

Abstract
Mitochondrial dysfunction and axonal degeneration are early pathological features of Alzheimer’s disease (AD)-affected brains. The underlying mechanisms and strategies to rescue it have not been well elucidated. Here, we evaluated axonal mitochondrial transport and function in AD subject-derived mitochondria. We analyzed mitochondrial transport and kinetics in human trans-mitochondrial “cybrid” (cytoplasmic hybrid) neuronal cells whose mitochondria were derived from platelets of patients with sporadic AD and compared these AD cybrid cell lines with cybrid cell lines whose mitochondria were derived from age-matched, cognitively normal subjects. Human AD cybrid cell lines, when induced to differentiate, developed stunted projections. Mitochondrial transport and function within neuronal processes/axons was altered in AD-derived mitochondria. Antioxidants reversed deficits in axonal mitochondrial transport and function. These findings suggest that antioxidants may be able to mitigate the consequences of AD-associated mitochondrial dysfunction. The present study provides evidence of the cause/effect of AD specific mitochondrial defects, which significantly enhances our understanding of the AD pathogenesis and exploring the effective therapeutic strategy for AD.
Keywords: Alzheimer’s disease, antioxidants, cybrid cells, mitochondrial dysfunction, mitochondrial transport
INTRODUCTION
Mitochondria are energy producing organelles found in most eukaryotic cells [1]. Mitochondria are particularly important in neuronal metabolism, function, and neurotransmitter synthesis and transport. As post-mitotic cells, neurons are especially susceptible to mitochondrial dysfunction [2, 3]. Their highly branched and extended processes are exceptionally susceptible to disruptions in mitochondrial transport. Dendritic mitochondria may in fact play a pivotal role in the morphogenesis and plasticity of neuronal spines and synapses [2, 4, 5]. For instance, an increased number and improved plasticity of spines and synapses have been correlated with increased dendritic mitochondrial content and mitochondrial activity [5, 6].
Evidence of mitochondrial dysfunction has been well documented in Alzheimer’s disease (AD) pathological samples, animal models, and in vitro cell cultures [7–10]. Abnormal mitochondrial morphology has also been reported in neurons from AD brains [11, 12]. Moreover, alterations in mitochondrial distribution seem to associate with impaired synaptic terminal formation, axonal process growth, synaptic loss, and even neuronal death in AD brains [13, 14].
Cytoplasmic hybrid (“cybrid”) cell models have been used to investigate the role of dysfunctional mitochondria in AD pathogenesis [7, 15, 16]. To generate AD and control (non-AD) cybrid cells, platelet mitochondria from human AD and age-matched non-AD subjects are transferred to human neuroblastoma (SH-SY5Y) cells depleted of endogenous mitochondrial DNA (mtDNA) [17]. AD cybrid cells recapitulate AD pathological features, especially mitochondrial defects observed in AD brain; these include increased oxidative stress, cytochrome oxidase defects, decreased membrane potential, impaired energy metabolism, and alterations in mitochondrial dynamics [7, 15, 16, 18]. However, little is known on the cause and effect of AD-derived mitochondria on axonal mitochondrial transport.
In the presence of staurosporine, 12-O-tetradecanoyl phorbol-13 acetate (TPA), or retinoic acid (RA), human SH-SY5Y neuroblastoma cells can differentiate into a mature neuronal phenotype with the appearance of neurite-like processes as the most prominent alterations [19]. Differentiated cybrid cells containing patient-derived mitochondria may provide a model for studying the effects of AD-specific axonal mitochondria function and transport on neuron differentiation.
Using differentiated human neuronal cybrid cell lines from AD and non-AD subjects, we evaluated axonal mitochondrial transport and function following induction by the chemical differentiation. We also examined the effect of antioxidant on mitochondrial defects in AD cybrid cells. Our study provides evidence of deficits in AD mitochondrial transport and the contribution of oxidative stress to these defects.
MATERIALS AND METHODS
Human subjects and creation of cybrid cell lines
Both AD patients and non-AD controls were recruited from the University of Kansas Alzheimer’s Disease Center (KUADC). Subjects with AD met the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria [20]. Non-AD subjects were cognitively normal and age-matched to AD subjects. This study was approved by the University of Kansas Medical Center (KUMC) Institutional Review Board. All subjects provided written informed consent to participate in the study. The ages of AD and non-AD subject platelet donors were 73.6 ± 2.96 and 75.8 ± 5.04 years, respectively. Gender, age and disease status of donor patients are presented in Supplementary Table 1.
Cybrid cell lines were created on the human neuroblastoma cell (SH-SY5Y) nuclear background (by the KU ADC Mitochondrial Genomics and Metabolism Core) [21]. To create the cybrid cell lines used for this study, SH-SY5Y cells that were previously depleted of endogenous mtDNA (Rho0 cells) were fused with platelet cytoplasm from human subjects, and repopulated with mitochondria containing mtDNA from patients or controls as previously described [22]. Quantitative real-time polymerase chain reaction (PCR) showed that intact mtDNA copies were present in all cybrids without detectable large-scale deletion in the cells used in the present study (Supplementary Figure 1).
