

Abstract
Chronic inflammation can be a major driver of the failure of a variety of organs, including chronic kidney disease (CKD). The NLR family pyrin domain-containing 3 (NLRP3) inflammasome has been shown to play a pivotal role in inflammation in a mouse kidney disease model. Nuclear factor erythroid 2-related factor 2 (Nrf2), the master transcription factor for anti-oxidant responses, has also been implicated in inflammasome activation under physiological conditions. However, the mechanism underlying inflammasome activation in CKD remains elusive. Here, we show that the loss of Nrf2 suppresses fibrosis and inflammation in a unilateral ureter obstruction (UUO) model of CKD in mice. We consistently observed decreased expression of inflammation-related genes NLRP3 and IL-1β in Nrf2-deficient kidneys after UUO. Increased infiltration of M1, but not M2, macrophages appears to mediate the suppression of UUO-induced CKD symptoms. Furthermore, we found that activation of the NLRP3 inflammasome is attenuated in Nrf2-deficient bone marrow–derived macrophages. These results demonstrate that Nrf2-related inflammasome activation can promote CKD symptoms via infiltration of M1 macrophages. Thus, we have identified the Nrf2 pathway as a promising therapeutic target for CKD.
Introduction
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master transcription factor for anti-oxidant and detoxification responses1, 2. Cellular Nrf2 levels are precisely regulated by transcriptional and post-transcriptional mechanisms to ensure adequate responses to oxidative stresses. Under physiological conditions, adaptor protein kelch-like ECH-associated protein 1 (Keap1) binds to the Nrf2 protein and accelerates proteasomal degradation of Nrf2. With oxidative stress, Keap1 releases Nrf2, which allows Nrf2 to translocate into the nucleus to activate the expression of anti-oxidant-related genes, such as NAD(P)H quinone oxidoreductase (NQO1), heme oxygenase 1 (HO-1), and glutamate-cysteine ligases (GCLM and GCLC)3. These anti-oxidant genes, especially HO-1, are protective against kidney dysfunction, including chronic kidney disease (CKD) and acute kidney injury4–6.
Nrf2 has been reported to play a role in a variety of pathologic kidney conditions. However, recent reports have also demonstrated that Nrf2 is required for activation of the nucleotide-binding domain, leucine-rich repeat containing protein (NLRP3) inflammasome7.
Inflammasomes are multimeric protein complexes that form in the cytosol in response to either exogenous pathogens or endogenous danger signals and induce proinflammatory effects. Although inflammasomes are an innate immune response to eliminate infectious pathogens from tissues, chronic inflammation can be a major driver of the failure of a variety of organs, including the development of CKD. Activation of inflammasomes is a key mediator in chronic inflammation, but aberrant inflammasome activation is involved in many different diseases, including atherosclerosis and heart failure8–10. NLRP3, a Nod-like receptor family member, and an adaptor apoptosis-associated speck-like protein with a caspase recruitment domain (ASC) within inflammasomes, activate caspase-1, which in turn promotes maturation of the proinflammatory cytokines interleukin-1 beta (IL-1β) and IL-1811, 12. Recently, the NLRP3 inflammasome has been shown to play a pivotal role in inflammation in murine kidney disease models13–16. Of note, Nrf2 knockout has led to the amelioration of several metabolic disorders and cardiovascular diseases, including atherosclerosis17–21, obesity22, and type 2 diabetes23, all of which are typically associated with chronic inflammation. These observations raise the intriguing possibility that Nrf2 may affect kidney diseases via inflammasome activation in addition to, or in lieu of, its classic role as an oxidative stress response factor. However, the role of Nrf2-related inflammasome activation in kidney disease is still unclear.
To better understand the role of Nrf2 in the progression of kidney diseases, we generated a unilateral ureter-ligated mouse model in wild-type (WT) and Nrf2-knockout (KO) backgrounds. We showed that Nrf2-dependent inflammasome activation was important for maintaining inflammatory macrophages, such as M1 macrophages, but not M2 macrophages. We also found that caspase-1 inhibitors were able to suppress M1 macrophage infiltration. These data increase our understanding of the role of Nrf2 in kidney diseases.
Results
Nrf2 deficiency suppresses inflammation and fibrosis in a UUO model
The murine unilateral ureter obstruction (UUO) model is one of the most useful models of kidney inflammation and fibrosis. First, we examined the kinetics of the mRNA levels of inflammasome-related genes and fibrosis genes during UUO-induced kidney injury. Although the inflammasome-related genes NLRP3 and IL-18 peaked 7 days after UUO, the induction of fibrosis genes was delayed; 14 days after UUO, the fibrosis genes remained upregulated (Fig. 1a). These results are consistent with a model in which inflammation is instructive for kidney fibrosis. Interestingly, the increased levels of the macrophage marker F4/80 coincided with the upregulation of Nrf2 during this time, which suggests that Nrf2 may function through macrophages in UUO-induced inflammation (Fig. 1b). To test the role of Nrf2, we induced UUO in WT and Nrf2-KO mice, in which the Nrf2 exon 4 was deleted24. We then performed a series of histological assessments after the UUO procedure. The extent of fibrosis was successively evaluated with Masson’s trichrome staining (Days 0, 3, 7, and 14). Although the WT and Nrf2-KO mice showed similar UUO-induced fibrosis by Day 7, we noted a statistically significant decrease in fibrosis, measured by fibrosis area, in the Nrf2-KO kidneys compared with the WT kidneys (Fig. 2a,b). Consistently, collagen IV staining also indicated a suppression of fibrosis in the Nrf2-KO mice (Fig. 2c).
