

Abstract
We have recently demonstrated that RUNX2 promoted, and 17β-Estradiol (E2) diminished, association of RANKL with the cell membrane in pre-osteoblast cultures. Here we show that, similar to E2, dihydrotestosterone (DHT) diminishes association of RANKL, and transiently transfected GFP-RANKL with the pre-osteoblast membrane without decreasing total RANKL mRNA or protein levels. Diminution of membrane-associated RANKL was accompanied with marked suppression of osteoclast differentiation from co-cultured pre-osteoclasts, even though DHT increased, not decreased, RANKL concentrations in pre-osteoblast conditioned media. A marked decrease in membrane-associated RANKL was observed after 30 min of either E2 or DHT treatment, and near-complete inhibition was observed by 1 hr, suggesting that the diminution of RANKL membrane association was mediated through non-genomic mechanisms. Further indicating dispensability of nuclear action of estrogen receptor, E2-mediated inhibition of RANKL membrane association was mimicked by an estrogen dendrimer conjugate (EDC) that cannot enter the cell nucleus. Finally, the inhibitory effect of E2 and DHT on RANKL membrane association was counteracted by the MMP inhibitor NNGH, and the effect of E2 (and not DHT) was antagonized by the Src inhibitor SU6656. Taken together, these results suggest that estrogens and androgens inhibit osteoblast-driven osteoclastogenesis through non-genomic mechanism(s) that entail, MMP-mediated RANKL dissociation from the cell membrane.
Keywords: membrane-initiated estrogen signaling, MMP, RANKL presentation, sex steroids, Src
1 INTRODUCTION
The RANKL/RANK/OPG system plays pivotal roles in regulating osteoclastogenesis driven by cells of the osteoblast and other lineages (Kearns, Khosla, & Kostenuik, 2008; Nakashima, Hayashi, & Takayanagi, 2012). In the osteoblast lineage, RANKL is biosynthesized by cells at various maturation stages, from mesenchymal stem cells (MSCs) to matrix embedded osteocytes. To fulfill its function of promoting osteoclast differentiation, activation, and survival, RANKL is presented to osteoclast lineage cells as either a membrane-bound or a soluble protein. Both forms bind and stimulate RANK on the target cell membrane, and this binding can be blocked by OPG, a secreted protein homologous to RANKL that acts as a decoy receptor (Kearns et al., 2008; Nakashima et al., 2012). It has been suggested that membrane-bound RANKL is more potent than soluble RANKL in promoting osteoclastogenesis (Douni et al., 2012; Kearns et al., 2008; Nakashima et al., 2000). Accordingly, while osteoblast- and osteocyte-borne RANKL robustly promoted osteoclastogenesis in co-culture assays, it failed to do so when presented to osteoclast precursors in conditioned media or in transwells that facilitated RANKL diffusion but not cell–cell contact (Jimi et al., 1996; Nakashima et al., 2000, 2011). In fact, it has been proposed that proteolytic cleavage of RANKL ectodomain (shedding) functions to dampen osteoclastogenesis in vitro and bone resorption in vivo (Hikita et al., 2006).
The bone-sparing property of sex steroids relies to a large extent on the attenuation of bone turnover, which is achieved via diverse mechanisms. Sex steroids inhibit osteoblast differentiation from their mesenchymal precursors (Di Gregorio et al., 2001; Jilka et al., 1998), they restrain osteoclastogenic signals emanating from mesenchymal/osteoblast-lineage cells, and they directly suppress osteoclast survival and function (Almeida et al., 2017; Frenkel et al., 2010; Khosla, Oursler, & Monroe, 2012; Vanderschueren et al., 2014). The best evidence for involvement of the RANKL/RANK/OPG system in the bone-sparing effects of sex steroid hormones is the capability of estrogens to stimulate OPG mRNA and protein biosynthesis in cells of the osteoblast lineage in vitro (Bord, Ireland, Beavan, & Compston, 2003; Hofbauer et al., 1999; Saika, Inoue, Kido, & Matsumoto, 2001) as well as in human subjects (Khosla, Atkinson, Dunstan, & O’Fallon, 2002). While estrogen-mediated OPG stimulation may contribute to inhibition of bone resorption, such a mechanism is not employed by androgens (Hofbauer, Hicok, Chen, & Khosla, 2002; Khosla et al., 2002). Additionally, there is little evidence that estrogens or androgens inhibit RANKL expression.
RUNX2, best known for its pivotal role in osteoblast differentiation and bone formation (Banerjee et al., 1997; Ducy, Zhang, Geoffroy, Ridall, & Karsenty, 1997; Komori et al., 1997; Otto et al., 1997), also controls osteoblast-driven osteoclastogenesis (Baniwal et al., 2012; Geoffroy, Kneissel, Fournier, Boyde, & Matthias, 2002; Liu et al., 2001; Maruyama et al., 2007). This property was attributed to RUNX2-mediated protein trafficking (Little et al., 2012), in particular increased RANKL membrane association (Martin et al., 2015). Because Estrogen Receptor (ER)α physically interacts with and inhibits RUNX2 (Khalid et al., 2008), we have recently tested the hypothesis that 17β-estradiol (E2) antagonized RUNX2-mediated RANKL trafficking, and the associated differentiation of co-cultured osteoclast precursors. We found that E2 indeed antagonized RUNX2-mediated osteoblast-driven osteoclastogenesis (Martin et al., 2015). This effect of E2 was not associated with alterations to RANKL expression assessed by either RT-qPCR or Western analysis of whole cell extracts. Instead, E2 diminished RUNX2-driven RANKL association with the osteoblast membrane. Surprisingly, however, E2 did not affect RANKL levels in osteoblast conditioned medium (Martin et al., 2015). How E2 decreases membrane-bound RANKL but not secreted RANKL remains to be clarified.
The bone sparing effects of androgens are largely mediated through aromatization to estrogens, but several lines of evidence suggest additional, aromatization-independent mechanisms (Frenkel et al., 2010; Vanderschueren et al., 2014). For example, orchiectomy of aromatase knockout male mice results in trabecular and cortical bone loss beyond that attributable to ERα inactivity (Matsumoto, Inada, Toda, & Miyaura, 2006). Given that the androgen receptor (AR) and RUNX2 usually inhibit each other (Baniwal et al., 2009; Kawate, Wu, Ohnaka, & Takayanagi, 2007; Little et al., 2014), we initiated the present study by testing whether the non-aromatizable androgen dihydrotestosterone (DHT) antagonized RUNX2-mediated osteoblast-driven osteoclastogenesis. We found that like E2, DHT antagonized RUNX2-mediated RANKL membrane association and the differentiation of co-cultured osteoclast precursors. We further present evidence suggesting that inhibition of RANKL membrane association by either E2 or DHT is mediated through non-genomic post-translational mechanisms.