Cybrid growth and differentiation
AD and non-AD cybrids were grown in T75 tissue culture flasks in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose (5.5 mM), 10% characterized fetal bovine serum (FBS; Gibco BRL, Logan, Utah), 100 μg/ml pyruvate, 50 μg/ml uridine, antibiotic-antimycotic, 100 Units/ml penicillin G, and 100 μg/ml streptomycin [7]. Medium was changed every 2 days. Twelve mm plastic coverslips and cell culture plates were coated with 1.5 mg/ml poly-D-lysine (Sigma-Aldrich, St. Louis, Missouri, USA, dissolved in sterile H2O) for 2 h at room temperature, and were rinsed twice with sterile H2O before use. Harvest of proliferating cybrid cells was initiated using 0.1% trypsin (Invitrogen, Carlsbad, CA) in phosphate-buffered saline (PBS, Invitrogen, Carlsbad, CA) for 5 min at 37◦C. Trypsin activity was inactivated by an equal volume of culture media containing serum. Cells were then harvested by centrifugation and re-suspended in culture media. Three thousand cells in 0.25 ml culture media were added to each well of a 24-well-plate with coverslips inside or 40,000 cells to each well of a 6-well-plate. One day later, culture media were changed to the differentiation media [neurobasal media supplemented with 1× B27 (Invitrogen, Carlsbad, CA) and 0.5 mM glutamine, and antibiotic-antimycotic] with 10 nM staurosporine (SAT, Sigma-Aldrich Corp, St Louis, Missouri, USA). We made 25 μM stock SAT in dimethyl sulfoxide (DMSO) and stored it at −80°C). Approximately, half of the differentiation media was replaced (with 10 nM SAT) every day as previously described [23, 24]. Based on the measurement of length of neuronal processes, day-14 cybrid cells reached plateau after differentiation in day-35 (data not shown), indicating that differentiation was considered to complete on day 14 of SAT treatment. To assess the effects of antioxidant on AD cells, 10 μM probucol (Sigma-Aldrich Corp, St Louis, Missouri, USA) or 200 μM ascorbic acid (AA, Sigma-Aldrich Corp, St Louis, Missouri, USA) were added to the media along the differentiation.
Respiratory chain complexes enzyme activities and ATP levels measurement
Enzyme complex I (NADH-ubiquinone reductase), complex IV (cytochrome c oxidase, CcO) activity, and ATP levels were determined as described previously [7]. Briefly, cybrid cells were washed with ice-cold PBS, and then harvested, centrifuged, and suspended in 50 l of isolation buffer containing 250 mM sucrose, 20 mM HEPES, and 1 mM EDTA. Cell suspensions (containing ~3–4 mg of protein/ml) were added to a cuvette containing 0.95 ml of 1 × assay buffer (10 mM Tris-HCl, and 120 mM KCl), and the reaction volume was brought to 1.05 ml with the addition of 1× enzyme dilution buffer (10 mM Tris-HCl, pH 7.0). The reaction was then initiated by the addition of 50 μl of ferrocytochrome substrate solution (0.22 mM). The change in absorbance of cytochrome c at 550 nm was measured using a Shimadzu (Kyoto, Japan) UV1200 spectrophotometer. Activity is expressed as micromoles of cytochrome oxidized per min−1 mg−1 protein using an extinction coefficient of 18.64 mM−1 cm−1. ATP levels were determined using an ATP Bioluminescence Assay Kit (Roche) following the manufacturer’s instruction [9, 25]. Briefly, cells were harvested, incubated on ice for 15 min, and centrifuged at 13,000 g for 10 min. ATP levels were measured using a Luminescence plate reader (Molecular Devices) with an integration time of 10 s.
Determination of ROS generation with electron paramagnetic resonance (EPR) analysis
The oxidation of the cyclic hydroxylamine 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethyl-pyrrolidine (CMH) was used to measure the ROS levels as described in our previous study [26, 27]. Cybrid cells cultured on 6-well plates were washed with PBS and then incubated with 100 μM CMH at 37°C for 30 min, followed by one wash of cold PBS. After that, cybrid cells were washed with ice-cold PBS, and then harvested with 100 μl of PBS for each well.
EPR measurements were performed using a Bruker EMXplus X-band EPR spectrometer running Bruker Xenon acquisition/processing software and equipped with a dual mode cavity (ER4116DM) and Oxford ESR900 cryostat. For each sample ~50 μl was loaded into a 1.5 mm O.D. micropipette (Blaubrand intraMARK), sealed with Bruker X-Sealant and placed into a 4 mm O.D. EPR tube (Wilmad Lab-glass 707-SQ-250M). The spectra were recorded at room temperature with the following acquisition parameters: microwave frequency 9.64 GHz; center magnetic field, 3440 gauss; scan range, 100 gauss; microwave power, 2 mW; modulation frequency, 100 kHz; modulation amplitude, 1 gauss and time constant, 82 ms. Signal intensity was determined as the height of the central peak on the up-field side of the midpoint.
Determination of mitochondrial ROS generation with MitoSox Red and mitochondrial membrane potential (ΔΨ) with TMRM
Cybrid cells were seeded at low density onto Lab-Tek eight-well chamber slides (10000 cells/well). Mitochondrial ROS generation was determined using MitoSox Red (Invitrogen), a unique fluorogenic dye highly selective for detection of superoxide production in live cell mitochondria. Cells were incubated with fresh medium containing 2.5 μM MitoSox for 30 min. To detect mitochondrial membrane potential, cells were co-stained with TMRM (100 nM; Invitrogen) for 30 min. Mitochondria were labeled with Mitotracker Red (MTRed, 100 nM, Invitrogen) or and MitoTracker Green without interference of mitochondrial membrane potential (MTGreen, 100nM, Invitrogen) for 30 min at 37°C before fixation to visualize mitochondrial morphology.
Fluorescence images were acquired on a Leica SP5 confocal microscope and analyzed using Leica LAS AF software (Leica Wetzlar). Excitation wavelengths were 543 nm for MitoSox, TMRM or MTRed, and 488 nm for MTGreen, respectively. Fluorescent signals were quantified using NIH Image J software. Post-acquisition processing was performed using MetaMorph (Molecular Devices) and NIH Image J software for quantification of fluorescent signals and measurement of mitochondrial density. Fluorescence intensity was quantified from the same sized mitochondrial area. Mitochondrial density and fluorescent intensity were quantified by an investigator blinded to experimental groups. More than 50 clearly identifiable mitochondria from randomly selected cells were measured and quantified.
