

Abstract
The biological basis underlying differentiation of naïve (NAI) T cells into effector (EFFE) and memory (MEM) cells is incompletely understood. Furthermore, whether NAI T cells serially differentiate into EFFE and then MEM cells (linear differentiation) or whether they concurrently differentiate into either EFFE or MEM cells (parallel differentiation) remains unresolved. We isolated NAI, EFFE, and MEM CD8+ T cell subsets from human peripheral blood and analyzed their gene expression by using microarrays. We identified 156 genes that strongly differentiate NAI, EFFE, and MEM CD8+ T cells; these genes provide previously unrecognized markers to help identify each cell type. Using several statistical approaches to analyze and group the data (standard heat-map and hierarchical clustering, a unique circular representation, multivariate analyses based on principal components, and a clustering method based on phylogenetic parsimony analysis), we assessed the lineage relationships between these subsets and showed that MEM cells have gene expression patterns intermediate between NAI and EFFE T cells. Our analysis suggests a common differentiation pathway to an intermediate state followed by a split into EFFE or MEM cells, hence supporting the parallel differentiation model. As such, conditions under which NAI T cells are activated may determine the magnitude of both EFFE and MEM cells, which arise subsequently. A better understanding of these conditions may be very useful in the design of future vaccine strategies to maximize MEM cell generation.
Keywords: CD8+ T cells, phylogeny, microarray analysis, lineage relationship, differentiation
To maintain an efficient T cell repertoire potentially capable of responding to any pathogen, T cells of diverse specificities are continually produced within the bone marrow throughout life. Those that survive thymic selection populate the periphery initially in a naïve (NAI) state. NAI T cells require encounters with cognate antigens of sufficient magnitude and appropriate additional signals to undergo clonal expansion and differentiation into effector (EFFE) and memory (MEM) cells. The genetic events that underlie this differentiation process are not fully understood. Furthermore, it is unclear whether NAI T cells first differentiate to EFFEs, which then differentiate into MEM cells (linear differentiation), or whether NAI T cells directly differentiate into EFFE and MEM cells simultaneously (parallel differentiation). Although the linear differentiation model is widely accepted (1), recent data appear to support the parallel differentiation model (2, 3).
Microarray technology represents a powerful tool in studying the gene expression changes that underlie the differentiation of NAI to EFFE and MEM T cells. Microarray analysis of murine CD8 T cell subsets has indeed provided unique insights into the process of MEM formation upon virus challenge (4, 5). In humans, the gene expression profiles of peripheral blood mononuclear cells have been analyzed through serial analysis of gene expression (6) and DNA microarrays (7). However, CD8 T cell subsets have not been similarly analyzed to elucidate the gene expression changes that drive MEM differentiation in human lymphocytes.
Recently, microarray analysis of hematopoietic cells demonstrated that gene expression data implicitly contain information about developmental relationships amongst cell types and lineage discrimination (8). Hence, we hypothesized that systematic comparisons of the gene expression patterns of NAI, EFFE, and MEM T cells might also shed light on the issue of linear versus parallel differentiation of T cells. We isolated NAI, EFFE, and MEM (based on expression of CD27 and CD45RA) CD8+ T cell subsets from the peripheral blood and analyzed these subsets on DNA microarrays. As expected, there are significant gene expression differences between these cell types. If each gene within a cell works independently, one could consider genes showing significant expression changes as “votes” in the differentiation process. In the differentiation of NAI to EFFE and MEM T cells, each differentially expressed gene could vote for one of the six possible permutations of gene-expression ordering for the three cell types: (EFFE, MEM, NAI), (EFFE, NAI, MEM), (NAI, MEM, EFFE), (NAI, EFFE, MEM), (MEM, NAI, EFFE), or (MEM, EFFE, NAI).
Our data showed that most of the differentially expressed genes place MEM cells as intermediary between the other two cell types and support the parallel differentiation model.
