

Abstract
The tetratricopeptide repeat (TPR) is a 34-aa α-helical motif that occurs in tandem arrays in a variety of different proteins. In natural proteins, the number of TPR motifs ranges from 3 to 16 or more. These arrays function as molecular scaffolds and frequently mediate protein–protein interactions. We have shown that correctly folded TPR domain proteins, exhibiting the typical helix–turn–helix fold, can be designed by arraying tandem repeats of an idealized TPR consensus motif. To date, three designed proteins, CTPR1, CTPR2, and CTPR3 (consensus TPR number of repeats) have been characterized. Their high-resolution crystal structures show that the designed proteins indeed adopt the typical TPR fold, which is specified by the correct positioning of key residues. Here, we present a study of the thermodynamic properties and folding kinetics of this set of designed proteins. Chemical denaturation, monitored by CD and fluorescence, was used to assess the folding and global stability of each protein. NMR-detected amide proton exchange was used to investigate the stability of each construct at a residue-specific level. The results of these studies reveal a stable core, which defines the intrinsic stability of an individual TPR motif. The results also show the relationship between the number of tandem repeats and the overall stability and folding of the protein.
Keywords: protein, folding, stability, hydrogen exchange
Repeat proteins contain tandem arrays of modules with similar amino acid sequences. The modules range in length from ≈20 to 40 aa and comprise a variety of simple structural motifs such as α/β, all α, and all β (1, 2). These motifs stack in tandem to form elongated nonglobular folds that differ radically in structure from globular proteins. The recurring nature of repeat proteins gives rise to a pattern of short-range regularized interactions, whereas globular proteins have more complex topologies that frequently contain many long-range interactions. Repeat proteins often function as mediators of protein–protein interactions (2), although the relationship between the number of repeats and functional avidity is far from clear.
The tetratricopeptide repeat (TPR) is a sequence of 34 aa encoding an α-helix–turn–α-helix motif (3, 4). The individual TPRs stack on top of each other, with between 3 and 16 direct repeats in a given protein domain (5). The regular, elongated structure that results can be visualized as a spiral staircase in which the individual TPR motifs are the steps (Fig. 1). TPRs have been identified in >300 proteins, whose functions range from cell-cycle control to transcriptional regulation, protein transport, protein folding, and neurogenesis (3, 5). The uniform secondary structure and regular tertiary structure of TPR domains makes them of particular interest with respect to both their folding and stability and the specificity of their binding interactions.
Fig. 1.

Structure of the consensus TPR proteins. (A) CTPR1. (B) CTPR2. (C) CTPR3. Repeat 1 is shown in yellow, repeat 2 is red, repeat 3 is blue, and the solvating helix is green. CTPR1 is a model based on the CTPR2 structure. CTPR2 and -3 are crystal structures (7). (D) The amino acid sequences of CTPR proteins (7). The N cap and consensus TPR motif (yellow) and solvating helix (green) sequence are displayed with the corresponding sequence underneath. The highly conserved residues are boxed in yellow (>50% conservation of an identical residue) and orange (50% of similar residue type).
To explore how the amino acid sequence specifies both the fold and stability of TPR proteins, we designed an idealized 34-aa repeat (6). We used a statistical approach to design a consensus TPR and repeated this motif one, two, and three times in tandem to create the designed proteins CTPR1, CTPR2, and CTPR3 (consensus TPR number of repeats) (Fig. 1). All three proteins are well folded in solution, and the high-resolution x-ray crystal structures of CTPR2 and -3 demonstrate that they adopt the TPR fold (6) (Fig. 1). Interestingly, in natural proteins a unit containing three tandem TPR motifs is the smallest and most prevalent in all organisms, which we suggest may reflect a functional rather than a structural requirement (5).
Our initial characterizations showed that in addition to achieving the desired fold, our designs generated proteins that are substantially more thermally stable than their natural counterparts. CTPR3 has a thermal melting point of 83°C, whereas the related and similarly sized 3-TPR domain from the protein PP5 has a melting temperature of 47°C (L. D'Andrea and L.R., unpublished observations). As the number of TPR repeats in the CTPR series increases, the thermal stability of the proteins also increases, with melting temperatures of 49, 74, and 83°C, for CTPR1, -2, and -3, respectively.
In this study, we investigate in greater detail the stability and folding kinetics of individual and repeated arrays of identical TPR motifs, by using chemical denaturation and NMR-detected hydrogen/deuterium (H/D) exchange. The results of these studies reveal a stable core of an individual TPR motif and illustrate how the stability of an individual motif is modulated as the number of flanking TPR motifs increases. These studies, to our knowledge, are the first to explore in detail the relationship between the number of repeats and both folding kinetics and global/local stability in a linear repeat protein.