Measurement of mitochondrial movement in AD and non-AD cybrid cells
To measure mitochondrial movement, both AD and non-AD cybrid cells were incubated with 50 nM MitoTrackerRed (MTRed; Molecular Probes, Invitrogen) for 10 min at 37°C. Time-lapse recordings of labeled mitochondrial movement were acquired on a Carl Zeiss (Axiovert 200) microscope with an incubation system (PeCon) to maintain cybrid cells at 37°C during image collection. Collection of image stacks and velocity measurements were made using Axiovision Software as previously described [8, 25]. For standard recordings, images of mitochondria in one axon per cybrid cell were collected every 3 s for 2 min.
Mitochondria in each frame of every video recording were individually tracked using AxioVision Software. The average velocity was calculated during the 2-min recording period. Then, the velocities of mitochondria in the process were averaged to obtain the average velocity for mitochondrial movement. In addition, the percentage of movable mitochondria, a total of mitochondrial traveling distance (total distance traveled regardless of direction during the recording period), and mitochondrial density in each process were determined according to the previous studies with modifications [7, 15]. Exposure periods (30–50 ms) were kept to a minimum to limit the phototoxicity. All data were obtained from five cell lines of either AD or non-AD cybrids.
Statistical analysis
Data are presented as mean ± SEM. Statistical analysis was performed using Statview software (SAS Institute, Version 5.0.1). Differences between means were assessed by Student’s t-test or one-way analysis of variance (ANOVA) with Fisher post hoc test. p < 0.05 was considered significant.
RESULTS
Differentiation of AD and non-AD cybrid cells
We observed morphological changes in staurosporine (SAT)-treated non-AD (Fig. 1A II) and AD cybrid cells (Fig. 1A IV) under non-serum conditions. SAT serves as a strong inducer of differentiation in SH-SY5Y cells. SAT concentrations between 4 and 25 nM are not toxic [19, 24, 28], so we treated cybrid cells with 10 nM SAT for 14 days. Differentiated cells began to appear 3 days after treatment and differentiation was complete on day 14. Over the course of a month, no cell detachment or apoptosis (based on stains to detect apoptosis) was observed. Typical differentiated morphology appeared as long and straight neuronal-like processes stretched out in non-AD cybrid cells (Fig. 1A II). In contrast, differentiated AD cells revealed much shorter neuronal like processes than differentiated non-AD cells (Fig. 1A IV). Neuronal process length showed a 3.6-fold increase in differentiated non-AD cybrid cells compared to non-differentiated non-AD cells, whereas we observed only a 1.4-fold increase in the processes in the differentiated AD cybrid cells (Fig. 1B). Process lengths between the undifferentiated non-AD and AD cybrid cells were comparable (p > 0.05). These results suggest that SH-SY5Y AD cybrid cells do not differentiate normally when induced by SAT.
Fig. 1.
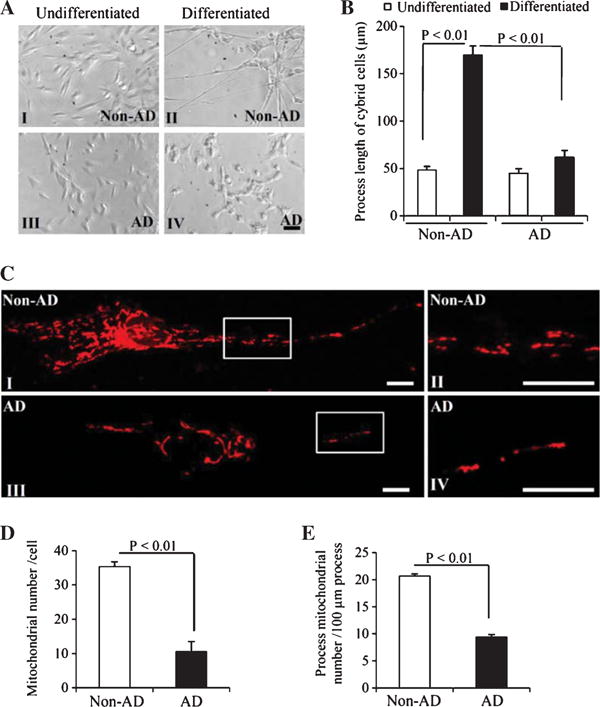
Comparison of differentiation status and mitochondrial density in neuronal processes between non-AD and AD cybrid cells during differentiation. A) Representative morphological images from non-AD and AD cybrid cells under undifferentiated conditions or induced by staurosporine (14 days after SAT treatment). Scale bar = 50 μm. B) Quantification of neuronal process length of cybrid cells using the image J program. Black columns denote the process length of cybrid cells following SAT treatment in both non-AD and AD groups. White open columns represent process lengths of undifferentiated cybrid cells without SAT treatment. C) Images show morphology of mitochondria in cybrid cells with MitoRed staining. In the processes of differentiated non-AD cybrid cells (I) and the enlarged images for processes (II), more mitochondria were observed than in the AD cells (III, and the enlarged images for processes in IV). Mitochondrial density (D, E) in the processes of differentiated non-AD and AD cybrid cells were quantified. Total numbers of mitochondria were counted within the processes per cybrid cell (D) and the numbers of mitochondria were counted per 100 μm process length (E). Data were collected from 20–25 processes from each non-AD or AD cybrid cell line.