Materials and Methods
Cell Isolation and Microarray Analysis. Details of the experimental procedures are described in ref. 9. Briefly, peripheral blood mononuclear cells from 10 subjects (5 healthy and 5 melanoma patients) were stained with antibodies specific for CD8, CD27, and CD45R A. Cells were selected for CD8+ and CD27+CD45RA+, CD27+CD45RA–, or CD27–CD45RA+ by means of FACS sorting (FACSVantage, Becton Dickinson). At least 100,000 cells from each group were isolated. Total RNA was extracted by using TRIzol (Invitrogen) and then underwent linear amplification with the Agilent linear amplification kit (Agilent Technologies, Palo Alto, CA). Amplified RNA was quantitated, and quality was determined by using a BioAnalyzer (Agilent Technologies). RNA was labeled by using the Agilent labeling kit and probed onto Agilent human 1 cDNA microarrays. Universal total RNA (Strategene) was used as reference. Hybridized arrays were scanned with the Agilent scanner, and data were extracted by using Agilent feature extraction software (version 7). Thirty arrays (10 of each cell type) were hybridized in five batches of 6 arrays.
Details of the Statistical Analysis. The data were first renormalized, and batch medians were removed so that each batch had the same median (details in ref. 9). Then the multiple testing r (10, 11) package multtest (12) was used to filter the genes that showed statistically significant differential expression patterns between the three cell types. A total of 168 features qualified as statistically significant after adjusting for multiple testing and choosing only the genes significant at the 0.05 adjusted P-value level, meaning that gene expression levels were significantly different across cell types. Note that 24 features were, in fact, duplicates, so they were reduced to single “gene expressions” by taking means. The remaining 156 genes were the main basis for our follow-up univariate and multivariate analyses (see Table 1, which is published as supporting information on the PNAS web site). To describe these differing patterns quantitatively, we developed an angular representation.
The Angular Representation. For each gene, values were categorized into three groups according to cell type: EFFE, MEM, and NAI T cells. Each gene may be visualized in a “box-and-whiskers” plot, or “boxplot,” which captures the 25th, 50th, and 75th quantile of values (the “box”) and the extreme values (“whiskers”). Because there are three groups, there are three boxplots for each gene. The boxplots thus exhibit relative expression changes in the three cell types. Fig. 1 shows boxplots for selectin-L, the IL-7 receptor gene (IL7R), and granzyme B.
Fig. 1.

Boxplots of sample genes demonstrating expression ranges in each cell type: selectin-L (A), IL-7R (B), and granzyme. B (C).
Each boxplot shows how the three cell categories are assigned “up,” “intermediary,” or “down” parity. The angular representation quantifies the boxplots as an (x, y) point and an angle by the following procedure:
Consider the medians of the gene expression levels for EFFE, MEM, and NAI cells.
Take x = median(MEM) – median(EFFE) and y = median(NAI) – median(MEM). These values are the effective “slopes.”
Combine into the coordinate (x, y) and normalize to length 1; i.e., multiply by a constant c so that
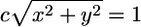 . Alternatively, we also normalize the distance by the standard deviation of the genes, which produces the scatter plot in Fig. 2B.
. Alternatively, we also normalize the distance by the standard deviation of the genes, which produces the scatter plot in Fig. 2B.Map the point onto the unit circle. This mapping results in a continuum of relations between the expression levels of EFFE, MEM, and NAI T cells (Fig. 2A). Note that genes exhibiting similar expression patterns are clumped together on the circle, and genes behaving in an opposite pattern are located diametrically opposite on the circle. See that granzyme B (GZMH) and selectin-L (SELL) are located on opposite sides of the circle.
Attached to each point is an angle starting at 0° at the point (1, 0) and increasing 360° counterclockwise around the circle.
Fig. 2.

Graphical representations of patterns of differential expression for the set of 156 significant genes: angular representation of genes (A), standardized scatter plot (B), and histogram of angles (C).
For the IL-7 receptor as an example, we describe the details of the computation of the angle statistic. Here are the three medians for each class for this gene: EFFE, 2.30; MEM, 3.44; NAI, 2.88. The difference between the median of type MEM and type EFFE is x = 3.44 – 2.30 = 1.14, and the difference between types NAI and MEM is y = 2.88 – 3.44 = –0.56.
