

Abstract
The capsids of neurotropic herpesviruses have the remarkable ability to move in specific directions within axons. By modulating bidirectional capsid transport to favor either retrograde (minus-end) or anterograde (plus-end) motion, these viruses travel to sensory ganglia or peripheral tissue at specific stages of infection. By using correlative motion analysis to simultaneously monitor the trafficking of distinct viral proteins in living neurons, we demonstrate that viral “tegument” proteins are complexed to capsids moving in axons. The removal of a subset of tegument proteins from capsids invariably preceded retrograde transport to the cell body in sensory ganglia, whereas addition of these proteins was coupled to anterograde transport of progeny capsids to the distal axon. Although capsid transport never occurred without associated tegument proteins, anterograde-specific tegument proteins were competent to travel to the distal axon independent of capsids. These findings are compatible with a model of viral bidirectional transport in which tegument proteins direct capsid traffic to specific intracellular locations during the infectious cycle.
Keywords: neuron, virus
After infection at exposed body surfaces, α-herpesviruses spread to sensory neurons innervating the infected peripheral tissue (i.e., neurons within trigeminal or dorsal root ganglia), where life-long latent infection is established. The reactivation of latent infection results in the production of progeny virus particles that transport in axons to the periphery, ultimately transmitting virions to new hosts. In the case of human α-herpesviruses, reactivated infection can present as cold sores (herpes simplex virus) and shingles (varicella zoster virus).
The structure of an extracellular herpesvirus particle consists of a DNA genome encased in an ≈125-nm-diameter icosahedral capsid. The capsid is surrounded by a layer of proteins called the tegument that resides between the capsid and a membrane envelope (1–3). Although herpesviruses, like all viruses, undergo stages of disassembly (after initial infection of a cell) and assembly (before exiting cells), how these processes impact viral intracellular transport is unclear.
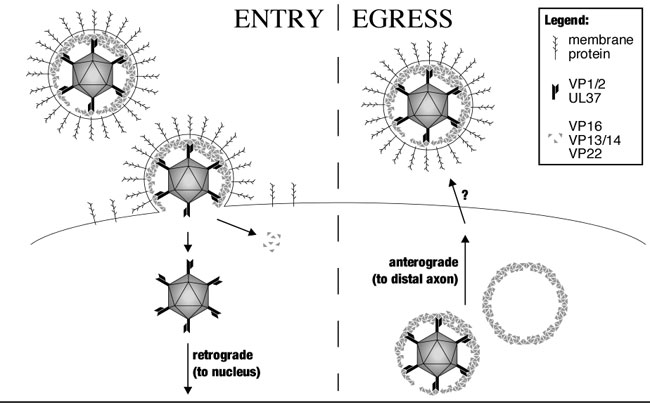
Upon contacting the axon terminal of a sensory neuron, the herpesvirus envelope fuses with the host cell membrane, and the capsid and tegument proteins are deposited into the cytosol (4). Cytosolic capsids initiate fast axonal transport toward the cell body of the neuron, ultimately docking at nuclear pores and injecting viral genomes into the nucleus (4–6). Notably, capsid transport toward the cell body is saltatory and bidirectional: dominant processive retrograde motion is frequently interrupted by brief intervals of anterograde motion (7). After viral replication in the nucleus, progeny capsids move to the axon and rapidly transport to the terminal (6, 8). Capsid transport is, again, saltatory and bidirectional; however, anterograde transport is favored, resulting in the efficient egress of capsids (9). Capsid targeting to the cell body or axon terminal correlates with changes in the anterograde component of bidirectional motion, whereas the retrograde component remains constant (7). Therefore, modulation of the anterograde component of capsid transport is fundamental to the complex α-herpesvirus infectious cycle in the vertebrate nervous system.