2 MATERIALS AND METHODS
2.1 Animals and reagents
C57BL/6 mice were obtained from the Jackson Laboratory, Sacramento, CA. All experimental procedures with animals were approved by the Institutional Animal Care and Use Committee at either USC or UTHSC. Tissue culture reagents including αMEM, phenol red-free αMEM, and trypsin were purchased from Gibco (Carlsbad, CA), fetal bovine serum (FBS), and charcoal-stripped serum (CSS) were purchased from Gemini (Sacramento, CA). Doxycycline (dox) was purchased from Calbiochem (La Jolla, CA) and administered at a final concentration of 0.5 μg/ml. A 17-β estradiol (E2) and dihydrotestosterone (DHT) were purchased from Sigma–Aldrich (St. Louis, MO) and administered at a final concentration of 10 nM. N-Isobutyl-N-(4-methoxyphenylsulfonyl) glycyl hydroxamic acid (NNGH) and SU6656 were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX) and administered at a final concentration of 5 μM. Estrogen dendrimer conjugates (EDC) and control dendrimers (CD) were synthesized as previously described (Harrington et al., 2006; Kim and Katzenellenbogen, 2006).
2.2 Cell culture
Newborn Mouse Calvarial Osteoblasts (NeMCOs) were prepared from 1- to 2-day-old newborn mice by digestion of parietal bones with 1 mg/ml collagenase P and 1.25% trypsin in PBS (Gabet, Noh, Lee, & Frenkel, 2011). Cells were maintained in αMEM supplemented with 20% FBS and 1% penicillin-streptomycin and used within two passages. For treatment with sex steroid hormones, cells were cultured in phenol red-free αMEM supplemented with 10% CSS. Primary osteoclast progenitors were prepared from spleens of 4–6-week-old mice by digestion with 1 mM Tris–HCl lysis buffer containing 0.74% NH4Cl as previously described (Udagawa et al., 1989). Co-cultures of NeMCOs with splenocytes were established and osteoclastogenesis was assessed by counting TRAP-positive cells with ≥3 nuclei as previously described (Martin et al., 2015). For functional analysis of osteoblast-driven activation of NFκB in osteoclasts, a RAW264.7/NFκB-Luc reporter cell line was used as previously described (Martin et al., 2015).
2.3 Plasmids and transfection
Transduction of NeMCOs with lentiviral particles encoding dox-inducible FLAG-RUNX2 was previously described (Baniwal et al., 2010; Martin et al., 2015). The GFP-Rankl plasmid (Kariya, Honma, Aoki, Chiba, & Suzuki, 2009), a gift from Dr. Hiroshi Suzuki, University of Tokyo, was introduced into NeMCO along with a RUNX2 expression vector (Ducy and Karsenty, 1995) or pcDNA3.0 using the jetPRIME transfection reagent (Polyplus-Transfection, New York, NY) and buffer according to the manufacturer’s protocol.
2.4 RNA extraction and analysis
Total RNA was extracted using Aurum™ total RNA mini kit (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s protocol and 1 μg RNA was reverse-transcribed using qScript™ cDNA supermix (Quanta Bioscience, Gaithersburg, MD). Real-time qPCR analysis was performed using the CFX96TM RT-PCR system (Bio-Rad Laboratories) and the iQ™ SYBR® Green Supermix (Bio-Rad Laboratories) according to the manufacturer’s protocol. The primers used for qPCR are listed in Table 1. Data were normalized for the mRNA levels of 18S, which themselves were not significantly affected by treatment.
TABLE 1.
Primers used for RT-qPCR
| Gene | Sequence (5′ to 3′) | |
|---|---|---|
| Osteocalcin | F | ACACTCCTCGCCCTATTGGC |
| R | TGCTTGGACACAAAGGCTGC | |
| Osterix | F | GTACGGCAAGGCTTCGCATCTG |
| R | CTGA TGTTTGCTCAAGTGGTCGC | |
| Rankl | F | GGGGGCCGTGCAGAAGGAAC |
| R | CTCAGGCTTGCCTCGCTGGG | |
| Runx2 | F | ATCACGCCGACCACCCGGC |
| R | GGCTACCACCTTGAAGGCCACG | |
2.5 Fluorescence imaging
Primary NeMCO cultures co-transfected with the GFP-RANKL and FLAG-RUNX2 expression vectors were fixed with freshly made 4% formaldehyde in PBS and incubated with mouse monoclonal antibodies against FLAG® epitope (Sigma–Aldrich) followed by secondary goat anti-mouse IgG (Abcam, Cambridge, MA). Cells were then mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) for fluorescence analysis. Fluorescence was detected using Nikon Eclipse Ti microscope. For endogenous Rankl immunofluorescence analysis, fixed cells were incubated with the FL-317 primary antibody (1:50) followed by the DyLight 488 secondary antibody (1:200) and mounted with Vectashield mounting medium (Vector Laboratories) containing DAPI, and images were captured using a ZEISS LSM 510 confocal system. Percentage of cell perimeter occupied by RANKL was determined by dividing the labeled portion of the cell perimeter by the total cell perimeter, which were tracked using the NIS-Element AR3.2 software.
2.6 Western blot analysis
Cells were lysed in a 50 mM Tris–HCl buffer (pH 7.4) containing 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and fresh protease inhibitor cocktail. Protein concentrations were measured by Bio-Rad Protein Assay Kit (Bio-Rad Laboratories). Equal amounts of protein were resolved by SDS–PAGE and transferred to Amersham Hybond™-PVDF membranes (GE Healthcare, Piscataway, NJ) and detected by rabbit polyclonal antibody to RANKL (FL-317) or Actin (I-19) from Santa Cruz Biotechnology.
2.7 ELISA
NeMCO/Rx2dox were cultured in 10 cm plates (500,000 cells/plate), initially in 10 ml of 10% CSS for 48 hr and then in 5 ml of 1% CSS for 12 hr. ELISA was performed using a mouse RANKL single plex Milliplex kit (MBN-41K-1RANKL; Millipore; Billerica, MA).
2.8 Statistical analysis
Effects of sex hormones on osteoclastogenesis, gene expression (RT-qPCR), RANKL secretion, NFκB-luciferase, and RANKL membrane association were determined in triplicate cultures and the results are presented as the mean ± SD. Differences were tested for significance using Student’s t-test and p < 0.05 (marked with *) was considered significant.