We solve 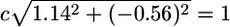 to obtain the normalization factor c = 0.79. Our new normalized point then becomes c · (x, y) = (0.90, –0.44), which is given on the unit circle by the coordinates and defines a unique angle on the circle, 334° (Fig. 2A). In Fig. 2B, the differences in medians between the groups MEM and EFFE, and EFFE and NAI, is divided by the gene's standard deviation. The histogram of the angles represented in Fig. 2A appears in Fig. 2C and is discussed below.
to obtain the normalization factor c = 0.79. Our new normalized point then becomes c · (x, y) = (0.90, –0.44), which is given on the unit circle by the coordinates and defines a unique angle on the circle, 334° (Fig. 2A). In Fig. 2B, the differences in medians between the groups MEM and EFFE, and EFFE and NAI, is divided by the gene's standard deviation. The histogram of the angles represented in Fig. 2A appears in Fig. 2C and is discussed below.
Multivariate Analysis. In the previous analyses, we detected distinct patterns by considering the genes individually. In the multivariate analysis, we consider the interactions and correlations between all genes and cell types. We use principal components analysis to provide simple, low-dimensional maps of the cell types and genes. This technique finds linear combinations of the original variables [called principal components (13)] that have a higher variance than the original variables; thus, with only a few principal components, we can represent a large proportion of the variability in the data.
Results
Significant Gene Expression Differences Among NAI, EFFE, and MEM CD8+ T Cells. As previously reported (9), NAI, EFFE, and MEM CD8+ T cell subsets were segregated based on expression of CD27 and CD45RA and sorted to >95% purity. Total RNA were extracted from these cells and analyzed on cDNA microarrays after linear amplification. Cells from 10 individuals (5 with stage III/IV melanoma and 5 healthy controls) were analyzed. At the scale of gene expression differences between cell types, the differences between melanoma and healthy T cells are imperceptible. Thus, it was reasonable to combine data from the melanoma and healthy groups for the purpose of studying differences between T cell subsets.
We filtered the genes that show statistically significant differences between gene expressions and among the three cell-type groups. The groups were NAI (CD27+ and CD45RA+), EFFE (CD27– and CD45RA+), and MEM (CD27+ and CD45RA–). A univariate analysis of the data showed 156 genes that were significantly differentially expressed amongst the three subsets.
Angular Representation. Using the angular representation, we have made a point on the circle for every significant gene. A histogram of the angles shows that 62 genes have angles between 0° and 90° (Fig. 2C). In particular, we take note of the following groups of genes (see the supporting information for complete lists).
EFFE down, MEM intermediary, NAI up. See Table 2, which is published as supporting information on the PNAS web site. Selectin-L (CD62L) and CC chemokine receptor 7 (CCR7) are both molecules that facilitate T cell homing to lymphoid tissues; they are expressed predominantly by NAI T cells and are expressed at intermediate levels by MEM T cells (14).
EFFE down, MEM up, NAI down. See Table 3, which is published as supporting information on the PNAS web site. Ahmed and coworkers (2) recently showed that the IL-7 receptor is selectively expressed in MEM T cells. The T cell receptor-interacting molecule is a recently identified transmembrane adaptor protein that is exclusively expressed in mature T cells (15).
EFFE up, MEM intermediary, NAI down. See Table 4, which is published as supporting information on the PNAS web site. Granzyme B is a cytolytic mediator that is expressed at high levels by EFFE CD8 T cells and also is expressed at intermediate levels in MEM CD8 T cells (16). It was recently shown that T cells express certain natural killer receptors, such as CD94 (labeled KLRD1 from hereon), upon activation (14), which is thought to modulate T cell activity.
EFFE up, MEM up, NAI down. See Table 5, which is published as supporting information on the PNAS web site. CD58 (LFA-3) is an adhesion molecule that is known to be expressed on activated and MEM T cells (4). HLA-DR is a major histocompatibility complex (MHC) class II molecule that is normally expressed on antigen-presenting cells; it is also expressed in T cells after activation (17).
EFFE up, MEM down, NAI up. See Table 6, which is published as supporting information on the PNAS web site. None of the significant genes had an expression pattern in which the MEM group is less expressed than the other two groups. This observation is illustrated in the upper-left quadrant of Fig. 2A, and the gap in the histograms of the angles of Fig. 2C.