Figure 1.

Trends of inflammatory and fibrogenic gene expression in unilateral ureter obstruction. These data were obtained from whole kidney tissue from 5 mice in each group. (a) Expression of inflammatory genes (nucleotide-binding domain, leucine-rich repeat containing protein [NLRP3] and interleukin-18 [IL-18]) and fibrogenic genes (connective tissue growth factor [CTGF] and alpha smooth muscle actin [α-SMA]) were assessed with quantitative reverse transcription PCR (qRT-PCR). The mRNA expression levels of each gene were normalized to the expression of 18S. *p < 0.05 relative to Day 0. (b) Representative western blotting experiments and analysis data for F4/80, nuclear factor erythroid 2-related factor 2 (Nrf2) and GAPDH.
Figure 2.
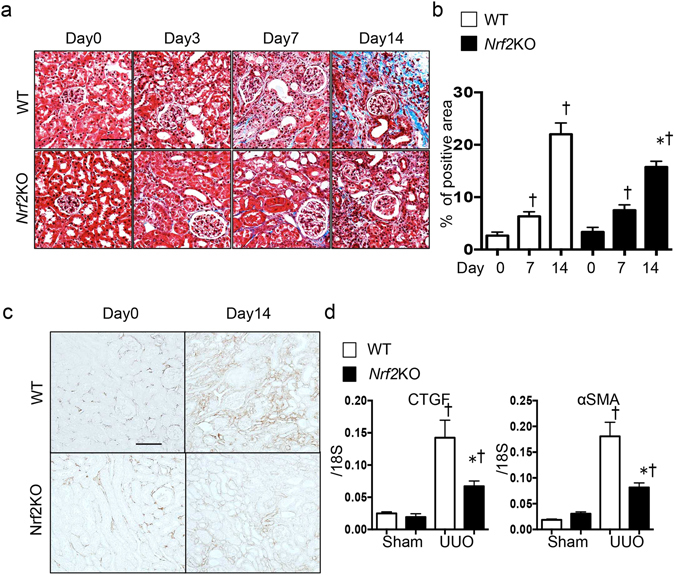
Fibrosis in wild-type (WT) and nuclear factor erythroid 2-related factor 2-knockout (Nrf2-KO) mice after unilateral ureter obstruction (UUO). These data were obtained from whole kidney tissues from 10 or more mice in each group. (a) Fibrosis was assessed with Masson’s trichrome staining. Representative images are shown. (b) The tubulointerstitial fibrosis area (%) was evaluated as the blue-stained positive area in the Masson’s trichrome staining. Scale bar = 50 μm. *p < 0.05 relative to WT-UUO on the same day after UUO. †p < 0.05 relative to Sham. (c) Collagen IV staining was performed to evaluate interstitial fibrosis. Representative images are shown. Scale bar = 100 μm. (d) The fibrosis-associated genes (connective tissue growth factor [CTGF] and alpha smooth muscle actin [α-SMA]) were assessed with quantitative reverse transcription PCR (qRT-PCR). *p < 0.05 relative to WT-UUO. †p < 0.05 relative to Sham.
When the mRNA levels of two fibrosis-related genes, connective tissue growth factor (CTGF) and alpha smooth muscle actin (α-SMA), were evaluated on Day 14 after UUO, the genes showed significantly elevated expression after UUO in the WT kidneys, whereas the gene induction was significantly suppressed in the Nrf2-KO-UUO mice compared with the WT mice (Fig. 2d). We also evaluated the amount of the fibrosis protein α-SMA and the inflammation protein IL-1β by western blot analysis (Fig. 3a,b). Although the α-SMA and IL-1β proteins increased markedly upon UUO in the WT mice, the increase in protein levels was less evident in the Nrf2-KO-UUO mice compared with the WT-UUO mice (Fig. 3a,b).
Figure 3.

Inflammasome activation in wild-type (WT) and nuclear factor erythroid 2-related factor 2-knockout (Nrf2-KO) mice after unilateral ureter obstruction. These data were obtained from whole kidney tissues from 6 or more mice in each group. (a) and (b) Representative western blot data. The following statistical analysis data are shown: *p < 0.05 relative to WT-UUO. †p < 0.05 relative to Sham. (c) Expression levels of inflammasome-related genes (Toll-like receptor 4 [TLR4], nucleotide-binding domain, leucine-rich repeat containing protein [NLRP3], caspase-1, and interleukin-18 [IL-18]) were assessed with quantitative reverse transcription PCR (qRT-PCR). The mRNA expression level of each gene was normalized to the expression of 18 S. *p < 0.05 relative to WT-UUO. †p < 0.05 relative to Sham.
Inflammasome activation has been shown to be involved in the progression of inflammation and fibrosis induced by UUO10. This finding led us to examine the mRNA levels of inflammation-related genes, including Toll-like receptor 4 (TLR4), NLR family pyrin domain-containing 3 (NLRP3), caspase 1, and IL-18. Similar to the previous sets of genes, the UUO-induced upregulation of TLR4 and NLRP3 was attenuated in the Nrf2-KO mice (Fig. 3c). These results indicate that the loss of Nrf2 suppresses inflammation in UUO.
M1 macrophages infiltrate the kidney after UUO
We focused on the role of Nrf2 in macrophages in kidney disease because the number of infiltrated macrophages decreased in the Nrf2-KO mice compared with the WT mice by Day 14 after UUO (Supplementary Fig. S1). These data encouraged us to focus on the characteristics of macrophages. Two macrophage subtypes, M1 and M2, are known to reciprocally control inflammatory responses. To understand the role of Nrf2 in macrophage function, we sought to characterize the behaviors of these macrophage subtypes during UUO-induced kidney fibrosis using WT mice.