3 RESULTS
3.1 DHT antagonizes Runx2-mediated osteoblast-driven osteoclastogenesis
NeMCO/Rx2dox are primary newborn mouse calvarial osteoblasts transduced with a lentivirus encoding doxycycline (dox)-inducible FLAG-tagged RUNX2 (Baniwal et al., 2010; Martin et al., 2015). As recently reported (Martin et al., 2015), induction of RUNX2 in NeMCO/Rx2dox results in robust stimulation of osteoclast differentiation from co-cultured splenocytes used as a source of osteoclast precursors (Figure 1a,b). Treatment of the co-cultures with DHT diminished RUNX2-mediated osteoblast-driven osteoclastogenesis (Figure 1a,b). Importantly, DHT did not decrease RUNX2 expression in NeMCO/Rx2dox cultures. In fact, while Runx2 mRNA levels did not change (Figure 1d), its protein levels increased (Figure 1c). However, consistent with antagonism of RUNX2 activity (Baniwal et al., 2009; Kawate et al., 2007), DHT inhibited expression of Bglap (Osteocalcin; Oc) (Figure 1e) and several other RUNX2 target genes. Antagonism of both RUNX2-driven gene expression and RUNX2-mediated osteoblast-driven osteoclastogenesis by DHT is the effects of E2 in the same NeMCO/Rx2dox culture system (Martin et al., 2015). Interestingly, however, RUNX2-mediated stimulation of Sp7 (Osterix; Osx) was not affected by DHT (Figure 1f).
FIGURE 1.
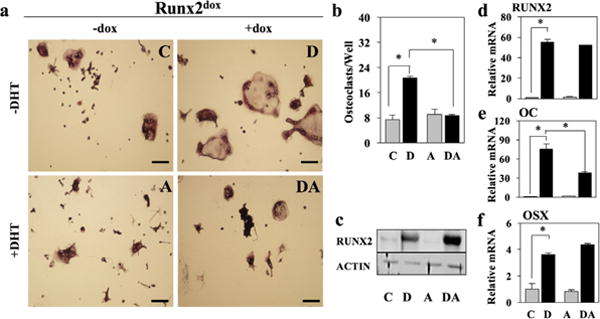
DHT counteracts osteoclastogenesis driven by forced RUNX2 expression. Newborn Mouse Calvarial Osteoblasts (NeMCO) were transduced with lentiviruses encoding dox-inducible RUNX2. NeMCO/Rx2dox were co-cultured with primary murine splenocytes in the presence of dox and/or DHT as indicated. (a and b) Osteoclasts were identified based on TRAP staining (a; scale bar = 100 μm) and enumerated (b). Results are from one of three experiments with similar results (Mean ± SD; n = 3; *p < 0.05). (c) Western blot analyses with anti-RUNX2 and anti-ACTIN antibodies. (d–f) Levels of the indicated mRNAs were determined by RT-qPCR (Mean ± SD; n = 3; *p < 0.05). C, control; D, dox; A, DHT; DA, dox plus DHT
3.2 DHT antagonizes RUNX2-mediated association of RANKL with the osteoblast membrane
We next investigated the possible involvement of RANKL in the inhibition RUNX2-mediated osteoblast-driven osteoclastogenesis by DHT. Consistent with our published work (Martin et al., 2015), RUNX2 (dox) did not change RANKL mRNA or protein levels (Figure 2a,c), but instead had the following effects that could account for its osteoclastogenic effect: (i) it increased by fourfold association of RANKL (as well as transiently transfected GFP-RANKL) with the osteoblast membrane (Figure 3); (ii) it increased by 10.5-fold the concentration of RANKL in the conditioned medium (Figure 2d); (iii) it decreased by 55% OPG mRNA levels (Figure 2b); and (iv) it stimulated by 12-fold the activity of NFκB in co-cultured RAW264.7 reporter cells (Figure 2e).
FIGURE 2.
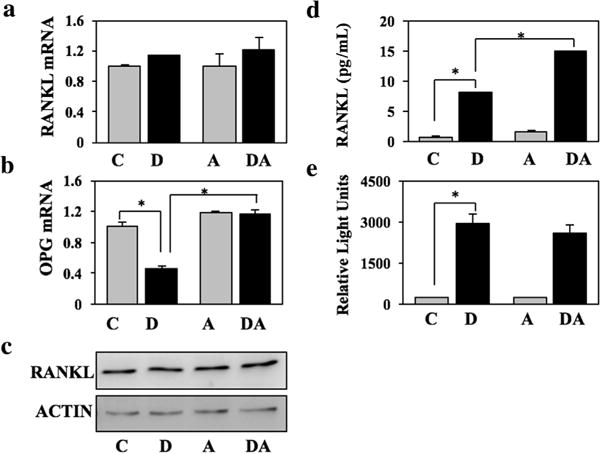
DHT does not inhibit RANKL levels. NeMCO/Rx2dox were treated for 48 hr with dox and/or DHT as indicated. (a and b) RANKL and OPG mRNA levels were measured by RT-qPCR. (c) RANKL expression was assessed by Western blot analysis using ACTIN as loading control. (d) RANKL concentrations in conditioned media were determined by ELISA. (e) RAW264.7/NFκB-Luc reporter cells were added to NeMCO cultures for additional 24 hr and luciferase assay was performed as previously described (Martin et al., 2015). C, control; D, dox; A, DHT; DA, dox plus DHT
FIGURE 3.
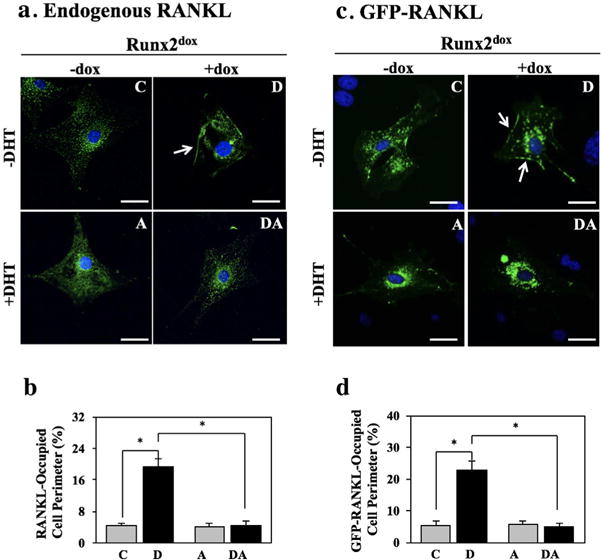
RUNX2 promotes and DHT antagonizes RANKL association with the osteoblast membrane. NeMCO/Rx2dox were treated for 48 hr with dox and/or DHT as indicated, and endogenous RANKL (a and b) or transiently expressed GFP-RANKL (c and d) were imaged by indirect and direct immunofluorescence, respectively. (a and c) Representative micrographs (Scale bar = 50 μm), with arrows marking membrane-associated RANKL. (b and d) Percentage of cell perimeter containing RANKL or GFP-RANKL was determined for ≥10 randomly selected cells per condition (Mean ± SD; n ≥ 10; *p < 0.05). C, control; D, dox; A, DHT; DA, dox plus DHT
Co-treatment with DHT revealed selective antagonism of the four aforementioned effects of RUNX2. DHT completely reversed RUNX2-mediated RANKL (and GFP-RANKL) membrane association (Figure 3) and the inhibition of OPG mRNA expression (Figure 2b). These anti-RUNX2 activities of DHT could account for the diminution of RUNX2-mediated osteoblast-driven osteoclastogenesis (Figure 1). Interestingly, DHT antagonized neither RUNX2-mediated stimulation of RANKL secretion (Figure 2d) nor the increase in NFκB-Luc activity in the RAW264.7 reporter cells (Figure 2e).