Materials and Methods
Materials. High-purity GuHCl was obtained from American Bioanalytical (Natick, MA). All other chemicals were from American Bioanalytical, Sigma, or Fluka.
Methods. Cloning, protein production, and purification. The designed proteins CTPR1, -2, and -3 were cloned, expressed, and purified as described in ref. 6.
Equipment and general procedures. In all equilibrium, kinetic, and NMR experiments, 50 mM phosphate (pH 6.8)/150 mM NaCl (CTPR buffer) was used. The molarity of the stock solutions of both urea and GuHCl were determined by using a refractometer (model NAR-1T, Atago, Tokyo).
Equilibrium experiments. Fluorescence and far-UV CD equilibrium unfolding measurements were performed and analyzed as described in ref. 7. Fluorescence measurements were made on a Series 2 Luminescence Spectrometer (SLM–Aminco Bowman, Urbana, IL); the excitation wavelength was 280 nm, and the emission at 337 nm (for CTPR1) or 335 nm (for CTPR2 and CTPR3) was recorded. Ellipticity at 222 nm was measured by using an Aviv CD spectrometer (model 215, Aviv Biomedical, Lakewood, NJ). Protein concentrations were typically 2–30 μM.
Kinetic experiments. Kinetic experiments were performed on an Applied Photophysics SX 18MV-R Stopped Flow Spectrometer (Surrey, U.K.) as described in ref. 8. Both unfolding and folding phases fitted well to a single-exponential process. No slow, Pro isomerization phases were observed in the refolding experiments over a 200-s time scale. The rate constants obtained were measured as a function of denaturant concentration, and the whole data set fit as described in ref. 8.
NMR H/2H exchange. Details of the assignment strategy and spectra for CTPR2 and CTPR3 are given in Supporting Materials and Methods, which is published as supporting information on the PNAS web site. NMR samples were lyophilized from CTPR buffer and resuspended in an equal volume of D2O at room temperature. HSQC experiments were collected at 20°C (CTPR3, 800 MHz; CTPR2, 600 MHz) for the first 24 h, then the number of scans was doubled, and data were collected over the next 5–40 days. The time base for the decay of the signals was taken to be the middle of the acquisition time.
NMR H/2H exchange analysis. The proportion of each proton that remained unexchanged with D2O at each time point was determined by calculating the height of the appropriate peak by using azara (9). The decay of signal for each proton was then fitted to a single exponential to obtain rate constants for the observed exchange reaction (kex) by using the program kaleidagraph (Synergy Software, Reading, PA). To quantitatively compare exchanges rates for each position in the CTPR proteins, protection factors at each amino acid residue were calculated (10) by using the program sphere (11). At sites in the CTPR proteins that are equivalent, we also can obtain ΔΔGex, the change in free energy for exchange upon extension of a TPR repeat, by using Eq. 1 as follows:
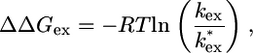 |
[1] |
where kex is the rate constant for exchange of the proton in one CTPR protein and k*ex is the exchange rate for the equivalent proton in a larger CTPR protein. In this analysis, we make the assumption that, under these relatively benign conditions (20°C, pH 6.8), protons in the CTPR proteins exchange by the commonly observed EX2 mechanism in which the exchange rate is proportional to the equilibrium between exchange-competent and -incompetent forms (12, 13). Such an assumption is reasonable because kF (kcl) is ≈20,000–35,000 s–1 (for CTPR2 and -3, respectively), whereas kint of the slowest exchanging residues is ≈1–10 s–1. In this case, the calculated global free energy of unfolding, ΔGappex (Eq. 2), can be calculated from the most slowly exchanging backbone amide protons of CTPR2 and -3.
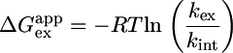 |
[2] |
Results
Design and Structure. The 34-aa repeat of the TPR was constructed by defining consensus residues from a statistical analysis, by using a database of >2,000 aligned TPR sequences. By using a consensus design, the key fold-determining amino acids, which specify the 3D structure of the protein, were incorporated. The full details of the design and structural characterization are described in ref. 6. Fig. 1 shows the designed sequences, a model of CTPR1, and the x-ray crystal structures of CTPR2 and -3, which adopt the typical TPR fold. We are using the same nomenclature for residues within the CTPR proteins as described in ref. 6. For example, a residue called Trp4(A1) represents a Trp at the fourth residue in helix A of the first TPR motif.