Mitochondrial density in differentiated AD and non-AD cybrid cells
We compared mitochondrial morphology in differentiated AD and non-AD cybrid cells by staining with a mitochondrial marker. In non-AD cybrid cells, mitochondria were shorter and scattered along the differentiated neuronal processes (Fig. 1C I and II), whereas in AD cells the mitochondria were concentrated within the shortened processes (Fig. 1C III and IV). After 14 days of SAT treatment, the number of mitochondria within the processes was significantly lower in AD cybrid cells than it was in non-AD cybrid cells (Fig. 1D). Each of non-AD cybrid cells contained about 35 mitochondria within its differentiated processes, whereas each of the AD cells contained about 11 mitochondria in its less differentiated processes (Fig. 1D). To confirm that the increased numbers of mitochondria in processes of non-AD cells represented a higher mitochondrial density, and was not simply due to the longer length of the differentiated processes in non-AD cells, we also calculated the number of mitochondria per 100 μm of process. Indeed, mitochondrial density was significantly higher in non-AD cells than AD cells (Fig. 1E), indicating that the content of mitochondria within AD cybrid processes was reduced.
Mitochondrial dysfunction in differentiated AD cybrid cells
We next evaluated mitochondrial function by measuring respiratory chain enzyme activities, mitochondrial membrane potential, and reactive oxygen species (ROS) generation in differentiated AD and non-AD cybrid cells. The enzymatic activities of complex I and IV were significantly reduced in differentiated AD cybrid cells compared to non-AD cybrids (Fig. 2A–B). Similarly, ATP levels in AD cybrid cells were lower than that in non-AD cells (Fig. 2C).
Fig. 2.
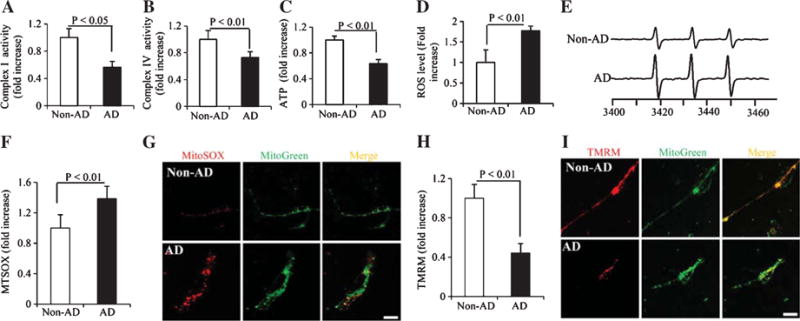
Comparison of mitochondrial function, mitochondrial membrane potential, and reactive oxygen species (ROS) levels between differentiated non-AD and AD cybrid cells. Enzymatic activity of complex I (A), IV (B), and cellular ATP levels (C) were determined in cell lysates from indicated cell groups. Data are expressed as fold increase relative to non-AD cybrid cells. Generation of ROS was detected by electron paramagnetic resonance (EPR) spectra (D, E) and mitochondrial ROS levels were measured by MitoSox staining intensity (F, G). Mitochondrial membrane potential was measured by TMRM staining intensity (H, I). Quantifications were determined of the signal intensity of EPR (D), immunofluorescent intensity for MitoSox (F) and TMRM (H) in mitochondria of the indicated cybrid cells. The representative EPR and staining images were shown for EPR (E), and red fluorescence for Mitosox (G) and TMRM (I), respectively. Data were collected from 20–25 processes from each non-AD or AD cybrid cell line.
ROS levels was detected by EPR studies using the cyclic hydroxylamine 1-hydroxy-3-methoxy-carbonyl-2,2,5,5-tetramethyl-pyrrolidine (CMH) as a spin trapping agent. Substantial ROS production was detected in AD cybrid cells compared to non-AD cybrid cells (Fig. 2D). The amount of the ROS level was quantified by measuring the height of the middle EPR peak from each spectrum (representative images are shown in Fig. 2E).
Given that mitochondria are a major source of ROS generation and that ROS accumulation affects mitochondrial function, we measured mitochondrial ROS generation. MitoSox staining intensity, which reflects mitochondrial ROS production, was increased in AD cybrid cells (Fig. 2F, G). Mitosox signal was enhanced and aggregated within the terminals of the differentiated AD neurons, suggesting that abnormal ROS accumulation in neuronal process terminals impair process differentiation in AD neurons.
We next assessed inner mitochondrial membrane potential in differentiated AD and non-AD cybrid cells using the fluorescent probe TMRM. Mitochondrial depolarization (disrupting or decreasing the membrane potential) reduces the mitochondrial staining intensity. Compared to non-AD mitochondria, the intensity of TMRM staining was significantly lower in AD cybrid cells (Fig. 2H, I). Decreased TMRM signal was observed in the axon-like neuronal processes of AD cybrid neurons (Fig. 2I). These results demonstrate impaired mitochondrial function and increased mitochondrial ROS generation/accumulation in differentiated cybrids that contain AD-derived mitochondria.
Impairment of mitochondrial motility in axons of AD cybrid neurons
After differentiation, both human AD and non-AD cybrid neuronal cells formed axon-like neuronal processes, which allowed us to directly evaluate the effects of AD subject-derived mitochondria on axonal mitochondrial transport and movement. We assessed mitochondrial motility by measuring the percentage of movable and stationary mitochondria, travel distance, and velocity. The percentage of moving mitochondria was significantly reduced in AD neuronal processes compared to non-AD processes (Fig. 3A). Conversely, increased numbers of stationary mitochondria were observed within AD neuronal processes (Fig. 3B), indicating defects of mitochondrial movement in AD cybrids in addition to impaired differentiation and lower mitochondrial process density of mitochondria. The velocity and total travel distance of mitochondria were also significantly decreased in AD neuronal processes (Fig. 3C–D). Quantification of mitochondrial movement and kymography revealed significantly reduced anterograde and retrograde mitochondrial movement in AD neuronal processes compared to non-AD cybrids (Fig. 3E–G). These data demonstrate mitochondrial transport and trafficking is impaired within AD-derived neuronal processes.
Fig. 3.