3.3 Sex steroid hormones unlikely antagonize RUNX2-mediated osteoblast-driven RANKL membrane association at the transcriptional level
RUNX2 and steroid hormone receptors typically exhibit mutual transcriptional inhibition (Baniwal et al., 2009; Chimge et al., 2011; Kawate et al., 2007; Khalid et al., 2008; Koromila et al., 2014; Little et al., 2014; Martin et al., 2015), which could potentially explain antagonism of RUNX2-driven osteoblast-mediated osteoclastogenesis by E2 (Martin et al., 2015) and by DHT (Figure 1). There are many exceptions, however, to the general mutual antagonism between RUNX2 and steroid hormone receptors. For example, a sizable proportion of DHT-stimulated genes in prostate cancer cells escaped antagonism by RUNX2 (Little et al., 2014) and several RUNX2-stimulated genes in ST2 mesenchymal stem cells escaped antagonism by glucocorticoids (Morimoto et al., 2016). Notably, RUNX2-mediated stimulation of Osx was antagonized by E2 (Martin et al., 2015) but not by DHT (Figure 1f). Conversely, RUNX2-mediated inhibition of OPG mRNA expression was antagonized by DHT (Figure 2b) and not by E2 (Martin et al., 2015). We also noted that attenuation of the transcriptional activity of RUNX2 at other target genes in osteoblasts (e.g., Osteocalcin) by either E2 or DHT is typically in the order of 50%, while antagonism of RUNX2 with regard to the effect on RANKL membrane association by both E2 (Martin et al., 2015) and DHT is complete (Figure 3). Finally, the inconsistency between effects of DHT on RUNX2-mediated RANKL membrane association versus RANKL secretion (Figure 3 vs. Figure 2d), and the similar observations with E2 (Martin et al., 2015) were difficult to explain solely based on transcriptional antagonism of RUNX2.
Given the arguments against antagonism of RUNX2-mediated RANKL membrane association at the transcriptional level, we considered the possibility that sex steroid hormones decreased RANKL membrane association post-transcriptionally. We first tested the timeframe within which sex steroid hormones cause RANKL to disappear from the membrane. In these experiments we transiently co-transfected NeMCOs with a plasmid expressing GFP-RANKL along with either pcDNA (vector control) or a plasmid constitutively expressing RUNX2. We first confirmed that GFP-RANKL membrane association increased (from 2% to 18%) when co-transfected with RUNX2 versus the pcDNA vector control (Figure 4 vs. supplementary Figure S1). We then followed the disappearance of GFP-RANKL from the cell perimeter after different time periods of treatment with either E2 or DHT. Remarkably, 30 min of treatment with either E2 or DHT was sufficient for a ∼50% reduction in RANKL association with the cell perimeter (Figure 4). The antagonism of RANKL membrane association was nearly complete within 1–2 hr (Figure 4). These rapid responses suggest that sex steroid hormones diminish association of RANKL with the osteoblast membrane through a non-genomic mechanism(s).
FIGURE 4.
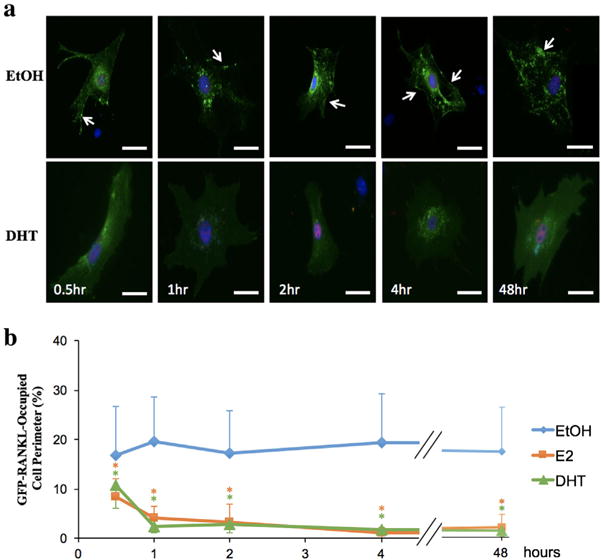
Rapid dissociation of RANKL from the pre-osteoblast membrane in response to E2 and DHT. NeMCO were transiently transfected with plasmids encoding GFP-RANKL and FLAG-RUNX2. Two days later, cultures were treated with ethanol (EtOH) vehicle or E2 or DHT for indicated time periods. (a) Representative DHT-treated and control cells are shown with direct fluorescence imaging of GFP-RANKL and immunofluorescence imaging of FLAG-RUNX2 (red). Scale bar, 50 μm. Arrows mark membrane-associated RANKL. (b) The percentage of cell perimeter occupied by GFP-RANKL was determined in a double-blinded fashion (Mean ± SD; n > 10; *p < 0.05)
3.4 Evidence that E2 and DHT diminish RANKL presentation through membranous, but distinct mechanisms
Given the rapidity with which GFP-RANKL was eliminated from the cell surface, we considered the possibility that these effects of E2 and DHT were mediated locally at the membrane level. We tested this contention in cells transfected as in Figure 4 and treated with an estrogen dendrimer conjugate (EDC) that activates membrane-associated ER and is incapable of entering the cell nucleus (Harrington et al., 2006; Kim et al., 2015). Remarkably, EDC diminished association of GFP-RANKL with the cell membrane with a time course indistinguishable from that of E2 (Figure 5a,b), suggesting that activation of membrane-localized ER indeed mediates the disappearance of RANKL from the cell surface. Because MMPs have been implicated in RANKL shedding from the cell membrane (Hikita et al., 2006; Schlondorff, Lum, & Blobel, 2001), we went on to test their involvement in the effects of sex hormones on RANKL membrane association. To this end, we repeated the co-transfection experiments in the presence of the broad MMP inhibitor NNGH. Indeed, neither E2 (Figure 5c) nor DHT (Figure 5d) decreased association of GFP-RANKL with the osteoblast membrane in the presence of NNGH. Finally, based on the involvement of c-Src in non-genomic signaling by E2 (Evinger and Levin, 2005) and in MMP activation (Yang, Bai, Yin, Wu, & Zhang, 2014), we also tested its involvement in the effects of sex steroid hormones on RANKL membrane association using the Src inhibitor SU6656. As shown in Figure 5c, SU6656 prevented E2 from diminishing the association of GFP-RANKL with the cell surface. However, SU6656 did not prevent the inhibition of RANKL membrane association by DHT (Figure 5d). Based on these results, we propose a working model, whereby bone loss at menopause is attributable in part to a decrease in ER-mediated MMP-dependent RANKL shedding (Figure 6); AR may engage a similar bone-sparing mechanism, which, unlike ER, is independent of Src.