Aggregate Expression Analyses Reveal Nonuniform Distribution. If we considered the genes as independent “voters,” the overwhelming vote was for the MEM cells as an intermediary type between NAI and EFFE. However, because genes do not act independently, a multivariate analysis that accounts for the correlations between genes was performed. The multivariate eigenanalysis approach shows that we can represent >80% of the information in the 156-row matrix of significant genes in a map on one principal plane chosen by principal components analysis (Fig. 3). A three-pronged structure emerges, suggesting a central intermediary state (InterMEM) between the three cell types that is closer to the MEM state than to the NAI or EFFE states.
Fig. 3.

First principal plane from the principal components analysis of the matrix of significant genes in all 30 samples.
Hierarchical Relationships. We also performed hierarchical clustering of these data, as shown in Fig. 4A. Almost all genes show the MEM group as intermediary between the other two very distinct cell types, with either higher expressions for NAI or EFFE. In the same spirit as ref. 8, we performed a phylogenetic analysis, considering the genes to be either expressed, underexpressed, or off. These three categories of expression are considered ordinal. Genes move from not expressed to expressed in one step, and from underexpressed to expressed in two steps. Using this assumption, we built the maximum parsimony tree of all of the arrays by using the 156 significant genes with the phylip* function dnapars. Fig. 4B shows such a tree. As in the case of hierarchical clustering, the tree shows how the cells group together according to type.
Fig. 4.

Tree representations of arrays: hierarchical clustering of arrays (A) and parsimony tree (B).
We identified several genes that, on their own, allow for the discrimination between the three groups. These genes add to the currently accepted markers of CD27 and CD45RA and include: CD164L1, syntaphilin, and IFN-γ receptor 2 for NAI cells; vinculin, granzyme B, and KLRD1 for EFFE cells; and T cell receptor-interacting molecule and granzyme K for MEM cells.
Discussion
In this study, NAI, EFFE, and MEM T cells were identified and sorted based on their expression of two surface markers, CD27 and CD45RA, an accepted combination of markers to delineate these subsets (18). Using DNA microarray analysis, we identified 156 genes that strongly differentiate NAI, EFFE, and MEM CD8+ T cells in the peripheral blood. NAI T cells are known to express CD45RA, CCR7, CD62L, CD27, and CD28; activated T cells, however, lose expression of CD62L and CD45RA and up-regulate activation markers such as HLA-DR and effector molecules such as granzyme (18). Importantly, although NAI T cells were sorted based on CD27 and CD45RA, these cells were found to have elevated expression of CCR7 and CD62L, providing an internal validation. Similarly, sorted EFFE cells were found to express HLA-DR and granzyme B. It should be pointed out that in the phenotypic analysis of circulating CD8+ T cells to identify unique functional subsets, many intermediate phenotypes exist, which may represent transient states. Nonetheless, gene expression data generated from the three subsets defined by CD27 and CD45RA fit well with known properties of NAI, EFFE, and MEM T cells and therefore represent reasonable composites of these three idealized states. This analysis focused on genes differentially expressed in all three cell types and thus does not include genes that are expressed similarly in two cell types and are different in the other.
The hallmarks of MEM T cells include longevity and more rapid proliferation and acquisition of EFFE function upon antigen encounter than NAI T cells (19). It is now known that MEM T cells are not uniform with respect to function, proliferative potential, trafficking patterns, and, hence, gene expression. MEM T cells may be divided into central and effector subtypes, based on expression of the chemokine receptor CCR7 (20). Central MEM T cells home to and mainly reside within lymph nodes; EFFE MEM cells home to target tissues. Indeed, data from this study demonstrate that circulating MEM CD8+ T cells (based on CD27+ CD45RA–) express intermediate levels of CCR7, suggesting that these cells constitute a mixture of EFFE and central MEM T cells. Although analysis of MEM cells further segregated into these two subtypes could yield additional information, the gene expression patterns we identified for MEM cells represent a composite of the two MEM subtypes, thus focusing on genes that underlie the memory state.