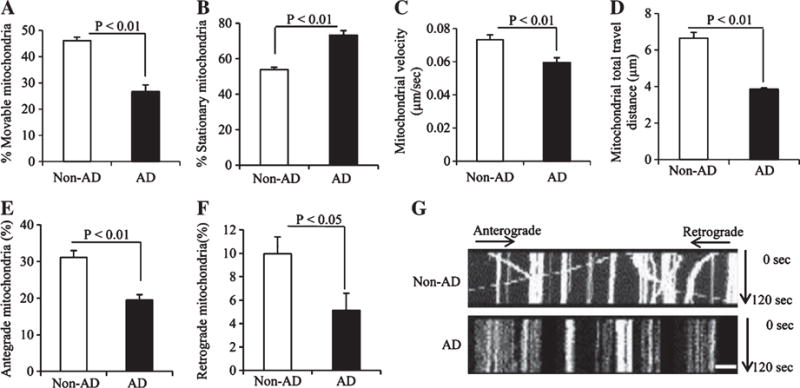
Comparison of mitochondrial movement parameters in the neuronal process of differentiated non-AD and AD cybrid cells. A, B) The percentage of movable (A) and stationary mitochondria (B) in the processes of cybrid cells. C) Average mitochondrial travel velocity and (D) mitochondrial travel distance were calculated. E, F) AD cybrid cells showed significant decreases in anterograde and retrograde mitochondrial movement compared to non-AD controls. Data were collected from 25 processes of each non-AD or AD cybrid cell line. G) Kymographs generated from the live imaging movies showing non-AD and AD cybrid cells, respectively. In the kymographs, the X axis is mitochondrial position and the Y axis represents the time lapse (0–120 s). Vertical white lines represent stationary mitochondria and diagonal lines represent moving mitochondria. Anterograde movements are from left to right and retrograde movements are from right to left. Scale bars = 10 μm.
Effects of antioxidants on cell differentiation, mitochondrial function, and axonal transports
Given that elevated ROS levels (Fig. 2D–G) might contribute to defective differentiation (Fig. 1A, B) as well as impaired mitochondrial morphology (Fig. 1C–E) and function (Fig. 2A–C, H, I) in AD cybrid cells, we examined whether antioxidants could rescue AD cybrid differentiation defects. Treatment with two antioxidants, probucol and ascorbic acid (AA) significantly increased neuronal processes (Fig. 4A, B) and mitochondrial density (Fig. 4C–E) within processes in AD cybrid cells compared to vehicle-treated control cells under normal differentiation condition. Furthermore, in the presence of antioxidants, AD cybrid cells displayed an improvement in mitochondrial function as shown by increased complex I and IV activity, ATP levels (Fig. 5A–C), and mitochondrial membrane potential ((Fig. 5G, I). Treatment of probucol or AA completely abolished ROS production/accumulation in AD cybrid cells (Fig. 5D–F, H). These data suggest that antioxidant treatment reverses perturbed AD-specific mitochondrial morphology and function in AD cybrid neurons.
Fig. 4.
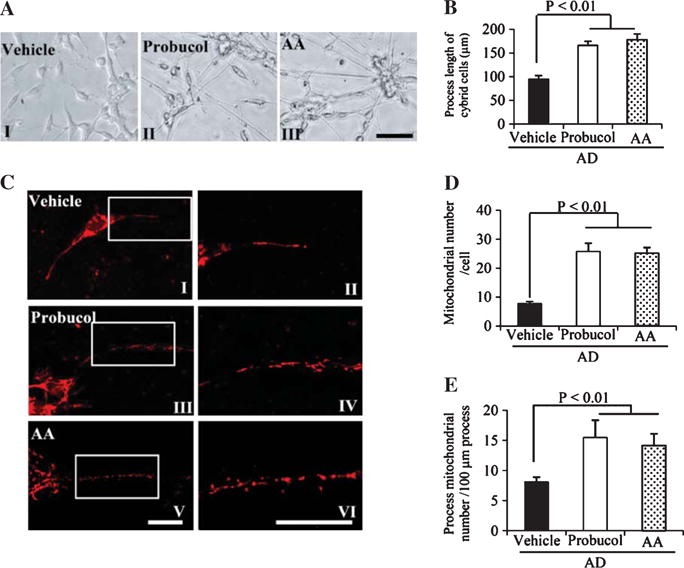
Effects of antioxidant treatment on differentiation status and mitochondrial density in differentiated AD cybrid cells. A) Representative morphological images from differentiated AD cybrid cells with the addition of antioxidant (10 μM probucol in AII, 200 μM ascorbic acid/AA in AIII or vehicle in AI) or vehicle treatment. Scale bar = 50 μm. B) Quantification of neuronal process length of cybrid cells using the image J program. C) Representative morphological images of mitochondria in the processes of above differentiated AD cybrid cells with MitoRed staining (I, III, and V) and the corresponding enlarged images of processes (II, IV, and VI). I and II: AD cells under differentiation with the addition of antioxidants probucol (III and IV) or AA (V and VI), respectively. Mitochondrial density (D, E) was quantified in the processes of differentiated non-AD and AD cybrid cells. Total numbers of mitochondria were counted within processes per cybrid cell (D) and numbers of mitochondria were counted per 100 μm process length (E).
Fig. 5.