It is important to emphasize one striking and unique feature of our design, the identical modular nature of the CTPR proteins. When each of the TPR repeats of CTPR2 and -3 are aligned, the rmsdeviationoftheirCα atoms is <0.4 Å. We have produced arrays that have identical interactions both between different TPR motifs within the same CTPR protein and between the same TPR repeats in different CTPR proteins.
Apparent Equilibrium Stability from Chemical Denaturation Studies. To determine how the apparent global stability of the CTPR proteins varies with the number of tandem TPR motifs, chemical denaturation studies were performed.
For each protein, when the fluorescence or ellipticity was monitored as a function of denaturant concentration, a reversible and sharp denaturation transition was observed (Fig. 2 A and B; see also Fig. 4, which is published as supporting information on the PNAS web site). Data were fit as described in ref. 7 to yield apparent values for [D]50% (midpoint of unfolding), mD–N, and 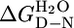 (the apparent free energy of unfolding in water) (see Table 1). As the number of repeats increases, both the [D]50% and, hence, apparent global stability increase. For example, the
(the apparent free energy of unfolding in water) (see Table 1). As the number of repeats increases, both the [D]50% and, hence, apparent global stability increase. For example, the 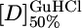 values at 20°C for CTPR1, -2, and -3 are 0.9, 2.6, and 3.4 M, respectively. For each protein, the apparent
values at 20°C for CTPR1, -2, and -3 are 0.9, 2.6, and 3.4 M, respectively. For each protein, the apparent 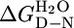 calculated from different denaturants were essentially the same, showing that the apparent
calculated from different denaturants were essentially the same, showing that the apparent 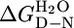 of the CTPR proteins is not affected by the increased salt concentration present when using GuHCl.
of the CTPR proteins is not affected by the increased salt concentration present when using GuHCl.
Fig. 2.

Global and residue-specific stability of CTPR1, -2, and -3. (A) GuHCl-induced equilibrium unfolding monitored by CD (CTPR1, ○; CTPR2, ▴; CTPR3, ▪) and fluorescence (CTPR2, ▵; CTPR3, □) at 25°C. (B) Urea-induced equilibrium unfolding of CTPR1 (□), CTPR2 (•), and CTPR3 (○) monitored by fluorescence at 20°C. (C) [GuHCl]-dependence of lnkF and lnkU for CTPR2 (•) and CTPR3 (○) at 20°C. The continuous curves show the best fit of the kinetic data (8). (D) Typical HSQC spectra of CTPR3 in H2O (black) with the HSQC spectra of CTPR3 after 2 h and 26 min of exchange (red) superimposed over it. The resonances of the side-chain indole NHε1 of the Trp residues are highlighted at the bottom left of the spectra. (E) Histogram showing the protection factors calculated from the exchange data for amides protons of CTPR2 (blue) and CTPR3 (red) vs. residue number.
Table 1. Thermodynamic parameters for equilibrium unfolding of CTPR proteins.
| Experiment | [D]50%,† M | mD—N,† kcal·mol-1·M-1 |
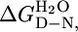 ,‡ kcal·mol-1 ,‡ kcal·mol-1
|
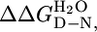 ,§ kcal·mol-1 ,§ kcal·mol-1
|
|---|---|---|---|---|
| 25°C GuHCl denaturation, CD and fluorescence as structural probes | ||||
| CTPR1, CD | 0.8 ± 0.01 | 1.9 ± 0.1 | 1.5 ± 0.1 | — |
| CTPR2, CD | 2.6 ± 0.01 | 2.6 ± 0.1 | 6.8 ± 0.2 | 5.3 ± 0.2 |
| CTPR2, Fluorescence | 2.6 ± 0.01 | 2.6 ± 0.1 | 6.6 ± 0.2 | 5.1 ± 0.2 |
| CTPR3, CD¶ | 3.4 ± 0.01¶ | 3.1 ± 0.1¶ | 10.7 ± 0.2 | 3.9 ± 0.3 |
| CTPR3, Fluorescence | 3.4 ± 0.01 | 3.1 ± 0.1 | 10.5 ± 0.4 | 3.9 ± 0.5 |
| 20°C GuHCl denaturation, fluorescence as structural probe | ||||
| CTPR1 | 0.9 ± 0.02 | 1.8 ± 0.1 | 1.5 ± 0.1 | — |
| CTPR2 | 2.6 ± 0.01 | 2.7 ± 0.1 | 7.1 ± 0.3 | 5.5 ± 0.3 |
| CTPR3 | 3.4 ± 0.01 | 3.0 ± 0.1 | 10.1 ± 0.4 | 3.1 ± 0.5 |
| 20°C Urea denaturation, fluorescence as structural probe | ||||
| CTPR1 | 1.9 ± 0.06 | 1.0 ± 0.1 | 1.9 ± 0.2 | — |
| CTPR2 | 4.9 ± 0.03 | 1.4 ± 0.1 | 6.9 ± 0.3 | 5.1 ± 0.4 |
| CTPR3 | 6.2 ± 0.02 | 1.5 ± 0.1 | 9.3 ± 0.5 | 2.3 ± 0.6 |
| 4°C GuHCl and urea denaturation, fluorescence as structural probe | ||||
| CTPR1—GuHCl | 1.1 ± 0.02 | 2.0 ± 0.2 | 2.1 ± 0.2 | — |
| CTPR1—Urea | 2.2 ± 0.06 | 1.0 ± 0.1 | 2.2 ± 0.2 | — |
The errors reported are from the fitting of the experimental data.