We used several statistical approaches to analyze and group data from these arrays: standard heat-maps and hierarchical clustering, a unique circular representation, multivariate analyses based on principal components, and a clustering method based on phylogenetic parsimony analysis. For the 156 genes found to differentiate between the three subsets, we assigned each gene a value of 0–360, corresponding to degrees in a circle, to characterize its expression in NAI, EFFE, and MEM cells. When the value for each gene is plotted onto a circle, a dramatic absence of genes in an entire region emerged (Fig. 2), which corresponds to genes underexpressed in MEM cells in relation to NAI and EFFE cells. These data suggest that of genes that differentiate NAI, EFFE, and MEM T cells, once a gene is turned on within MEM cells, it does not get turned off.
Another striking observation arose from principal components analysis. Of a total of 30 samples analyzed, 80% of the variability fell onto only two dimensions, suggesting a very highly ordered structure at play in regulating NAI, EFFE, and MEM differentiation. From all of these analyses, MEM cells consistently fall between NAI and EFFE, being closer to EFFE than to NAI. This observation is also confirmed by phylogenetic tree and heat-map analysis, suggesting a biological explanation of why MEM cells can differentiate much more rapidly into EFFEs than NAI T cells (19). These data are also in agreement with the elegant data in mice from Ahmed and coworkers (4) that showed that upon activation, a fraction of NAI T cells acquire IL-7R expression, and these cells differentiate directly into MEM cells without going through an EFFE stage.
The analysis of lineage relationships between cells represents a previously unrecognized use of microarray data. A recent report (8) used phylogenetic analysis of gene expression data to show that mRNA implicitly contained information about developmental relationships among cell types. Our phylogenetic tree analysis shows that MEM cells have gene expression patterns between NAI and EFFE T cells. Furthermore, principal component analysis shows that an intermediate state (InterMEM) exists that is closest to a MEM type, suggesting a model in which, upon activation, NAI T cells first go to this intermediary state. Most cells then go on to become EFFEs, but some become MEM cells. This model is consistent with that proposed by Ahmed and colleagues (2). Furthermore, MEM cells may go back to this intermediary state subsequent to activation to become EFFEs. The finding that gene expression differences between MEM cells and the intermediary state are much less than those between NAI cells and the intermediary state is consistent with the long-held observations that MEM T cells can differentiate into EFFEs much more rapidly than NAI T cells (19). Another finding of our data that strongly supports the notion that MEM T cells are intermediate between NAI and EFFE cells is that there are only eight genes whose expression patterns are not intermediary between those of NAI and EFFEs. Hence, these eight genes may serve as an important basis for the memory T cell state.
A theoretical concern may be that if genes in MEM cells consist of two subgroups, ones that group with NAI cells and ones (e.g., those responsible for proliferation) that group with EFFEs, then the composite gene expression pattern would appear as intermediate. If this were the case, the boxplots would show MEM at a similar level to one of the other types (either like NAI or EFFE), and the angles of the boxplots would be concentrated only at the four extremes of the circle: (0, 0), (0, 1), (–1, 0), and (0, –1). To address this possibility, we performed simulations generating 100 genes with a pattern tailored to have similar genewise medians and standard deviations as the data, but showing this double grouping. These simulated genes produce heat-map and angular plots that are not at all comparable to the ones from the actual data, thus making this scenario very unlikely (see Fig. 5, which is published as supporting information on the PNAS web site).
Taken together, our results confirm that a significant number of differentially expressed genes underlie the differentiation from NAI to EFFE and MEM T cells. Many genes in each group fit well with known biological differences between these subsets, providing internal validation. Additional genes not previously known to be uniquely associated with one subtype over the others were discovered. These genes may serve as previously unrecognized markers to delineate subsets. Importantly, to our knowledge, this is the first study to use gene expression patterns to determine lineage relationship between T cell subsets. Through several methods, including angular presentation and phylogenetic tree analysis, MEM T cells consistently show gene expression patterns intermediate to NAI and EFFE T cells. These findings fit known properties of MEM T cells to rapidly acquire EFFE functions and suggest that NAI T cells may differentiate into EFFE and MEM cells concurrently, hence supporting the parallel differentiation model. As such, these data also suggest that conditions under which NAI T cells are activated may determine the ratio between EFFE and MEM cells that arises subsequently. A better understanding of these conditions may be very useful in the design of future vaccine strategies in which the goal is to maximize MEM cell generation.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation Division of Mathematical Sciences Grant 0241246, the American Cancer Society, and the Stanford Vice Provost for Undergraduate Education Summer Research program.