FIGURE 5.
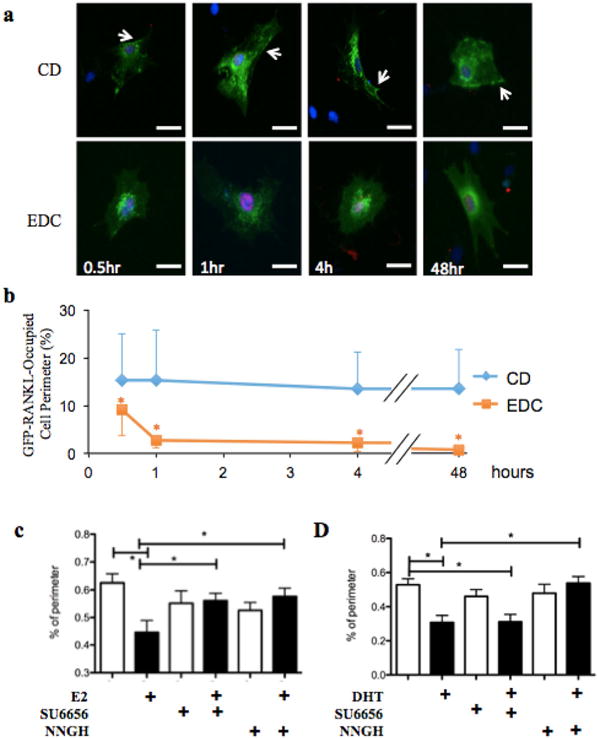
Sex steroids dissociate RANKL from the pre-osteoblast membrane through non-genomic mechanisms. (a and b) GFP-RANKL membrane association was determined in NeMCO as in Figure 4 after treatment with 10 nM estrogen-dendrimer conjugates (EDC) or control dendrimers (CD) for the indicated time periods. Representative images (bar = 50 μm) show GFP-RRANKL in green and FLAG-RUNX2 in red. Arrows mark membrane-associated GFP-RANKL. (b) RANKL membrane association was assessed as in Figure 4b (c and d) GFP-RANKL and RUNX2 were transiently expressed in NeMCO cultures and GFP-RANKL membrane association was assessed as in a and b after 24 hr of treatment with E2 (c) or DHT (d) along with NNGH or SU6656. Quantitative data are Mean ± SD; n > 10; *p < 0.05
FIGURE 6.
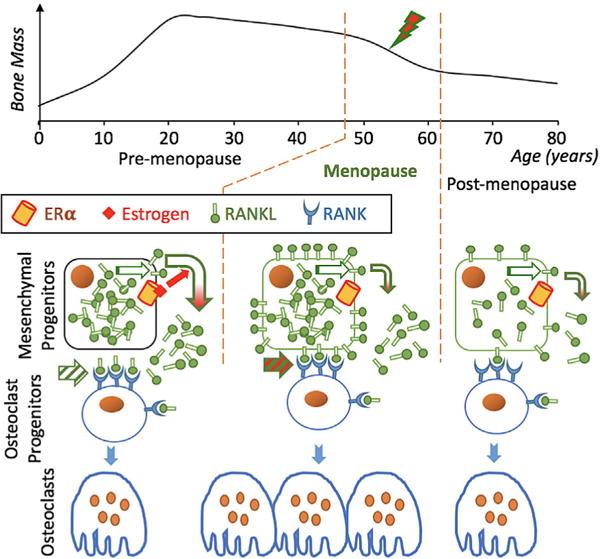
Working model for increased osteoblast-driven osteoclastogenesis upon menopause. Top, A typical time course describing age-dependent women’s bone mass, with increased rate of bone loss at early menopause (lightning bolt). Bottom, Schematic illustration of a shift from engagement of RANK by soluble RANKL to membrane-bound RANKL (striped arrows). This shift, caused by loss of E2-mediated RANKL shedding (redheaded arrows), results in excessive osteoclastogenesis and rapid bone loss. Compensatory feedback mechanisms activated at late menopause may include decreased RANKL (illustrated on the right) and/or increased OPG biosynthesis (LaCroix et al., 2013; Liu et al., 2005)
4 DISCUSSION
Despite the well-established bone-sparing properties of sex steroid hormones, the underlying molecular mechanisms remain poorly understood. Likely contributing to their anti-resorptive properties, we have now demonstrated that both E2 (Martin et al., 2015) and DHT (Figure 3) diminish RANKL association with the pre-osteoblast membrane. Because RANKL is most active in promoting osteoclastogenesis while bound to the membrane of presenting cells (see Introduction), its depletion from this compartment in the presence of either E2 (Martin et al., 2015) or DHT (Figure 1), even without a decrease in soluble RANKL, explains the observed diminution of osteoclast differentiation from their co-cultured precursors. Interestingly, the inhibition of osteoclastogenesis by E2 (Martin et al., 2015) and DHT (Figure 1) was not paralleled by decreased NFκB activity in the RAW 264.7 reporter cells (Figure 2e), possibly reflecting dependence of the former only on cell–cell contact. The disappearance of RANKL from the cell membrane in response to E2 (Martin et al., 2015) or DHT (Figure 2) did not result from inhibition of RANKL biosynthesis because neither its RNA, nor its intracellular, nor its extracellular protein levels was decreased. In the case of DHT, extracellular RANKL levels were not only uninhibited, but in fact increased (Figure 2d). Our working model assigns the inhibition of osteoclastogenesis by E2 (Martin et al., 2015) and DHT (Figure 1) to a specific decrease in membrane-associated RANKL, possibly through MMP-mediated ectodomain shedding. Clinical relevance of our work is suggested by the increased presentation of RANKL on the surface of bone marrow stromal cells (as well as B and T cells) in postmenopausal compared to premenopausal women and its decreased presentation on these cells in estrogen-treated versus untreated postmenopausal women (Eghbali-Fatourechi et al., 2003).