Calculated as described in ref. 7.
The difference in stability between two CTPR proteins, calculated as 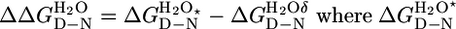 &
& 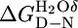 are the stability of the two different CTPR proteins.
are the stability of the two different CTPR proteins.
When repeated in triplicate, [D]50% = 3.4 ± 0.01 M, mD—N = 3.0 ± 0.1 kcal·mol-1M-1 (mean value ± 1 SD of the mean). Thus, [D]50% and mD—N are in the order 0.05 and 0.1, respectively.
Unfolding/Refolding Kinetics of CTPR Proteins. To investigate the origin of the increase in stability between CTPR1, -2, and -3, their folding and unfolding kinetics were studied. These kinetics were observed to be extremely rapid, which limited our study to the CTPR2 and -3 proteins and to GuHCl concentrations between ≈1.5 and 4.5 M. Fig. 2C shows the chevron plots of lnk vs. [GuHCl] for CTPR2 and -3. These plots illustrate that the addition of a TPR motif affects the rates of both folding and unfolding. However, although the folding rate does increase slightly (factor of 1.8), the greater effect on moving from CTPR2 to CTPR3 is a considerable decrease of the unfolding rate (factor of 36). Thus, as each TPR motif is added to the CTPR proteins, the increase in global stability stems primarily from a decrease in the rate of unfolding (see Tables 2 and 3 and Fig. 2C; ref. 8).
Table 2. Comparison of thermodynamic parameters measured from equilibrium, kinetic, and H/D exchange experiments.
| Equilibrium, GuHCl
|
Kinetic, GuHCl
|
H/D exchange, 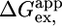 , kcal·mol-1 , kcal·mol-1
|
|||
|---|---|---|---|---|---|
| Experiment |
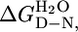 , kcal·mol-1 , kcal·mol-1
|
mD—N, kcal·mol-1·M-1 |
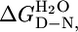 , kcal·mol-1 , kcal·mol-1
|
mKin,† kcal·mol-1·M-1 | |
| CTPR1 | 1.5 ± 0.1 | 1.8 ± 0.1 | N.D.‡ | N.D‡ | N.D.§ |
| CTPR2 | 7.1 ± 0.3 | 2.7 ± 0.1 | 6.4 ± 0.7 | 2.5 ± 0.3 | 7.8¶ |
| CTPR3 | 10.1 ± 0.4 | 3.0 ± 0.1 | 8.8 ± 0.5 | 2.6 ± 0.2 | 9.5¶ |
All experiments were carried out at 20°C and in CTPR buffer unless otherwise stated. Errors reported are from the fitting of experimental data.
Calculated using mKin = RT(mkF + mkF) where RT = 0.582 kcal·mol-1 at 20°C.
Not determined because folding and unfolding kinetics occurred within the deadtime of the stopped-flow spectrometer.
Not determined because exchange occurred more rapidly than data could be collected.
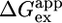 was calculated from the slowest exchanging residue within the protein (Fig. 4A).
was calculated from the slowest exchanging residue within the protein (Fig. 4A).