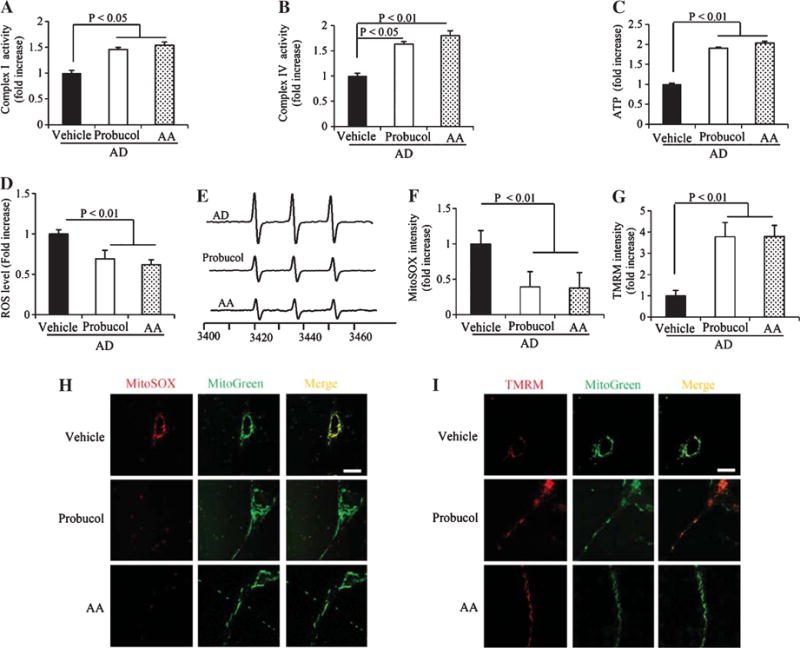
Effects of antioxidant on mitochondrial function, membrane potential and ROS levels in differentiated AD cybrid cells. Enzymatic activity of complex I (A), IV (B), and cellular ATP levels (C) were determined in cell lysates from differentiated AD cybrid cells with or without the addition of antioxidants. Data were expressed as fold increase relative to AD cybrid cells without the addition of antioxidant (vehicle treatment). Generation of ROS was detected by electron paramagnetic resonance (EPR) spectra (D, E) and mitochondrial ROS levels were measured by MitoSox staining intensity (F, H). Mitochondrial membrane potential was measured by TMRM staining intensity (G, I). Quantifications were determined of the signal intensity of EPR (D), immunofluorescent intensity for MitoSox (F) and TMRM (G) in mitochondria of the indicated cybrid cells. The representative EPR and staining images were shown for EPR (E), and red fluorescence for Mitosox (H) and TMRM (I). Data were collected from 20–25 processes of each AD cybrid cell line with different treatments.
Probucol and AA also improved mitochondrial transport and motility (Fig. 6). There was an increase in the percentage of movable mitochondria (Fig. 6A), as well as a decrease in the percentage of stationary ones in AD cybrid cells (Fig. 6B). Mitochondrial movement velocity and total travel distance were also rescued (Fig. 6C, D). Quantification of mitochondrial movement and kymography revealed that the antioxidant treatments increased both anterograde and retrograde mitochondrial movement in AD neuronal processes (Fig. 6E–G).
Fig. 6.
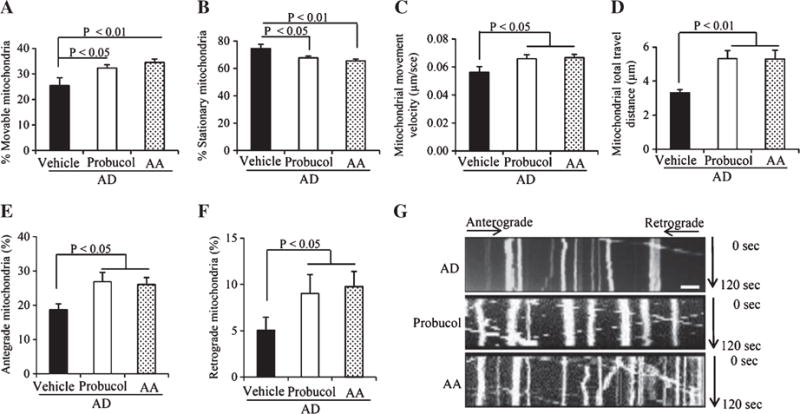
Effect of antioxidant treatment on mitochondrial movement parameters in the neuronal processes from differentiated AD cybrid cells. A, B) The percentage of mobile (A) and stationary mitochondria (B) in the neuronal processes of cybrid cells. C) Average mitochondrial travel velocity and (D) mitochondrial travel distance were calculated. Antioxidant treatment significantly rescues anterograde and retrograde mitochondrial movement in AD cybrid cells (E, F). G) Kymographs generated from live imaging movies represent differentiated AD cybrid cells with or without antioxidant treatment. In the kymographs, the X axis is mitochondrial position and the Y axis represents time lapse (0–120 s). Vertical white lines represent stationary mitochondria and diagonal lines represent mobile mitochondria. Anterograde movements are from left to right and retrograde movements are from right to left. Scale bars = 10 μm.
DISCUSSION
We investigated differentiation capacity, mitochondrial function, and mitochondrial dynamics in differentiated cybrid cell lines containing platelet-derived mitochondria derived from AD and normal subjects. AD cybrid cells exhibited impaired outgrowth of axonal processes based on the measurement of the development of thin and extended neuronal-like processes. This suggests that AD-derived mitochondria may adversely affect neuronal maturation and differentiation.
We also demonstrated defective mitochondrial respiration and energy metabolism in differentiated AD cybrid cells, as shown by decreased complex I and IV activity, and ATP levels. These results are consistent with changes previously reported in undifferentiated AD cybrid cells [7]. Prior studies have shown that ROS are essential for neuron differentiation [29], while overproduced ROS are cytotoxic [16]. The non-AD cybrids displayed increased mitochondrial ROS production and membrane potential following SAT-induced differentiation. SAT-differentiated AD cybrid neurons similarly showed higher levels of ROS, however, these cells did not demonstrate an increase in their mitochondrial membrane potential in response to SAT-induced differentiation (Supplementary Figure 2).
We observed the association between ROS overproduction and decreased membrane potentials in AD cybrid neuronal process, and speculate that these defects may arrest neuronal process outgrowth in AD cybrids. Substantial evidence supports the hypothesis that respiratory chain complex dysfunction and impairment of ATP production play a critical role in AD pathogenesis [30, 31]. Our differentiated AD cybrid cells recapitulate features observed in AD patients, showing impaired mitochondrial respiratory function and reduced electron respiratory chain enzyme activity.