The present study argues against our initial working hypothesis that sex steroid hormones decrease RANKL presentation on the pre-osteoblast membrane via transcriptional antagonism of RUNX2 (Frenkel et al., 2010; Martin et al., 2015). Instead, a non-genomic mechanism(s) is suggested by the rapid action of both E2 and DHT (Figure 4) and by faithful mimicry of E2 by EDC (Figure 5a,b), which is incapable of activating nuclear ER (Harrington et al., 2006). Among possible non-genomic mechanisms, sex steroid hormones may inhibit RANKL trafficking to the pre-osteoblast membrane or accelerate its internalization or shedding. In favor of the latter, RANKL shedding by MMPs, specifically MMP14 and ADAM10, has been described (Hikita et al., 2006; Schlondorff et al., 2001), and MMP(s) are required for E2-and DHT-mediated decrease in RANKL membrane-association (Figure 5c,d).
A series of mouse genetics studies performed during the past decade assigned the regulation of female trabecular bone mass to ERα in osteoclasts, while the regulation of cortical bone mass was assigned to ERα in mesenchymal cells early in the osteoblast lineage (Almeida et al., 2017; Vanderschueren et al., 2014). In particular, ovariectomy-induced endocortical osteoclastogenesis (as well as periosteal bone accrual in intact mice) was linked to estrogen signaling in cells expressing Prx1-Cre, but not more mature cells expressing Col1a1-Cre (Almeida et al., 2013). This suggests that loss of estrogen signaling in pre-osteoblasts at an early developmental stage, prior to Col1a1 expression, is involved in estrogen loss-induced endocortical bone resorption. The present study suggests that accumulation of RANKL at the membrane of such early osteoblast progenitors drives endocortical bone resorption in response to estrogen loss. Our data also suggest that a similar mechanism is activated upon loss of androgen signaling in pre-osteoblasts (regardless of aromatization); this may be relevant to aromatization-independent bone mass control by androgens in males (Almeida et al., 2017; Vanderschueren et al., 2014).
That E2 protected endocortical bone through a non-genomic mechanism was initially suggested based on indispensability of the nuclear coactivator SRC-1 for this bone sparing effect (Modder et al., 2004), and this concept was more recently reiterated by the ability of EDC to mimic the effect of E2 on cortical bone despite its incapability of entering the cell nucleus (Bartell et al., 2013). While these authors invoked regulation of oxidative stress as the underlying molecular mechanism (Bartell et al., 2013), the present study brings forth the regulation of RANKL presentation as an alternative/additional mechanism underlying the endocortical anti-resorptive property of estrogens.
In addition to regulating RANKL membrane association in pre-osteoblasts, as suggested by the present study, E2 also stimulates OPG expression ([Hofbauer et al., 1999; Saika et al., 2001] and Figure 2b) and inhibits expression of the osteoclastogenic factor Cxcl12 in MSCs (Ucer et al., 2016). It is also important to note the involvement of osteocyte RANKL in ovariectomy-induced cortical bone loss (Fujiwara et al., 2016). These and other mechanisms (Bellido et al., 1995; Garcia et al., 2013; Krum et al., 2008; Nakamura et al., 2007; Ucer et al., 2016) are not mutually exclusive; any combination or all of them may contribute to the regulation of endocortical bone resorption by estrogens.
In conclusion, we propose a model, whereby estrogen-loss at menopause leads to accumulation of RANKL on the pre-osteoblast membrane, likely through attenuation of RANKL shedding (Figure 6). We further speculate that the resulting osteoclastogenesis and rapid bone loss is attenuated later after menopause by compensatory mechanisms, for example, a decrease in RANKL (illustrated in Figure 6) or an increase in OPG biosynthesis. Indeed, epidemiological studies suggest that serum OPG increases (LaCroix et al., 2013; Liu et al., 2005) and serum RANKL decreases (Liu et al., 2005) as women age. These observations, as well as the positive association between serum OPG levels and hip fracture (LaCroix et al., 2013) led these authors to hypothesize that increased OPG biosynthesis was a response to elevated osteoclastogenesis and bone resorption. If increased RANKL presentation is indeed a major mechanism contributing to postmenopausal osteoporosis, then mimicry of sex steroid hormones in promoting attenuating RANKL presentation could ultimately lead to novel therapeutic approaches to disease management.
Supplementary Material
Acknowledgments
We thank Masashi Honma and Dr. Hiroshi Suzuki (University of Tokyo) for the GFP-RANKL plasmid (Kariya et al., 2009) and Paul Kostenuik (Phylon Pharma Services), and Chun-Ya Han (Amgen) for performing RANKL ELISA. This work was funded by grants 5R01AR064354 to SAK, DK015556 to JAK, and DK071122 to BF, who holds the J. Harold and Edna L. LaBriola Chair in Genetic Orthopaedic Research at the University of Southern California.