We first harvested bone marrow cells from green fluorescent protein (GFP) transgenic mice (GFPTG) to transplant into the tail veins of the WT mice. We then sorted macrophages from the whole kidney based on the cell-surface expression of the macrophage markers CD11b and F4/80. As shown in Supplementary Figure 2a, the CD11b+-F4/80low and CD11b+-F4/80high cell populations were GFP-positive, which indicated that these cells were bone marrow–derived macrophages. We noted that the two cell populations with CD11b+-F4/80low and CD11b+-F4/80high increased by Day 3 of UUO, as previously reported25. The CD11b+-F4/80high cell group increased further by UUO-Day 7. We sorted CD11b+-F4/80low and CD11b+-F4/80high with fluorescence-activated cell sorting (FACS) and examined the mRNA expression of the M1 marker, iNOS, and the M2 marker, CD206, with quantitative reverse transcription PCR (qRT-PCR). The higher iNOS mRNA expression in CD11b+-F4/80low indicated that this population represented M1 macrophages, whereas the higher CD206 expression in CD11b+-F4/80high suggested that these were M2 macrophages (Supplementary Fig. 2b). Based on these data, we concluded that M1 macrophages infiltrate the kidney tissue at an early stage (Day 3) and that M2 macrophages increase after Day 7 upon UUO.
Nrf2-dependent inflammasome activation contributes to maintaining M1 macrophages in UUO-induced kidney inflammation
We then examined the time course of M1 and M2 macrophage infiltration in the WT and Nrf2-KO mice. We did not detect a substantial difference between the genotypes in either the M1 or M2 macrophage numbers until Day 7 after UUO. However, the Nrf2-KO kidneys showed substantially lower numbers of M1 macrophages at Day 14 after UUO compared to the WT kidneys (Fig. 4a,b). When we sorted M1 macrophages from the WT and Nrf2-KO mice on Day 7 and investigated the expression of the inflammasome-related genes, the mRNA levels of IL-1β and caspase 1 in M1 macrophages were significantly suppressed in the Nrf2-KO samples compared with the WT samples (Fig. 4c), thus demonstrating that Nrf2 is necessary for the induction of inflammasome-related genes within M1 macrophages.
Figure 4.
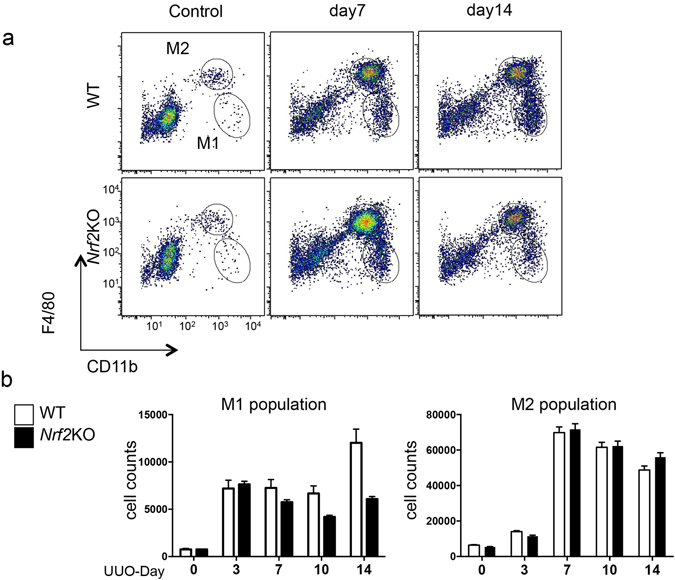
Macrophage polarization in wild type-unilateral ureter obstruction (WT-UUO) mice and nuclear factor erythroid 2-related factor 2-knockout unilateral ureter obstruction (Nrf2-KO-UUO) mice. These data were obtained from whole kidney tissues from 4 mice in each group. (a) Representative flow cytometry data are shown. (b) The absolute cell numbers of the M1 and M2 populations after live cell gating. *p < 0.05 relative to WT-UUO on the same day after UUO.
To determine the roles of inflammasome activation in M1 macrophage infiltration, we administered the caspase 1-specific inhibitor VX765 orally during the later phase of UUO (Days 7–14) and investigated the maintenance of the M1 macrophage population. The VX765 treatment in the later stage was sufficient to decrease the M1 macrophage population (Fig. 5a,b). The mRNA expression of IL-1β and α-SMA was also suppressed significantly by VX765 treatment (Fig. 5c). These results indicate that inflammasome activation is important for maintaining the M1 macrophages after their initial infiltration and that Nrf2 may contribute to this process.
Figure 5.