Author contributions: S.H. and P.P.L. designed research; S.H., M.H., T.X., and P.P.L. performed research; S.H., M.H., T.X., and P.P.L. contributed new reagents/analytic tools; S.H., M.H., and P.P.L. analyzed data; and S.H., M.H., and P.P.L. wrote the paper.
Abbreviations: NAI, naïve; EFFE, effector; MEM, memory; CCR7, CC chemokine receptor 7.
References
- 1.Opferman, J. T., Ober, B. T. & Ashton-Rickardt, P. G. (1999) Science 283, 1745–1748. [DOI] [PubMed] [Google Scholar]
- 2.Kaech, S., Tan, J., Wherry, E., Konieczny, B., Surh, C. & Ahmed, R. (2003) Nat. Immunol. 4, 1191–1198. [DOI] [PubMed] [Google Scholar]
- 3.Manjunath, N., Shankar, P., Wan, J., Weninger, W., Crowley, M. A., Hieshima, K., Springer, T. A., Fan, X., Shen, H., Lieberman, J. & von Andrian, U. H. (2001) J. Clin. Invest. 108, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaech, S., Hemby, S., Kersh, E. & Ahmed, R. (2002) Cell 111, 837–851. [DOI] [PubMed] [Google Scholar]
- 5.Manjunath, N., Shankar, P., Stockton, B., Dubey, P. D., Lieberman, J. & von Andrian, U. H. (1999) Proc. Natl. Acad. Sci. USA 96, 13932–13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hashimoto, S., Nagai, S., Sese, J., Suzuki, T., Obata, A., Sato, T., Toyoda, N., Dong, H.-Y., Kurachi, M., Nagahata, T., et al. (2003) Blood 101, 3509–3513. [DOI] [PubMed] [Google Scholar]
- 7.Panelli, M., Wang, E., Phan, G., Puhlmann, M., Miller, L., Ohnmacht, G., Klein, H. & Marincola, F. (2002) Genome Biol. 3, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kluger, Y., Tuck, D., Chang, J., Nakayama, Y., Poddar, R., Kohya, N., Lian, Z., Ben Nasr, A., Halaban, H., Krause, D., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 6508–6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu, T., Shu, C., Purdom, E., Dang, D., Ilsley, D., Guo, Y., Weber, J., Holmes, S. & Lee, P. (2004) Cancer Res. 64, 3661–3667. [DOI] [PubMed] [Google Scholar]
- 10.Ihaka, R. & Gentleman, R. (1996) J. Comput. Graphical Stat. 5, 299–314. [Google Scholar]
- 11.Dudoit, S., Gentleman, R. C. & Quackenbush, J. (2003) BioTechniques, Suppl., 45–51. [PubMed]
- 12.Ge, Y., Dudoit, S. & Speed, T. P. (2003) Test 12, 1–44. [Google Scholar]
- 13.Mardia, K. V., Kent, J. T. & Bibby, J. M. (1979) Multivariate Analysis (Academic, New York).
- 14.Moser, J., Gibbs, J., Jensen, P. & Lukacher, A. (2002) Nat. Immunol. 3, 189–195. [DOI] [PubMed] [Google Scholar]
- 15.Kirchgessner, H., Dietrich, J., Scherer, J., Isomaki, P., Korinek, V., Hilgert, I., Bruyns, E., Leo, A., Cope, A. & Schraven, B. (2001) J. Exp. Med. 193, 1269–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelso, A., Costelloe, E., Johnson, B., Groves, P., Buttigieg, K. & Fitzpatrick, D. (2002) Int. Immunol. 14, 605–613. [DOI] [PubMed] [Google Scholar]
- 17.Abbas, A. & Lichtman, A. (2003) Cellular and Molecular Immunology (Saunders, Philadelphia).
- 18.Hamann, D., Baars, P. A., Rep, M. H. G., Hooibrink, B., Kerkhof-Garde, S. R., Klein, M. R. & van Lier, R. A. W. (1997) J. Exp. Med. 186, 1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barber, D., Wherry, E. & Ahmed, R. (2003) J. Immunol. 171, 27–31. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. (1999) Nature 401, 708–712. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.