Impaired mitochondrial trafficking and motility were found in differentiated AD neuronal processes (axons) in our study. Compared to non-AD mitochondria, mitochondrial movement within neuronal processes was significantly reduced in AD cybrid cells and the percentage of stationary mitochondria increased. Within AD cybrid neuronal processes even non-stationary mitochondria moved more slowly than they did in control cybrid neuronal processes. Overall, these findings suggest that mitochondrial movement within AD cybrid cell lines is impaired. In view of the significance of axonal mitochondrial transport in neuronal transmission and synaptic function [8, 25], AD-associated aberrant axonal mitochondrial trafficking may have important implications in AD-associated neuronal and synaptic dysfunction.
Finally, we found that antioxidants mitigate perturbations and improve the axonal transport of mitochondria that contain AD-subject specific mitochondrial defects. Treatments with probucol, an antioxidant preventing protein or lipid peroxidation by stimulating the endogenous antioxidant enzymes [32], and ascorbic acid that interacts directly with the oxidizing radicals and effectively scavenges superoxide and other ROS [33], improved neuronal differentiation, mitochondrial respiratory function, and energy metabolism in AD cybrids. Given that mitochondrial permeability transition pore (mPTP) plays a key role in cell survival and death [34] via alterations in mitochondrial structure and function and ROS production/accumulation, mPTP-mediated mitochondrial perturbation could contribute to oxidative stress and aberrant mitochondrial transport. Indeed, blockade of cyclophilin D, a key component of mPTP, by genetic deletion of CypD or pharmacological inhibition of CypD, suppresses mitochondrial ROS production and protects against oxidative stress- and amyloid beta peptide (Aβ)-mediated mitochondrial and neuronal dysfunction in vitro cellular model and in vivo human disease mouse model [9, 25, 35]. Furthermore, depletion of CypD significantly reverses Aβ-induced axonal mitochondrial abnormalities, improves synaptic function, and attenuates loss of synapse [8, 36]. Oxidative stress- or CypD-mediated activation signal transduction including MAPkinase (p38 and ERK1/2) could also be involved in abnormalities in AD-derived mitochondria and axonal mitochondrial transport as shown in AD cybrid neurons [7, 37]. Alternatively, the calcium-binding protein EFhd2 modulates intracellular calcium response, actin dynamics, and microtubule transport. Recent study has demonstrated EFhd2 that EFhd2 modulates synapse formation in vitro, which is linked to neurodegenerative diseases including dementia and AD [38], therefore, dysregulation of EFhd2 in human dementia might play a role in AD mitochondrial perturbation and axonal degeneration. These studies strongly support that mitochondrial-generated oxidative stress accentuates mitochondrial dysfunction itself in this ex vivo model of AD-specific mitochondrial function relevant to AD mitochondrial and neuronal degeneration. This phenomenon is potentially reversible with antioxidant treatment.
In conclusion, we now provide evidence of mitochondrial defects in differentiated neuronal cells whose mitochondria originated from AD subject platelets. Abnormal mitochondrial transport and movement in AD neuronal/axon-like processes correlates with structural and functional mitochondrial alterations. Defects in AD-derived mitochondria might include the following mechanisms: 1) impaired mitochondrial respiration and energy metabolism; 2) increased mitochondrial oxidative stress; and 3) significant alterations in mitochondrial transport. Antioxidants can rescue defects in mitochondrial transport and function observed in this model. We thereby propose that increased ROS production in AD-derived mitochondria disrupts mitochondrial transport and movement, and interferes with normal mitochondrial function and neuron differentiation (Fig. 7). These abnormalities observed in AD-cybrid human neuronal cell lines recapitulate AD-associated mitochondrial pathology in an in vitro system, and demonstrate the potential of this approach to model AD mitochondrial dysfunction as well as the consequences of that dysfunction.
Fig. 7.
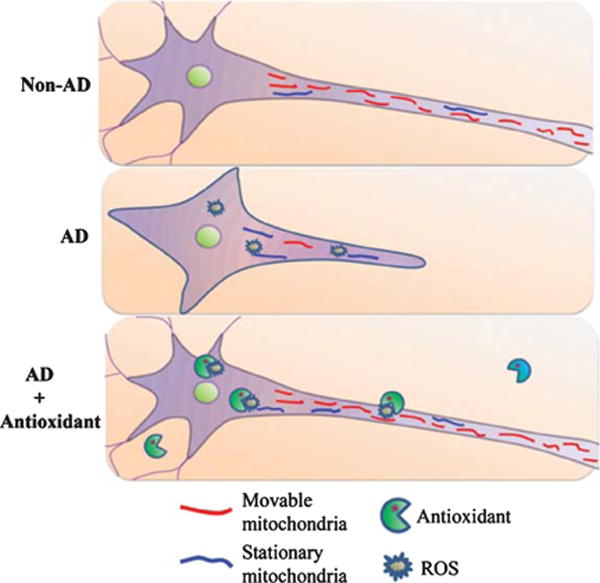
Schematic diagram depicting the role of antioxidant treatment on differentiation ability and axonal mitochondrial transport in AD cybrid cells. Compared to non-AD derived mitochondria (red), AD-derived mitochondria reveal higher levels of ROS, which arrests differentiation and mitochondria movement resulting in less movable mitochondria and more stationary mitochondria (blue) in AD cybrids. Antioxidants suppress ROS accumulation, which in turn rescue mitochondrial defects including axonal mitochondrial transport and neuronal differentiation.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Institute on Aging (R37AG037319 and R01AG044793), National institute of Neurological disorder and Stroke (NS R01NS65482), and the University of Kansas Alzheimer’s Disease Center (NIA P30AG035982). The EPR instrumentation was provided by NSF Chemical Instrumentation Grant (# 0946883).
Footnotes
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0532r1).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-160532.