Table 3. Kinetic parameters from unfolding and refolding experiments on CTPR proteins.
| Protein |
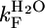 , s-1 , s-1
|
τ,* μs | mkF, M-1 |
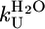 , s-1 , s-1
|
mkU, M-1 | βT kin† | βT eq.‡ |
|---|---|---|---|---|---|---|---|
| CTPR2 | 19,860 ± 6,692 | 50 | 2.24 ± 0.19 | 0.35 ± 0.21 | 2.06 ± 0.18 | 0.5 | 0.4 |
| CTPR3 | 35,032 ± 7,601 | 29 | 2.20 ± 0.10 | 0.010 ± 0.005 | 2.34 ± 0.11 | 0.5 | 0.4 |
CTPR1 was too fast to measure using stopped-flow spectrometer (deadtime was ≈0.5 ms).
Calculated from 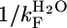 .
.
Calculated from kinetic data only, βT = mkF/(mkF + mkU).
Calculated from kinetic and equilibrium data, βT = RT (mkF/mD-N), where RT = 0.582 kcal·mol-1 at 20°C.
Hydrogen Exchange Kinetics. It is of considerable interest to also examine the stability of the CTPR proteins on a residue-specific basis. Such studies identify the stable core of an individual TPR motif and also report on how this stability changes when the TPR motif is part of a longer array.
In the CTPR proteins, only 12 different amino acid types are used in the consensus sequence, and just three residue types predominate: 15.2% Ala, 15.2% Asn, and 14.4% Tyr. Furthermore, because each of the TPR modules is near identical in sequence and structure, there is significant peak overlap (6). Nevertheless, we have assigned the majority of the backbone nitrogen and proton resonances of CTPR1, -2, and -3 (Fig. 2D; see also Fig. 5, which is published as supporting information on the PNAS web site). Unique probes are therefore available throughout the protein structure to report on the dynamics and stability on a residue basis. The exchange of individual backbone and side-chain amide protons with deuterated solvent was monitored by using NMR.
CTPR1. Under the conditions in which the exchange experiments were performed (20°C, pH 6.8), the amide protons of CTPR1 exchange with solvent more rapidly than the first 1D spectrum can be acquired (Fig. 5). For the purposes of this work, we did not explore further the effect of temperature and pH on the exchange behavior of CTPR1.
CTPR2 and CTPR3. Both CTPR2 and -3 possess a substantial number of protons that are protected from rapid exchange with D2O. These consist of 33 amide protons and 1 indole NHε1 for CTPR2 and 44 amide protons and at least one indole NHε1 for CTPR3 (Figs. 2D and 4). In addition, CTPR3 contains a further six protected sites that resonate at positions that consist of two overlapping residues. These sites are consistent with identical residues in the same positions in either the second or third TPR motif. Of these sites, two have different enough exchange rates that they fit sufficiently well to a double exponential, allowing the slower rate to be assigned to the residue in the second TPR motif and the faster to the third TPR motif. Finally, CTPR2 and -3 contain two and four protected sites, respectively, that cannot be assigned because of overlap.
When the protection factors of CTPR2 and -3 are studied, the overall pattern along the backbone was found to be similar, although the magnitude of the protection at different positions was different (Figs. 2E and 3). For both proteins, little or no protection is seen for the helices that start and terminate the structure (A-helix of TPR1 and the C-terminal solvating helix). Increasing protection from exchange is seen moving inward from both N and C termini. The highest protection is observed along the central helix within the array. This protection corresponds to helix A in the second TPR motif of CTPR2 and helix B in the second TPR motif of CTPR3. Thus, in CTPR2 and -3, the central, most stable, region of the protein is identified. The stability of CTPR2 and -3 that we calculate from the exchange rates of these slowest exchanging protons, assuming an EX2 exchange mechanism, is consistent with the global stability of the protein calculated from the GuHCl denaturation experiments (Table 2).
Fig. 3.