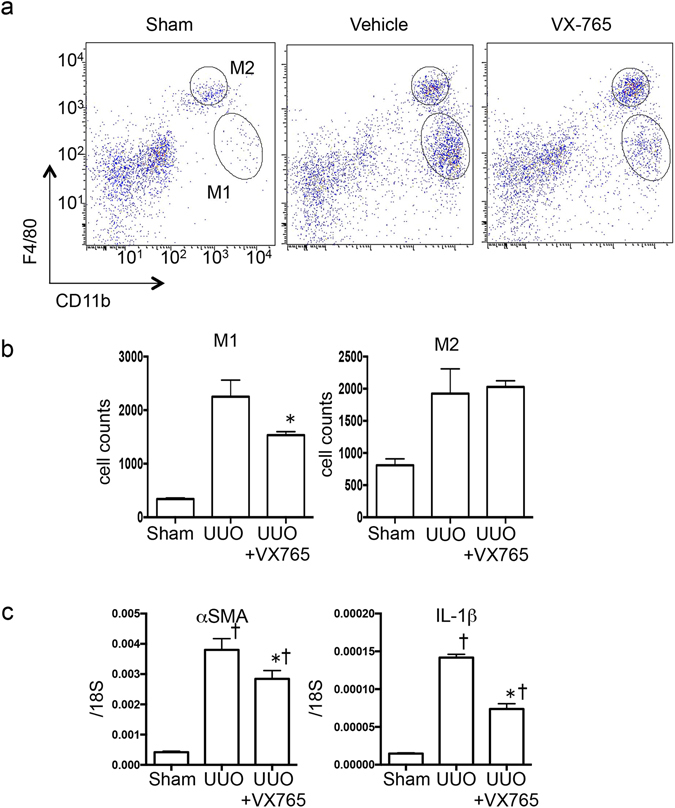
Infiltration of M1 was inhibited with a caspase 1-specific inhibitor, VX765, administered in unilateral ureter obstruction. These data were obtained from whole kidney tissues from 3–4 mice in each group. (a) Representative flow cytometry data are shown. (b) The absolute cell numbers of the M1 and M2 populations after the live cell gating. *p < 0.05 relative to vehicle group. †p < 0.05 relative to Sham. (c) RNA was isolated from whole kidney tissues from each group. Alpha smooth muscle actin (α-SMA) and interleukin-1β (IL-1β) were assessed with quantitative reverse transcription PCR (qRT-PCR). The mRNA expression level of each gene was normalized to the expression of 18S. *p < 0.05 relative to vehicle group. †p < 0.05 relative to Sham.
Next, we tested whether the role of Nrf2 in kidney inflammation is cell autonomous within macrophages. We transplanted cells from either Nrf2-KO or Asc-KO bone marrow into WT bone marrow. We then evaluated kidney fibrosis on Day 14 after UUO with Masson’s trichrome staining. The UUO-induced kidney fibrosis was significantly suppressed in the kidneys of the WT animals that received Nrf2-KO bone marrow cells (Fig. 6a,b). The mRNA levels of the fibrosis genes α-SMA and CTGF were significantly reduced in the animals with transplanted Nrf2-KO bone marrow cells compared with those with transplanted WT cells (Fig. 6c). We obtained similar results upon the transplantation of Asc-KO bone marrow cells (Supplementary Fig. S3a,b), which indicated that inflammasome activation drives the progression of fibrosis. To investigate the direct role of Nrf2 in the activation of NLRP3 inflammasomes, we isolated bone marrow–derived macrophages (BMDMs) from the WT and Nrf2-KO mice. We then treated the BMDMs with lipopolysaccharide-adenosine triphosphate (LPS-ATP) and measured the activation of NLRP3 inflammasomes by monitoring the protein levels of the cleavage of caspase 1 and IL-1β in the BMDMs via western blot analysis and by monitoring the secretion of IL-1β and IL-6 into the supernatant with enzyme-linked immunosorbent assay (ELISA). We found decreased caspase 1 cleavage and a reduced secretion of IL-1β from the Nrf2-KO BMDMs compared with the WT cells (Fig. 7a–c). However, the secretion of IL-6, which is expressed by the Nfκb pathway, did not differ between the genotypes, which suggests that Nrf2 plays a specific role in inflammasome activation. These data demonstrate that Nrf2 is directly involved in the activation of NLRP3 inflammasomes within macrophages.
Figure 6.
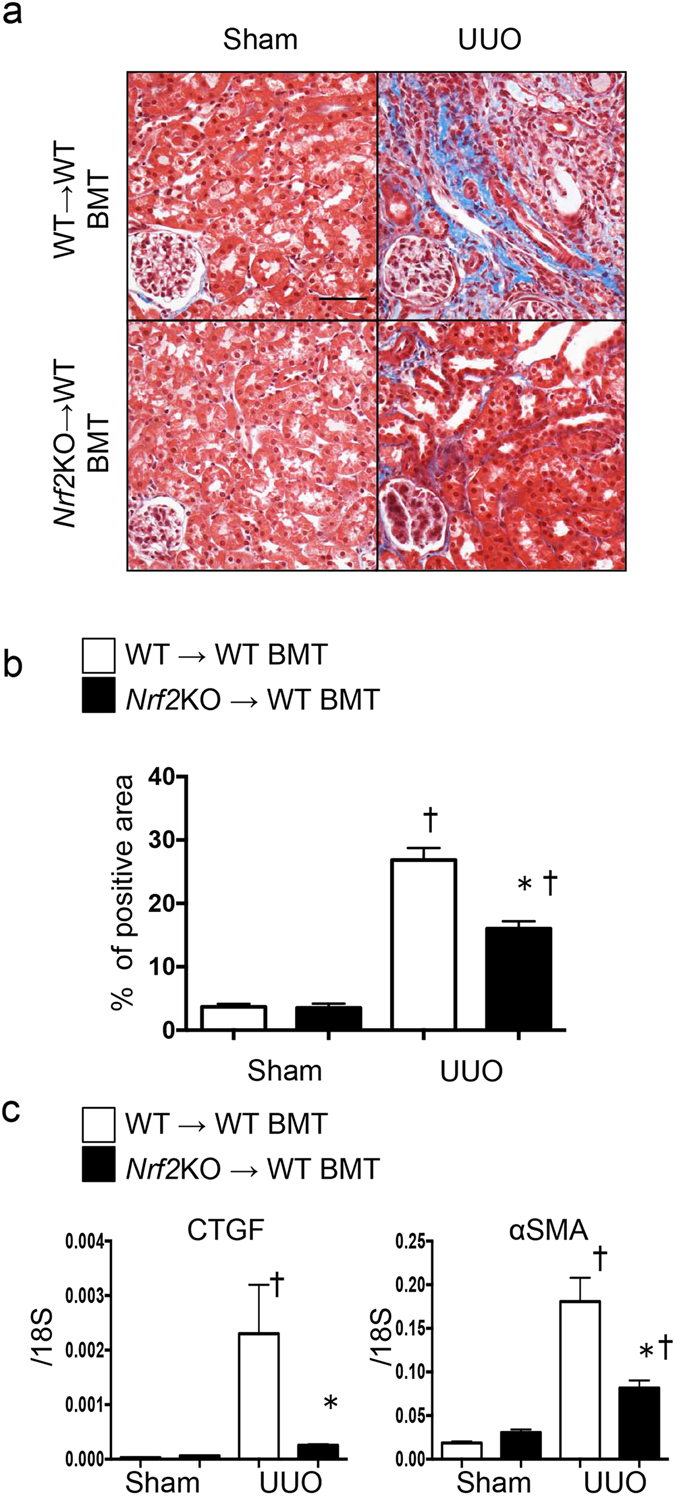
Fibrosis in unilateral ureter obstruction (UUO) after bone marrow transplantation (BMT). BMT was performed to determine the role of Nrf2 in macrophages. These data were obtained from whole kidney tissues from 6 or more mice in each group. (a) Fibrosis was assessed with Masson’s trichrome staining. Representative images are shown. (b) The tubulointerstitial fibrosis area (%) was evaluated as the blue-stain-positive area in Masson’s trichrome staining. Scale bar = 50 μm. *p < 0.05 relative to BMT (wild-type [WT] → WT)-UUO. †p < 0.05 relative to Sham. (c) Connective tissue growth factor (CTGF) and alpha smooth muscle actin (α-SMA) were assessed by quantitative reverse transcription PCR (qRT-PCR). *p < 0.05 relative to BMT (WT → WT)-UUO. †p < 0.05 relative to Sham.
Figure 7.

The role of nuclear factor erythroid 2-related factor 2 (Nrf2) in bone marrow–derived macrophages (BMDMs). The in vitro assays using bone marrow–derived macrophages (BMDMs) were performed at least 3 times. Each experiment was performed in duplicate. (a) and (b) Representative western blot data from cell lysates. The following statistical analysis data are shown: (c) The levels of interleukin-1β (IL-1β) and IL-6 were detected with enzyme-linked immunosorbent assay (ELISA) in supernatant. *p < 0.05 relative to lipopolysaccharide (LPS) and adenosine triphosphate (ATP) stimulation for wild-type (WT)-BMDMs.