Funding information
National Institute of Diabetes and Digestive and Kidney Diseases, Grant numbers: DK015556, DK071122; National Institute of Arthritis and Musculoskeletal and Skin Diseases, Grant number: 5R01AR064354
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, Manolagas SC. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. The Journal of Clinical Investigation. 2013;123:394–404. doi: 10.1172/JCI65910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, Laurent MR, Dubois V, Claessens F, O’Brien CA, Bouillon R, Manolagas SC. Estrogens and androgens in skeletal physiology and pathophysiology. Physiological Reviews. 2017;97:135–187. doi: 10.1152/physrev.00033.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee C, McCabe LR, Choi JY, Hiebert SW, Stein JL, Stein GS, Lian JB. Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. Journal of Cellular Biochemistry. 1997;66:1–8. doi: 10.1002/(sici)1097-4644(19970701)66:1<1::aid-jcb1>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Baniwal SK, Khalid O, Sir D, Buchanan G, Coetzee GA, Frenkel B. Repression of Runx2 by androgen receptor (AR) in osteoblasts and prostate cancer cells: AR binds Runx2 and abrogates its recruitment to DNA. Molecular Endocrinology. 2009;23:1203–1214. doi: 10.1210/me.2008-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baniwal SK, Khalid O, Gabet Y, Shah RR, Purcell DJ, Mav D, Frenkel B. Runx2 transcriptome of prostate cancer cells: Insights into invasiveness and bone metastasis. Molecular Cancer. 2010;9:258. doi: 10.1186/1476-4598-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baniwal SK, Shah PK, Shi Y, Haduong JH, Declerck YA, Gabet Y, Frenkel B. Runx2 promotes both osteoblastogenesis and novel osteoclastogenic signals in ST2 mesenchymal progenitor cells. Osteoporosis International. 2012;2012:1399–1413. doi: 10.1007/s00198-011-1728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartell SM, Han L, Kim HN, Kim SH, Katzenellenbogen JA, Katzenellenbogen BS, Manolagas C. Non-nuclear-initiated actions of the estrogen receptor protect cortical bone mass. Molecular Endocrinology. 2013;27:649–656. doi: 10.1210/me.2012-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido T, Jilka RL, Boyce BF, Girasole G, Broxmeyer H, Dalrymple SA, Manolagas SC. Regulation of interleukin-6, osteoclastogenesis, and bone mass by androgens. The role of the androgen receptor. The Journal of Clinical Investigation. 1995;95:2886–2895. doi: 10.1172/JCI117995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bord S, Ireland DC, Beavan SR, Compston JE. The effects of estrogen on osteoprotegerin, RANKL, and estrogen receptor expression in human osteoblasts. Bone. 2003;32:136–141. doi: 10.1016/s8756-3282(02)00953-5. [DOI] [PubMed] [Google Scholar]
- Chimge NO, Baniwal SK, Little GH, Chen YB, Kahn M, Tripathy D, Frenkel B. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: The role of SNAI2. Breast Cancer Research. 2011;13:R127. doi: 10.1186/bcr3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Gregorio GB, Yamamoto M, Ali AA, Abe E, Roberson P, Manolagas SC, Jilka RL. Attenuation of the self-renewal of transit-amplifying osteoblast progenitors in the murine bone marrow by 17 beta-estradiol. The Journal of Clinical Investigation. 2001;107:803–812. doi: 10.1172/JCI11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douni E, Rinotas V, Makrinou E, Zwerina J, Penninger JM, Eliopoulos E, Kollias G. A RANKL G278R mutation causing osteopetrosis identifies a functional amino acid essential for trimer assembly in RANKL and TNF. Human Molecular Genetics. 2012;21:784–798. doi: 10.1093/hmg/ddr510. [DOI] [PubMed] [Google Scholar]
- Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Molecular and Cellular Biology. 1995;15:1858–1869. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. The Journal of Clinical Investigation. 2003;111:1221–1230. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evinger AJ, 3rd, Levin ER. Requirements for estrogen receptor alpha membrane localization and function. Steroids. 2005;70:361–363. doi: 10.1016/j.steroids.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Frenkel B, Hong A, Baniwal SK, Coetzee GA, Ohlsson C, Khalid O, Gabet Y. Regulation of adult bone turnover by sex steroids. Journal of Cellular Physiology. 2010;224:305–310. doi: 10.1002/jcp.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O’Brien CA. RANKL (receptor activator of NFkappaB ligand) produced by osteocytes is required for the increase in B cells and bone loss caused by estrogen deficiency in mice. The Journal of Biological Chemistry. 2016;291:24838–24850. doi: 10.1074/jbc.M116.742452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabet Y, Noh T, Lee C, Frenkel B. Developmentally regulated inhibition of cell cycle progression by glucocorticoids through repression of cyclin A transcription in primary osteoblast cultures. Journal of Cellular Physiology. 2011;226:991–998. doi: 10.1002/jcp.22412. [DOI] [PubMed] [Google Scholar]
- Garcia AJ, Tom C, Guemes M, Polanco G, Mayorga ME, Wend K, Krum SA. ERalpha signaling regulates MMP3 expression to induce FasL cleavage and osteoclast apoptosis. Journal of Bone and Mineral Research. 2013;28:283–290. doi: 10.1002/jbmr.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy V, Kneissel M, Fournier B, Boyde A, Matthias P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Molecular and Cellular Biology. 2002;22:6222–6233. doi: 10.1128/MCB.22.17.6222-6233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington WR, Kim SH, Funk CC, Madak-Erdogan Z, Schiff R, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen dendrimer conjugates that preferentially activate extranuclear, nongenomic versus genomic pathways of estrogen action. Molecular Endocrinology. 2006;20:491–502. doi: 10.1210/me.2005-0186. [DOI] [PubMed] [Google Scholar]
- Hikita A, Yana I, Wakeyama H, Nakamura M, Kadono Y, Oshima Y, Tanaka S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. The Journal of Biological Chemistry. 2006;281:36846–36855. doi: 10.1074/jbc.M606656200. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. 1999;140:4367–4370. doi: 10.1210/endo.140.9.7131. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Hicok KC, Chen D, Khosla S. Regulation of osteoprotegerin production by androgens and anti-androgens in human osteoblastic lineage cells. European Journal of Endocrinology. 2002;147:269–273. doi: 10.1530/eje.0.1470269. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Takahashi K, Munshi M, Williams DC, Roberson PK, Manolagas SC. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. The Journal of Clinical Investigation. 1998;101:1942–1950. doi: 10.1172/JCI1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimi E, Nakamura I, Amano H, Taguchi Y, Tsurukai T, Tamura M, Suda T. Osteoclast function is activated by osteoblastic cells through a mechanism involving cell-to-cell contact. Endocrinology. 1996;137:2187–2190. doi: 10.1210/endo.137.5.8612568. [DOI] [PubMed] [Google Scholar]
- Kariya Y, Honma M, Aoki S, Chiba A, Suzuki H. Vps33a mediates RANKL storage in secretory lysosomes in osteoblastic cells. Journal of Bone and Mineral Research. 2009;24:1741–1752. doi: 10.1359/jbmr.090409. [DOI] [PubMed] [Google Scholar]