Comparison of the H/D exchange rates of CTPR2 and -3. (A) Histogram showing the stability (ΔGex–kcal·mol–1) vs. residue number for CTPR2 (blue) and CTPR3 (red). The two red bars indicate the stability of CTPR2 and -3 from equilibrium experiments. (B) Schematic representations of CTPR2 and -3 showing the stability of each residue (space fill of the backbone nitrogen atom) calculated from H/D exchange experiments. These representations are colored depending on their stability with white being least stable (≈4.5 kcal·mol–1) through to the most stable being purple/red (≈9.5 kcal·mol–1). (C) Representation of the interaction between Trp and Pro residues within CTPR2. The side chains of Trp, Pro, Leu, and Lys are shown as a transparent space fill and sticks; the hydrogen bond between the Trp side-chain indole NHε1 and backbone of Pro is represented by a black line. (D Left) Histogram showing the difference in stability (ΔΔGex) between CTPR2 and -3 at pH 6.8. The results include a direct comparison of the first and second repeats of CTPR2 with their counterparts in CTPR3 (blue) and a comparison of the second repeat and solvating helix of CTPR2 with the third repeat and solvating helix of CTPR3 (red). Areas of local, intermediate, and globally exchanging residues are shown. (Right) The structure of CTPR3 with locally, intermediate, and globally exchanging residues shown in white, yellow, and red, respectively. In A, B, and D and Fig. 2E, there are four cases in which a resonance could be assigned to residues in either the second or third TPR motif of CTPR3. In these cases, the resultant rate could be from either residue or the same for both residues. Therefore, we have indicated the resultant protection factor, ΔGex, or ΔΔGex for both positions. These residues are Trp-4, Asn-6, Lys-13, and Asn-34 in the second and third TPR motifs.
Although the pattern of protection from exchange seen along the sequence of CTPR2 and -3 is the same, the magnitudes are very different (Figs. 2E and 3). To show these changes clearly, the change in free energy for exchange between CTPR2 and -3 (ΔΔGex) was calculated for residues observable in both forms (Fig. 3). This analysis is possible because of the identical repeating nature of the motifs that causes identical structure and solvent accessibility for each repeat.
Discussion
Global Stability of the CTPR Proteins. The chemical denaturation data we present demonstrate that as the number of tandem TPR motifs increases, the apparent global stability of the protein increases. The predominant cause of the increased stability is a significant decrease in the rate of protein unfolding.
Local Stability: Residue-Specific Stability Determined by Hydrogen Exchange. Hydrogen exchange enables us to distinguish not only global effects but also the local effects of increasing the number of flanking TPR motifs in the CTPR proteins. We found that although CTPR1 is folded, as detected by both CD and NMR, and undergoes a global denaturation transition, all its amide protons are in rapid exchange with solvent. This result is a function of our performing the experiments at 20°C and pH 6.8, where exchange is rapid, rather than an indication that CTPR1 is unfolded under all conditions.
When additional repeats are added, to give CTPR2 and -3, many amide protons become protected from exchange. In both cases, increasing protection is seen on going from the ends to the center of the proteins (Figs. 2E and 3). Assuming an EX2 mechanism, both CTPR2 and -3 possess a set of amides, located in the most central TPR motif, that likely exchange with solvent by a global unfolding process (Fig. 3 and Table 2). Although the overall pattern of protection along the backbone of CTPR2 and -3 is the same, the magnitude of the protection at different positions differs dramatically. The effect of the addition of an extra repeat is mainly demonstrated by the change in stability of the amides of the second TPR motif. The extension makes them more stable to exchange, as well as increasing the stability of the amides in the third TPR motif that interact with the second TPR motif (Fig. 3). Although it might seem intuitively obvious that increasing the number of repeats will increase protein stability, and hence slow amide exchange rates, to our knowledge this work is the first explicit demonstration of this phenomenon for a repeat protein. There are few data on similar systems with which to compare our results. The closest is the study by Hamill and coworkers (13, 14) that showed that the β-sheet protein tenascin could be stabilized by an extension of two residues at the C terminus.
Specific Stabilization Motif Involving a Conserved Side Chain. The backbone amide proton exchange experiments discussed above report on the overall distribution of stability in the designed TPR repeat proteins. However, hydrogen exchange experiments also highlighted specific side-chain interactions that may play key roles in specifying the TPR fold. Interestingly, we found that the side-chain indole NHε1 of Trp4(A2) was protected from exchange in both CTPR2 and CTPR3 (Figs. 2D and 5). The Trp4(A3) in CTPR3 also may be protected; however, it overlaps with Trp4(A2), which precludes further analysis. The protection of the Trp side-chain indole NHε1 is not only highly unusual for a side chain, but it is also unusual for a side chain that is relatively solvent exposed (≈15%). Moreover, this NHε1 is within hydrogen-bonding distance from the backbone carbonyl group of the preceding highly conserved Pro32(Turn2) (Fig. 3C). It is this Pro that is crucial in breaking helix B and forming the turn between motifs: PNNAEAW. The hydrogen bond between the Trp NHε1 and backbone Pro is shielded from exchange by a sandwich of Leu residues [Leu28(B1) and Leu31(B2)] and a Lys residue above [Lys26(B2)]. The Leu residues are highly conserved as large hydrophobic residues (usually Leu, Ile, or Val).