Discussion
In this study, we elucidated two main points concerning the role of Nrf2 in the progression of kidney disease. First, Nrf2 activation of macrophages is essential for inflammasome activation. In an Nrf2-deficient murine kidney disease model, the prolongation of inflammation was suppressed, and fibrosis was ameliorated. Nrf2-related NLRP3 inflammasome activation was important for maintaining the M1 population in this disease model and, as a result, seems to contribute to the process by which organ inflammation becomes chronic inflammation.
The importance of inflammasome activation in the progression of kidney disease, including UUO, has already been reported26. Many studies have examined the role of inflammasome activation and an upstream pathway in the progression of kidney disease. We have reported that aldosterone caused inflammasome activation via macrophage mitochondrial dysfunction27. We also reported that the mitochondrial redox response could induce inflammasome activation in nephrotic syndrome mice28. However, no reports have suggested how inflammasome activation contributes to the progression of kidney disease in these disease states. Our data also show that M1 increases on UUO-Day 3 and that M2 was already prominent on Day 7. Fujiu et al. performed a similar study and showed that M2 becomes dominant at an early stage25. It is particularly interesting that a continued observation of the M1 population at Day 14 showed almost no changes in the WT mice but a clear decrease in the Nrf2-KO and Asc-KO mice. In the Nrf2-deficient cells, the NLRP3 inflammasome might be activated after UUO in vivo or after stimulation in vitro. This finding suggests that there is both Nrf2-related and Nrf2-unrelated NLRP3 inflammasome activation. Nrf2-dependent inflammasome activation may be involved in maintaining the M1 population. The mechanism is unclear, but it has been reported that high-mobility group protein box 1 (HMGB1) is involved in inducing the M1 population29, 30, and it is possible that HMGB1 and extracellular histones act as damage-associated molecular pattern (DAMP) molecules31. Although the association of cytokines such as IL-1β also plays an important role in inflammasome activation, it has become clear that DAMPs such as HMGB1 are extremely important in enhancing the progression of inflammation32. Because HMGB1 or extracellular histone activates TLR4 signaling33, this action may contribute to the induction of macrophages to M134. This kind of in vivo vicious cycle may be a factor underlying why pathologies become chronic, and it is an important discovery because the suppression of inflammasome activation may become a viable therapeutic target.
The results of this study suggest that Nrf2-dependent inflammasome activation plays an important role in the pathogenesis of kidney disease and that it may contribute to the chronicity of the disease. A particularly interesting result is that maintenance of the M1 population associated with inflammasome activation later causes fibrosis. However Nrf2 activators have the potential to be effective new drugs in various fields35, 36. The results of this study should not discourage therapeutic intervention with Nrf2-activating drugs. The mechanism of action of the Nrf2 activators has not been clarified in previous studies, and the possibility of direct involvement—that is, without the involvement of transcriptional activity—has also been suggested7.