- Kawate H, Wu Y, Ohnaka K, Takayanagi R. Mutual transactivational repression of Runx2 and the androgen receptor by an impairment of their normal compartmentalization. The Journal of Steroid Biochemistry and Molecular Biology. 2007;105:46–56. doi: 10.1016/j.jsbmb.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocrine Reviews. 2008;29:155–192. doi: 10.1210/er.2007-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalid O, Baniwal SK, Purcell DJ, Leclerc N, Gabet Y, Stallcup MR, Frenkel B. Modulation of Runx2 activity by estrogen receptor-alpha: Implications for osteoporosis and breast cancer. Endocrinology. 2008;149:5984–5995. doi: 10.1210/en.2008-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S, Atkinson EJ, Dunstan CR, O’Fallon WM. Effect of estrogen versus testosterone on circulating osteoprotegerin and other cytokine levels in normal elderly men. The Journal of Clinical Endocrinology and Metabolism. 2002;87:1550–1554. doi: 10.1210/jcem.87.4.8397. [DOI] [PubMed] [Google Scholar]
- Khosla S, Oursler MJ, Monroe DG. Estrogen and the skeleton. Trends in Endocrinology and Metabolism. 2012;23:576–581. doi: 10.1016/j.tem.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Katzenellenbogen JA. Hormone-PAMAM dendrimer conjugates: Polymer dynamics and tether structure affect ligand access to receptors. Angewandte Chemie International Edition in English. 2006;45:7243–7248. doi: 10.1002/anie.200601923. [DOI] [PubMed] [Google Scholar]
- Kim SH, Madak-Erdogan Z, Bae SC, Carlson KE, Mayne CG, Granick S, Katzenellenbogen JA. Ligand accessibility and bioactivity of a hormone-dendrimer conjugate depend on pH and pH history. Journal of the American Chemical Society. 2015;137:10326–10335. doi: 10.1021/jacs.5b05952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts [see comments] Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Koromila T, Baniwal SK, Song YS, Martin A, Xiong J, Frenkel B. Glucocorticoids antagonize RUNX2 during osteoblast differentiation in cultures of ST2 pluripotent mesenchymal cells. Journal of Cellular Biochemistry. 2014;115:27–33. doi: 10.1002/jcb.24646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO Journal. 2008;27:535–545. doi: 10.1038/sj.emboj.7601984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCroix AZ, Jackson RD, Aragaki A, Kooperberg C, Cauley JA, Chen Z, Wactawski-Wende J. OPG and sRANKL serum levels and incident hip fracture in postmenopausal Caucasian women in the Women’s Health Initiative Observational Study. Bone. 2013;56:474–481. doi: 10.1016/j.bone.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little GH, Noushmehr H, Baniwal SK, Berman BP, Coetzee GA, Frenkel B. Genome-wide Runx2 occupancy in prostate cancer cells suggests a role in regulating secretion. Nucleic Acids Research. 2012;40:3538–3547. doi: 10.1093/nar/gkr1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little GH, Baniwal SK, Adisetiyo H, Groshen S, Chimge NO, Kim SY, Frenkel B. Differential effects of RUNX2 on the androgen receptor in prostate cancer: Synergistic stimulation of a gene set exemplified by SNAI2 and subsequent invasiveness. Cancer Research. 2014;74:2857–2868. doi: 10.1158/0008-5472.CAN-13-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. The Journal of Cell Biology. 2001;155:157–166. doi: 10.1083/jcb.200105052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JM, Zhao HY, Ning G, Zhao YJ, Chen Y, Zhang Z, Chen JL. Relationships between the changes of serum levels of OPG and RANKL with age, menopause, bone biochemical markers and bone mineral density in Chinese women aged 20–75. Calcified Tissue International. 2005;76:1–6. doi: 10.1007/s00223-004-0007-2. [DOI] [PubMed] [Google Scholar]
- Martin A, Xiong J, Koromila T, Ji JS, Chang S, Song YS, Frenkel B. Estrogens antagonize RUNX2-mediated osteoblast-driven osteoclastogenesis through regulating RANKL membrane association. Bone. 2015;75:96–104. doi: 10.1016/j.bone.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama Z, Yoshida CA, Furuichi T, Amizuka N, Ito M, Fukuyama R, Komori T. Runx2 determines bone maturity and turnover rate in postnatal bone development and is involved in bone loss in estrogen deficiency. Developmental Dynamics. 2007;236:1876–1890. doi: 10.1002/dvdy.21187. [DOI] [PubMed] [Google Scholar]
- Matsumoto C, Inada M, Toda K, Miyaura C. Estrogen and androgen play distinct roles in bone turnover in male mice before and after reaching sexual maturity. Bone. 2006;38:220–226. doi: 10.1016/j.bone.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Modder UI, Sanyal A, Kearns AE, Sibonga JD, Nishihara E, Xu J, Khosla S. Effects of loss of steroid receptor coactivator-1 on the skeletal response to estrogen in mice. Endocrinology. 2004;145:913–921. doi: 10.1210/en.2003-1089. [DOI] [PubMed] [Google Scholar]
- Morimoto E, Li M, Khalid AB, Krum SA, Chimge NO, Frenkel B. Glucocorticoids hijack runx2 to stimulate Wif1 for suppression of osteoblast growth and differentiation. Journal of Cellular Physiology. 2016 doi: 10.1002/jcp.25399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Kato S. Estrogen prevents bone loss via estrogen receptor alpha and induction of fas ligand in osteoclasts. Cell. 2007;130:811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: Modulation of the expression by osteotropic factors and cytokines. Biochemical and Biophysical Research Communications. 2000;275:768–775. doi: 10.1006/bbrc.2000.3379. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Takayanagi H. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nature Medicine. 2011;17:1231–1234. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends in Endocrinology and Metabolism. 2012;23:582–590. doi: 10.1016/j.tem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development [see comments] Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Saika M, Inoue D, Kido S, Matsumoto T. 17beta-estradiol stimulates expression of osteoprotegerin by a mouse stromal cell line, ST-2, via estrogen receptor-alpha. Endocrinology. 2001;142:2205–2212. doi: 10.1210/endo.142.6.8220. [DOI] [PubMed] [Google Scholar]
- Schlondorff J, Lum L, Blobel CP. Biochemical and pharmacological criteria define two shedding activities for TRANCE/OPGL that are distinct from the tumor necrosis factor alpha convertase. The Journal of Biological Chemistry. 2001;276:14665–14674. doi: 10.1074/jbc.M010741200. [DOI] [PubMed] [Google Scholar]
- Ucer S, Iyer S, Kim HN, Han L, Rutlen C, Allison K, Manolagas SC. The effects of aging and sex steroid deficiency on the murine skeleton are independent and mechanistically distinct. Journal of Bone and Mineral Research. 2016 doi: 10.1002/jbmr.3014. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udagawa N, Takahashi N, Akatsu T, Sasaki T, Yamaguchi A, Kodama H, Suda T. The bone marrow-derived stromal cell lines MC3T3-G2/PA6 and ST2 support osteoclast-like cell differentiation in cocultures with mouse spleen cells. Endocrinology. 1989;125:1805–1813. doi: 10.1210/endo-125-4-1805. [DOI] [PubMed] [Google Scholar]
- Vanderschueren D, Laurent MR, Claessens F, Gielen E, Lagerquist MK, Vandenput L, Ohlsson C. Sex steroid actions in male bone. Endocrine Reviews. 2014;35:906–960. doi: 10.1210/er.2014-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Bai ZG, Yin J, Wu GC, Zhang ZT. Role of c-Src activity in the regulation of gastric cancer cell migration. Oncology Reports. 2014;32:45–49. doi: 10.3892/or.2014.3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.