It is interesting to postulate that the hydrogen bonding between Trp and Pro, shielded by the conserved hydrophobics, is important in stabilizing and specifying both the breaking of each B-helix and the turn between TPR motifs. There is precedence for such from similar interactions present in the “Trp-cage” motif (15, 16) and the “Tyr corner” of the Greek key β-sandwich proteins (17). The Trp-cage was first revealed in NMR studies on a 20-residue fragment of the extendin-4 peptide of Glia monster saliva (NLYIQWLKDGGPSSGRPPPS). This work showed a striking feature of close association of the Gly and three Pro residues with the Tyr and Trp side chains (underlined in the sequence) that was named the Trp-cage. In particular, there is a distinctive indole-NHε1 hydrogen bond to the Pro (i+10) backbone carbonyl. Further, in the Trp cage, Pro rings interact with the indole ring on both faces, reminiscent of the shielding provided by the Leu and Lys with the indole ring in the TPR motif. Although the interactions in the CTPR turns are not identical to those of the Trp cage, there are clear similarities, and we believe that the interactions represented here are of importance in specifying this design's TPR superrepeat structure.
Comparison of CTPR Folding with That of Other Proteins. In recent years, there have been many papers published that sought correlations between stability/size/topology and folding rate. The most successful has been a parameter called contact order (CO), which links folding rate with the topology of the native state (18, 19). If a protein has a low CO, its residues interact, on average, with others close in sequence (e.g., α-helical proteins) and folding is fast. Because the CTPR proteins have topologies that are dominated by identical modular short-range interactions, their CO and relative CO (CO normalized for protein length) are low (relative CO for CTPR2 = 8% and CTPR3 = 6%), suggesting that the proteins should fold very quickly. This idea is true, with CTPR2 and -3 having extrapolated folding rate constants in water of ≈20,000 and ≈35,000 s–1, respectively (Table 3). Moreover, their extremely fast folding is of a comparable speed to other fast-folding α-helical proteins such as the engrailed homeodomain and the λ-repressor (20).
Conclusions
Many linear repeat proteins with different repeated motifs, repeat numbers, structures, and functions have been identified over the past 10 years. In comparison with globular proteins, relatively little is known about the structure, stability, and folding of these repeat proteins. This study is, to our knowledge, one of the first to comprehensively investigate the stability and folding of repeat proteins based on the TPR motif. Our approach is to investigate the effect of different numbers of identical tandem repeats on protein stability and folding. Such studies complement the elegant work that has been performed on natural repeat proteins, in particular on ankyrin repeats (21–29). In this case, the thermodynamics of unfolding were studied by using a naturally occurring sequence, the protein being progressively truncated by the deletion of individual ankyrin repeats (21–25, 28, 29).
We have established some of the factors that govern the stability of TPR proteins. The CTPR proteins have been shown to fold very rapidly. The stable core of a TPR, comprising the central α-helices, has been identified. The apparent global stabilities of the CTPR proteins have been shown to increase with the number of tandem TPR motifs. We have determined that the enhancement of stability observed for the longer CTPR constructs is a consequence of slower rates of unfolding, rather than faster rates of folding.
H/D exchange experiments also have revealed an important structural feature, which we propose is a key determinant of the structure (and to some extent the stability) of these particular TPR motifs. The side chain of a buried Trp makes a critical hydrogen bond with a backbone amide group; the hydrogen-bonding being shielded from exchange by the presence of two bulky hydrophobic groups.
These studies not only establish some of the basic thermodynamic and kinetic properties of the system and therefore pave the way for more detailed future studies, they also provide an important starting point for further design projects aimed at building more complex architectures and functionalities.
Supplementary Material
Acknowledgments
We thank L. D'Andrea for initiating the TPR studies and for helpful discussions. We thank M. Cocco for NMR expertise and data collection and the groups of S.E.J. and L.R. for paper reading and insightful discussion. E.R.G.M. was a Wellcome Fellow and holds a Research Fellowship at Girton College. This work was partly funded by the Welton Foundation.
Author contributions: E.R.G.M. and L.R. designed research; E.R.G.M. and K.S. performed research; E.R.G.M., K.S., and L.R. analyzed data; and E.R.G.M., S.E.J., and L.R. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TPR, tetratricopeptide repeat; CTPR, consensus TPR.