References
- 1.Chan DC. Mitochondria: Dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Kann O, Kovacs R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292:C641–C657. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol. 2006;18:453–459. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Hollenbeck PJ. The pattern and mechanism of mitochondrial transport in axons. Front Biosci. 1996;1:d91–d102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Gan X, Huang S, Wu L, Wang Y, Hu G, Li G, Zhang H, Yu H, Swerdlow RH, Chen JX, Yan SS. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim Biophys Acta. 2014;1842:220–231. doi: 10.1016/j.bbadis.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo L, Du H, Yan S, Wu X, McKhann GM, Chen JX, Yan SS. Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One. 2013;8:e54914. doi: 10.1371/journal.pone.0054914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silva DF, Selfridge JE, Lu J, E L, Roy N, Hutfles L, Burns JM, Michaelis EK, Yan S, Cardoso SM, Swerdlow RH. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet. 2013;22:3931–3946. doi: 10.1093/hmg/ddt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis. 2006;9:119–126. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- 12.Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J Neurosci. 2013;33:17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuadrado-Tejedor M, Cabodevilla JF, Zamarbide M, Gomez-Isla T, Franco R, Perez-Mediavilla A. Age-related mitochondrial alterations without neuronal loss in the hippocampus of a transgenic model of Alzheimer’s disease. Curr Alzheimer Res. 2013;10:390–405. doi: 10.2174/1567205011310040005. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trimmer PA, Borland MK. Differentiated Alzheimer’s disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid Redox Signal. 2005;7:1101–1109. doi: 10.1089/ars.2005.7.1101. [DOI] [PubMed] [Google Scholar]
- 16.Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Jr, Miller SW, Davis RE, Parker WD., Jr Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- 17.King MP, Attardi G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 18.Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J Neurochem. 2004;89:1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 19.Prince JA, Oreland L. Staurosporine differentiated human SH-SY5Y neuroblastoma cultures exhibit transient apoptosis and trophic factor independence. Brain Res Bull. 1997;43:515–523. doi: 10.1016/s0361-9230(97)00328-6. [DOI] [PubMed] [Google Scholar]
- 20.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller SW, Trimmer PA, Parker WD, Jr, Davis RE. Creation and characterization of mitochondrial DNA-depleted cell lines with “neuronal-like” properties. J Neurochem. 1996;67:1897–1907. doi: 10.1046/j.1471-4159.1996.67051897.x. [DOI] [PubMed] [Google Scholar]
- 22.Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res. 2007;85:3416–3428. doi: 10.1002/jnr.21167. [DOI] [PubMed] [Google Scholar]
- 23.Leskiewicz M, Regulska M, Budziszewska B, Jantas D, Jaworska-Feil L, Basta-Kaim A, Kubera M, Lason W. Neurosteroids enhance the viability of staurosporine and doxorubicin treated differentiated human neuroblastoma SH-SY5Y cells. Pharmacol Rep. 2008;60:685–691. [PubMed] [Google Scholar]
- 24.Trimmer PA, Schwartz KM, Borland MK, De Taboada L, Streeter J, Oron U. Reduced axonal transport in Parkinson’s disease cybrid neurites is restored by light therapy. Mol Neurodegener. 2009;4:26. doi: 10.1186/1750-1326-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang D, Wang Y, Zhang Z, Du H, Yan S, Sun Q, Zhong C, Wu L, Vangavaragu JR, Yan S, Hu G, Guo L, Rabinowitz M, Glaser E, Arancio O, Sosunov AA, McKhann GM, Chen JX, Yan SS. Increased neuronal PreP activity reduces Abeta accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum Mol Genet. 2015;24:5198–5210. doi: 10.1093/hmg/ddv241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang D, Zhang Z, Li H, Yu Q, Douglas JT, Bratasz A, Kuppusamy P, Yan SS. Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer’s disease mouse brain. J Alzheimers Dis. 2016;51:571–580. doi: 10.3233/JAD-150917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borland MK, Trimmer PA, Rubinstein JD, Keeney PM, Mohanakumar K, Liu L, Bennett JP., Jr Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol Neurodegener. 2008;3:21. doi: 10.1186/1750-1326-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsatmali M, Walcott EC, Crossin KL. Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res. 2005;1040:137–150. doi: 10.1016/j.brainres.2005.01.087. [DOI] [PubMed] [Google Scholar]
- 30.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 31.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuzuya M, Kuzuya F. Probucol as an antioxidant and antiatherogenic drug. Free Radic Biol Med. 1993;14:67–77. doi: 10.1016/0891-5849(93)90510-2. [DOI] [PubMed] [Google Scholar]
- 33.Arrigoni O, De Tullio MC. Ascorbic acid: Much more than just an antioxidant. Biochim Biophys Acta. 2002;1569:1–9. doi: 10.1016/s0304-4165(01)00235-5. [DOI] [PubMed] [Google Scholar]
- 34.Rao VK, Carlson EA, Yan SS. Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta. 2014;1842:1267–1272. doi: 10.1016/j.bbadis.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32:398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du H, Guo L, Wu X, Sosunov AA, McKhann GM, Chen JX, Yan SS. Cyclophilin D deficiency rescues Abeta-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim Biophys Acta. 2014;1842:2517–2527. doi: 10.1016/j.bbadis.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan X, Wu L, Huang S, Zhong C, Shi H, Li G, Yu H, Howard Swerdlow R, Xi Chen J, Yan SS. Oxidative stress-mediated activation of extracellular signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfunction. Free Radic Biol Med. 2014;75:230–240. doi: 10.1016/j.freeradbiomed.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borger E, Herrmann A, Mann DA, Spires-Jones T, Gunn-Moore F. The calcium-binding protein EFhd2 modulates synapse formation in vitro and is linked to human dementia. J Neuropathol Exp Neurol. 2014;73:1166–1182. doi: 10.1097/NEN.0000000000000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.