It has been shown that the expression of many anti-oxidant and anti-inflammatory gene clusters is controlled by Nrf2 activation. It can be imagined that an upregulation of such gene cluster expression would have an organ-protective effect; Nrf2 activators are thus garnering attention as potential new drugs in various fields35, 36. In kidney diseases, several reports have shown that the activation of Nrf2 ameliorates kidney dysfunction by inhibiting inflammasome activation5, 37. The redox status that is produced by mitochondrial dysfunction and NAD(P)H oxidase activation could induce NLRP3 inflammasome activation. However, in this study, organ fibrosis was actually suppressed in Nrf2-deficient mice. One reason for this could be that the expression of anti-oxidant (e.g., SOD2, NQO1) clusters is already suppressed in this model. NQO1 and S100a9 are Nrf2-dependent genes because the expression of these genes disappeared in the Nrf2-KO mice before and after UUO (Fig. S5). There was no statistically significant difference between the WT-UUO and Nrf2-KO-UUO mice (Supplementary Figs S4 and S5). For S100a9, another Nrf2-related gene, the expression increased in related with Nrf2 protein expression (Supplementary Figs S4 and S5), and it was suggested that gene expression associated with Nrf2 activation may vary widely across different diseases.
Another possibility is that there might be several types of mechanism of NLRP3 inflammasome activation and that some – but not all –are dependent on Nrf2. CD36 could be a key molecule for understanding the relationship between Nrf2 and inflammasome activation. Nrf2 regulates the expression of CD36 in macrophages38. In addition, Sheedy et al. reported that CD36 has a pivotal role in NLRP3 inflammasome activation39. In kidney disease, a CD36 antagonist has been shown to prevent disease progression40.
Based on the results of this study, Nrf2-dependent inflammasome activation plays an important role in the progression of kidney disease. This activation had a particularly strong effect on the persistence of M1 macrophage infiltration. These novel results contribute to elucidating the pathology of chronic kidney disease, especially prolonged inflammation.
Materials and Methods
All methods were performed in accordance with the relevant guidelines and regulations.
Animals
The experimental protocols (no. 15–061, no. 15–077 and no. 15–112) were approved by the Animal Research Committee of Kawasaki Medical School, which is based on the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23, revised 1996). Eight-week-old male mice weighing 20 to 30 g at the beginning of the study were designated wild-type (WT). GFP-TG mice were kindly provided by Y. Sunada (Neurology Department, Kawasaki Medical University, Okayama, Japan)41. Asc homozygous knockout (Asc-KO) mice were kindly provided by M. Takahashi (Jichi Medical University, Shimotsuke, Japan)8. Nrf2 mutant (Nrf2-KO) mice were purchased from RIKEN (Ibaraki, Japan)24. All mice underwent unilateral ureteral obstruction (UUO) or a sham operation (sham). All mice were from the C57B/6 J background.
Bone marrow transplantation
Bone marrow transplantation (BMT) was performed according to a standard protocol described previously27, 41. Using this protocol, four types of chimeric mice were obtained as follows: BMT (WT → WT), BMT (GFP → WT), BMT (Asc-KO → WT), and BMT (Nrf2-KO → WT) mice.
Cell culture
Bone marrow–derived macrophages (BMDMs) were used for in vitro assays. Bone marrow cultures were prepared using Macrophage colony-stimulating factor (MCSF)-containing media42. To activate the NLRP3 inflammasome, BMDMs were primed for 3 hours with ultrapure LPS (InvivoGen, San Diego, CA, USA), followed by stimulation with ATP (5 mM) 30 min before the cell lysates and supernatants were harvested. IL-1β and IL-6 were detected using ELISA (R&D Systems, Minneapolis, MN, USA).
Western immunoblotting
Kidney and cell lysates were extracted with extraction buffer or sample buffer as described previously43. Protein samples were subjected to immunoblotting analysis with antibodies against α-SMA (100M4795; Sigma-Aldrich, St Louis, MO, USA), ASC (sc-22514-R; Santa Cruz Biotechnology, Dallas, TX, USA), caspase-1 (sc-514; Santa Cruz Biotechnology), F4/80 (MCA497GA; AbD Serotec, Raleigh, NC, USA), IL-1β (ab9722; Abcam, Cambridge, MA, USA), Nrf2 (sc-13032; Santa Cruz Biotechnology) and GAPDH (sc-25778; Santa Cruz Biotechnology). Signals were detected using an enhanced chemiluminescence system (GE Healthcare Japan, Tokyo, Japan). Each western blot was performed on a pooled sample of 4–6 mice from each group.
RNA extraction and quantitative reverse transcription PCR
The protocols for extracting RNA and making cDNA were described in a previous report44. The primers and probes for the TaqMan analysis were designed using sequence information from GenBank (National Institutes of Health, Bethesda, MD, USA)45 and Primer3 online software (http://frodo.wi.mit.edu/primer3/; accessed July 1, 2015). The primer and probe sequences are listed in the Supplementary Table in the supplemental information. TaKaRa Premix Ex Taq (Takara Bio, Inc., Otsu, Japan), with a final reaction volume of 20 μl, was used for the TaqMan probe-based quantitative reverse transcription PCR (qRT-PCR) reaction, which was performed on an Applied Biosystems 7500 Fast Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The level of mRNA expression in each sample was quantified using the absolute quantification standard curve method46. The plasmid cDNA of each gene was used to prepare the absolute standards. The concentration was measured using the A260, which was converted to the number of copies using the molecular weight of the DNA. Each mRNA expression level was normalized to that of the housekeeping 18 S ribosomal RNA gene. The levels of expression in each sample were normalized to that of 18S RNA.