References
- 1.Groves, M. R. & Barford, D. (1999) Curr. Opin. Struct. Biol. 9, 383–389. [DOI] [PubMed] [Google Scholar]
- 2.Kobe, B. & Kajava, A. V. (2000) Trends Biochem. Sci. 25, 509–515. [DOI] [PubMed] [Google Scholar]
- 3.Blatch, G. L. & Lassle, M. (1999) BioEssays 21, 932–939. [DOI] [PubMed] [Google Scholar]
- 4.Das, A. K., Cohen, P. W. & Barford, D. (1998) EMBO J. 17, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Andrea, L. D. & Regan, L. (2003) Trends Biochem. Sci. 28, 655–662. [DOI] [PubMed] [Google Scholar]
- 6.Main, E. R. G., Xiong, Y., Cocco, M. J., D'Andrea, L. & Regan, L. (2003) Structure (London) 11, 1–20. [DOI] [PubMed] [Google Scholar]
- 7.Main, E. R., Fulton, K. F. & Jackson, S. E. (1998) Biochemistry 37, 6145–6153. [DOI] [PubMed] [Google Scholar]
- 8.Jackson, S. E. & Fersht, A. R. (1991) Biochemistry 30, 10428–10435. [DOI] [PubMed] [Google Scholar]
- 9.Boucher, W. (1993–2002) azara, (Univ. of Cambridge, Cambridge, U.K.).
- 10.Clarke, J. & Itzhaki, L. S. (1998) Curr. Opin. Struct. Biol. 8, 112–118. [DOI] [PubMed] [Google Scholar]
- 11.Bai, Y., Milne, M., Mayne, L. & Englander, S. W. (1993) Proteins 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai, Y., Sosnick, T. R., Mayne, L. & Englander, S. W. (1995) Science 269, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamill, S. J., Meekhof, A. E. & Clarke, J. (1998) Biochemistry 37, 8071–8079. [DOI] [PubMed] [Google Scholar]
- 14.Meekhof, A. E., Hamill, S. J., Arcus, V. L., Clarke, J. & Freund, S. M. (1998) J. Mol. Biol. 282, 181–194. [DOI] [PubMed] [Google Scholar]
- 15.Barua, B. & Andersen, N. H. (2002) Lett. Pept. Sci. 8, 221–226. [Google Scholar]
- 16.Neidigh, J. W., Fesinmeyer, R. M. & Andersen, N. H. (2002) Nat. Struct. Biol. 9, 425–430. [DOI] [PubMed] [Google Scholar]
- 17.Hamill, S. J., Cota, E., Chothia, C. & Clarke, J. (2000) J. Mol. Biol. 295, 641–649. [DOI] [PubMed] [Google Scholar]
- 18.Plaxco, K. W., Simons, K. T., Ruczinski, I. & Baker, D. (2000) Biochemistry 39, 11177–11183. [DOI] [PubMed] [Google Scholar]
- 19.Plaxco, K. W., Simons, K. T. & Baker, D. (1998) J. Mol. Biol. 277, 985–994. [DOI] [PubMed] [Google Scholar]
- 20.Kubelka, J., Hofrichter, J. & Eaton, W. A. (2004) Curr. Opin. Struct. Biol. 14, 76–88. [DOI] [PubMed] [Google Scholar]
- 21.Bradley, C. M. & Barrick, D. (2002) J. Mol. Biol. 324, 373–386. [DOI] [PubMed] [Google Scholar]
- 22.Zweifel, M. E. & Barrick, D. (2001) Biochemistry 40, 14357–14367. [DOI] [PubMed] [Google Scholar]
- 23.Zweifel, M. E. & Barrick, D. (2001) Biochemistry 40, 14344–14356. [DOI] [PubMed] [Google Scholar]
- 24.Zhang, B. & Peng, Z. (2000) J. Mol. Biol. 299, 1121–1132. [DOI] [PubMed] [Google Scholar]
- 25.Mosavi, L. K., Minor, D. L., Jr., & Peng, Z. Y. (2002) Proc. Natl. Acad. Sci. USA 99, 16029–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang, K. S., Guralnick, B. J., Wang, W. K., Fersht, A. R. & Itzhaki, L. S. (1999) J. Mol. Biol. 285, 1869–1886. [DOI] [PubMed] [Google Scholar]
- 27.Tang, K. S., Fersht, A. R. & Itzhaki, L. S. (2003) Structure (London) 11, 67–73. [DOI] [PubMed] [Google Scholar]
- 28.Mello, C. C. & Barrick, D. (2004) Proc. Natl. Acad. Sci. USA 101, 14102–14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripp, K. W. & Barrick, D. (2004) J. Mol. Biol. 344, 169–178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.