J Biol Chem. 2006;281:33078. doi: 10.1074/jbc.M607904200. [DOI] [PubMed] [Google Scholar]
- 11.Kumar KA, La Clair JJ, Fuchs PL. Org Lett. 2011;13:5334. doi: 10.1021/ol202139z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Yu WL, Guizzunti G, Foley TL, Burkart MD, La Clair JJ. Nat Prod. 2010;73:1659. doi: 10.1021/np100371k. [DOI] [PubMed] [Google Scholar]; b) Álvarez-Micó X, Rocha DD, Guimarães LA, Ambrose A, Chapman E, Costa-Lotufo LV, La Clair JJ, Fenical W. Chembiochem. 2015;16:2002. doi: 10.1002/cbic.201500229. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Farnaes L, La Clair JJ, Fenical W. Org Biomol Chem. 2014;12:418. doi: 10.1039/c3ob41355a. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yu WL, Jones BD, Kang M, Hammons JC, La Clair JJ, Burkart MD. J Nat Prod. 2013;76:817. doi: 10.1021/np3004497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.GRP78 also known as BiP (immunoglobulin heavy-chain binding protein) is the endoplasmic reticulum (ER) orthologue of the Hsp70 family of molecular chaperones and is intricately involved in functions of the ER through its interactions with a variety of substrates and regulatory proteins, see: Behnke J, Feige MJ, Hendershot L. J Mol Biol. 2015;427:1589. doi: 10.1016/j.jmb.2015.02.011.Zhu G, Lee AS. J Cell Physiol. 2015;230:1413. doi: 10.1002/jcp.24923.Ni M, Zhang Y, Lee AS. Biochem J. 2011;434:181. doi: 10.1042/BJ20101569.
- 14.a) Finka A, Sharma SK, Goloubinoff P. Front Mol Biosci. 2015;2:29. doi: 10.3389/fmolb.2015.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stricher F, Macri C, Ruff M, Muller S. Autophagy. 2013;9:1937. doi: 10.4161/auto.26448. [DOI] [PubMed] [Google Scholar]
- 15.a) Kumari S, Mg S, Mayor S. Cell Res. 2010;20:256. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li D, Shao L, Chen BC, Zhang X, Zhang M, Moses B, Milkie DE, Beach JR, Hammer JA, III, Pasham M, Kirchhausen T, Baird MA, Davidson MW, Xu P, Betzig E. Science. 2015;349:aab3500. doi: 10.1126/science.aab3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spooner RA, Lord JM. Toxins (Basel) 2015;7:49. doi: 10.3390/toxins7010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Expression of GRP78 is directly related to stress with a complex feedback existing between GRP78 levels and the UPR, see: Wang Y, Wu H, Li Z, Yang P, Li Z. Exp Cell Res. 2016;S0014-4827:30433.
- 18.GRP78 is well documented to be involved in the UPR: Zhu G, Lee AS. J Cell Physiol. 2015;230:1413. doi: 10.1002/jcp.24923.de Ridder G, Ray R, Misra UK, Pizzo SV. Methods Enzymol. 2011;489:245–57. doi: 10.1016/B978-0-12-385116-1.00014-5.Sato M, Yao VJ, Arap W, Pasqualini R. Adv Genet. 2010;69:97. doi: 10.1016/S0065-2660(10)69006-2.
- 19.Korennykh A, Walter P. Annu Rev Cell Dev Biol. 2012;28:251. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- 20.Lee DH, Goldberg AL. Trends Cell Biol. 1998;8:397. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.