Flow cytometric analysis
Kidney cells were prepared according to a protocol described previously25. Kidney tissue was immediately homogenized using gentleMACS™ (Miltenyi Biotec, Tokyo, Japan) with collagenase-containing extraction buffer. The cells were dissolved in FACS buffer to perform FACS analysis using a FACS CantoIITM (BD Bioscience, Tokyo, Japan) and FlowJo software (Tree Star, Ashland, OR, USA). An allophycocyanin (APC)-labeled anti-mouse F4/80 antibody (B202565; BioLegend, San Diego, CA, USA) and a phycoerythrin (PE)-labeled anti-mouse/human CD11b antibody (B194776; BioLegend) were used. Cell sorting was performed with FACSAriaTM III (BD Bioscience) for RNA extraction using an RNeasy Kit (Qiagen, Germantown, MD, USA).
Histopathological examination
Half of the kidneys were fixed in 4% paraformaldehyde and embedded in paraffin for histological analysis. Masson trichrome–stained specimens were observed under an inverted microscope (BZ-9000, Keyence, Osaka, Japan). The percentage of blue-stained scarred areas was quantified using a color image analyzer (WinROOF; Mitani, Fukui, Japan)47.
Immunohistochemical staining
For immunohistochemical staining, serial cryostat sections (4 µm thick) of paraffin-embedded specimens were rehydrated in phosphate buffered saline (PBS) and subjected to antigen retrieval in a microwave. Antibodies against collagen IV (ab6586; Abcam, Cambridge, MA, USA) and F4/80 (MCA497GA; AbD Serotec) were used, and detection was carried out using the Histofine Simple StainTM MAX PO (MULTI) kit (Nichirei, Tokyo, Japan) and 3,3-diaminobenzidine (Sigma-Aldrich).
Statistical analyses
All values are expressed as the means ± standard error of the mean (SEM). Statistical analyses were calculated using GraphPad Prism6 software (GraphPad Software, La Jolla, CA, USA). Comparison between multiple groups was performed by using one-way ANOVA. P values less than 0.05 were considered statistically significant.
Electronic supplementary material
Acknowledgements
We thank Etsuko Yorimasa, Yuriko Katayama and Ayano Higashi for providing animal care, and Satomi Hanada, Keiko Shirakiya and Keiko Satoh for assistance with the in vitro assays. This work was supported in part by a JSPS KAKENHI Grant (No. 24591220 to HN, No. 21591047 and 24390218 to NK), the Uehara Memorial Foundation (to HN), Suzuken Memorial Foundation (to HN), and by a Research Project Grant from the Kawasaki Medical School (No. 23B-41 to NK).
Author Contributions
H.N. designed the study, performed the experiments and wrote the paper. Y.S. performed the experiments. I.S. wrote the paper. C.I., S.I., A.U. and K.K. performed the in vivo experiments. S.T., M.T. and H.T. developed the idea for the study and designed the inflammasome experiments. M.S., T.S. and N.K. designed the study. T.S. and M.Y. developed the idea and designed the Nrf2 experiments. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Yuji Sogawa and Hajime Nagasu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08054-2
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Yates MS, Kensler TW. Keap1 eye on the target: chemoprevention of liver cancer. Acta Pharmacol Sin. 2007;28:1331–1342. doi: 10.1111/j.1745-7254.2007.00688.x. [DOI] [PubMed] [Google Scholar]
- 2.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seng S, Avraham HK, Birrane G, Jiang S, Avraham S. Nuclear matrix protein (NRP/B) modulates the nuclear factor (Erythroid-derived 2)-related 2 (NRF2)-dependent oxidative stress response. J Biol Chem. 2010;285:26190–26198. doi: 10.1074/jbc.M109.095786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu CC, et al. Resveratrol ameliorates renal damage, increases expression of heme oxygenase-1, and has anti-complement, anti-oxidative, and anti-apoptotic effects in a murine model of membranous nephropathy. PLoS One. 2015;10:e0125726. doi: 10.1371/journal.pone.0125726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang SM, et al. Antroquinonol mitigates an accelerated and progressive IgA nephropathy model in mice by activating the Nrf2 pathway and inhibiting T cells and NLRP3 inflammasome. Free Radic Biol Med. 2013;61:285–297. doi: 10.1016/j.freeradbiomed.2013.03.024. [DOI] [PubMed] [Google Scholar]
- 6.Qin T, et al. Sinomenine activation of Nrf2 signaling prevents hyperactive inflammation and kidney injury in a mouse model of obstructive nephropathy. Free Radic Biol Med. 2016;92:90–99. doi: 10.1016/j.freeradbiomed.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Zhao C, Gillette DD, Li X, Zhang Z, Wen H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J Biol Chem. 2014;289:17020–17029. doi: 10.1074/jbc.M114.563114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawaguchi M, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi: 10.1161/CIRCULATIONAHA.110.982777. [DOI] [PubMed] [Google Scholar]
- 9.Mezzaroma E, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vilaysane A, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010;21:1732–1744. doi: 10.1681/ASN.2010020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 12.Srinivasula SM, et al. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277:21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 13.Komada T, et al. Role of NLRP3 Inflammasomes for Rhabdomyolysis-induced Acute Kidney Injury. Sci Rep. 2015;5:10901. doi: 10.1038/srep10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shahzad K, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87:74–84. doi: 10.1038/ki.2014.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuang Y, et al. NLRP3 inflammasome mediates albumin-induced renal tubular injury through impaired mitochondrial function. J Biol Chem. 2014;289:25101–25111. doi: 10.1074/jbc.M114.578260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abais JM, et al. Nod-like receptor protein 3 (NLRP3) inflammasome activation and podocyte injury via thioredoxin-interacting protein (TXNIP) during hyperhomocysteinemia. J Biol Chem. 2014;289:27159–27168. doi: 10.1074/jbc.M114.567537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sussan TE, et al. Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One. 2008;3:e3791. doi: 10.1371/journal.pone.0003791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barajas B, et al. NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler Thromb Vasc Biol. 2011;31:58–66. doi: 10.1161/ATVBAHA.110.210906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freigang S, et al. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol. 2011;41:2040–2051. doi: 10.1002/eji.201041316. [DOI] [PubMed] [Google Scholar]
- 20.Duewell P, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yajima N, et al. Critical role of bone marrow apoptosis-associated speck-like protein, an inflammasome adaptor molecule, in neointimal formation after vascular injury in mice. Circulation. 2008;117:3079–3087. doi: 10.1161/CIRCULATIONAHA.107.746453. [DOI] [PubMed] [Google Scholar]
- 22.Pi J, et al. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J Biol Chem. 2010;285:9292–9300. doi: 10.1074/jbc.M109.093955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chartoumpekis DV, et al. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes. 2011;60:2465–2473. doi: 10.2337/db11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 25.Fujiu K, Manabe I, Nagai R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J Clin Invest. 2011;121:3425–3441. doi: 10.1172/JCI57582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komada T, et al. ASC in renal collecting duct epithelial cells contributes to inflammation and injury after unilateral ureteral obstruction. Am J Pathol. 2014;184:1287–1298. doi: 10.1016/j.ajpath.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Kadoya H, et al. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J. 2015;29:3899–3910. doi: 10.1096/fj.15-271734. [DOI] [PubMed] [Google Scholar]
- 28.Nishi Y, et al. Selective estrogen receptor modulation attenuates proteinuria-induced renal tubular damage by modulating mitochondrial oxidative status. Kidney Int. 2013;83:662–673. doi: 10.1038/ki.2012.475. [DOI] [PubMed] [Google Scholar]
- 29.Su Z, et al. HMGB1 Facilitated Macrophage Reprogramming towards a Proinflammatory M1-like Phenotype in Experimental Autoimmune Myocarditis Development. Sci Rep. 2016;6:21884. doi: 10.1038/srep21884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Tian S, et al. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol. 2015;308:F69–75. doi: 10.1152/ajprenal.00484.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miao EA, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allam R, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. 2012;23:1375–1388. doi: 10.1681/ASN.2011111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai J, et al. HMGB1-Driven Inflammation and Intimal Hyperplasia After Arterial Injury Involves Cell-Specific Actions Mediated by TLR4. Arterioscler Thromb Vasc Biol. 2015;35:2579–2593. doi: 10.1161/ATVBAHA.115.305789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chertow GM, de Zeeuw D, Committee BS. Bardoxolone methyl in type 2 diabetes and advanced chronic kidney disease. N Engl J Med. 2014;370:1768. doi: 10.1056/NEJMc1400872. [DOI] [PubMed] [Google Scholar]
- 36.Shelton LM, Park BK, Copple IM. Role of Nrf2 in protection against acute kidney injury. Kidney Int. 2013;84:1090–1095. doi: 10.1038/ki.2013.248. [DOI] [PubMed] [Google Scholar]
- 37.Shin DH, et al. The NRF2-heme oxygenase-1 system modulates cyclosporin A-induced epithelial-mesenchymal transition and renal fibrosis. Free Radic Biol Med. 2010;48:1051–1063. doi: 10.1016/j.freeradbiomed.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishii T, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 39.Sheedy FJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Souza AC, et al. Antagonism of scavenger receptor CD36 by 5A peptide prevents chronic kidney disease progression in mice independent of blood pressure regulation. Kidney Int. 2016;89:809–822. doi: 10.1016/j.kint.2015.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagiwara H, et al. Bone marrow transplantation improves outcome in a mouse model of congenital muscular dystrophy. FEBS Lett. 2006;580:4463–4468. doi: 10.1016/j.febslet.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 42.Yu J, et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA. 2014;111:15514–15519. doi: 10.1073/pnas.1414859111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagasu H, et al. Endothelial dysfunction promotes the transition from compensatory renal hypertrophy to kidney injury after unilateral nephrectomy in mice. Am J Physiol Renal Physiol. 2012;302:F1402–1408. doi: 10.1152/ajprenal.00459.2011. [DOI] [PubMed] [Google Scholar]
- 44.Satoh M, et al. Mitochondrial damage-induced impairment of angiogenesis in the aging rat kidney. Lab Invest. 2011;91:190–202. doi: 10.1038/labinvest.2010.175. [DOI] [PubMed] [Google Scholar]
- 45.Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2016;44:D67–72. doi: 10.1093/nar/gkv1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dhanasekaran S, Doherty TM, Kenneth J, Group TBTS. Comparison of different standards for real-time PCR-based absolute quantification. J Immunol Methods. 2010;354:34–39. doi: 10.1016/j.jim.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 47.Nagasu H, et al. Azelnidipine attenuates glomerular damage in Dahl salt-sensitive rats by suppressing sympathetic nerve activity. Hypertens Res. 2012;35:348–355. doi: 10.1038/hr.2011.184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.