Induced changes in the cellular environment, possibly resulting from viral gene expression and cytopathic effect, do not play an obvious role in intracellular capsid targeting (7). Instead, capsids appear to carry determinants that interface with and regulate host bidirectional-transport complexes. The tegument proteins found around capsids in extracellular virions are promising candidates as transport effectors. However, assays for capsid–tegument interactions in cells have been limited to static imaging and purification studies. Whether any tegument proteins are associated with actively transported capsids is unknown. Using correlative motion analysis, we have investigated the composition of the capsid-transport complex in axons of primary sensory neurons. A collection of dual-fluorescent viruses that express capsids fused to a monomeric red fluorescent protein (mRFP1) and a tegument protein fused to the GFP were isolated (10). The diffraction-limited emissions of both viral components were tracked in axons, and correlation of the motion of both fluorophores was used to identify components of the capsid-transport complex. We show here that capsids transport in axons while complexed to tegument proteins. Furthermore, changes in the capsid–tegument composition are coupled to changes in capsid-transport direction in axons. These findings support a model in which tegument proteins affect both capsid transport and targeting in neurons.
Materials and Methods
Virus and Cells. All recombinant viruses were derived from the pBecker3 infectious clone of pseudorabies virus (PRV)-Becker, a virulent PRV isolate (11). Viruses were propagated in pig kidney epithelial cells (PK15). Viral growth rates were measured by single-step analysis, and titers were determined by plaque assay as described in ref. 12. Dissociated sensory neurons from chick dorsal root ganglia (DRG) were cultured as previously described, whereas DRG explants were grown on poly(dl)-ornithine and laminin (9, 13). Neurons were cultured for 2–3 d before viral infection.
Virus Construction. PRV-GS847 encodes mRFP1 fused to the VP26 capsid protein as described in ref. 7. The dual-fluorescent and FLAG-tagged viruses were derived from the infectious clone (pGS847) encoding PRV-GS847. The alleles encoding GFP-VP1/2 and FLAG-VP1/2 were recombined into pGS847 by RecA-dependent homologous recombination as described in ref. 12. This recombination resulted in pGS909 and pGS957, which produced the viruses PRV-GS909 (PRV-Becker mRFP1-VP26 GFP-VP1/2) and PRV-GS957 (PRV-Becker mRFP1-VP26 FLAG-VP1/2) upon transfection into PK15 cells. PRV-GS935 is a monofluorescent virus encoding GFP-VP1/2.
Dual-fluorescent viruses encoding mRFP1-VP26 and UL37-GFP (PRV-GS1022), VP16-GFP (PRV-GS1046), GFP-VP13/14 (PRV-GS1023), or GFP-VP22 (PRV-GS1215) were isolated by recombining a PCR product, encoding a kanamycin-resistance gene flanked by FLP-recombination target (FRT) sites and adjacent to the GFP coding sequence, into pGS847. The 5′ 40 nt of the PCR primers were homologous to the site of insertion in pGS847, and linear DNA recombination was achieved with the EL250 strain of Escherichia coli (14). The kanamycin cassette was subsequently removed with Flp recombinase, and a resulting clone was transfected into PK15 cells from which virus was harvested. In the case of GFP-VP13/14 and GFP-VP22, the remaining FRT site and the adjacent sequence in the viral genome coded for 15 aa at the GFP–tegument-protein fusion junction. The remaining FRT site in UL37-GFP and VP16-GFP were preceded by a stop codon and were noncoding.
Transfection of Infectious Clones. PK15 cells (≈2,000,000) were harvested with trypsin and centrifuged at 300 × g for 5 min. The pellet was washed twice with PBS, and the final pellet was resuspended in 0.25 ml of PBS. The cells were mixed with infectious clone DNA isolated from 1 ml of stationary E. coli culture then transfected by using an ECM 630 electroporation system (BTX Instrument Division, Harvard Apparatus). The cells were pulsed twice with the following settings: 425 V, 0 Ω, and 25 μF. The transfected cells were cultured in DMEM plus 2% FBS, and virus was harvested when all cells exhibited a cytopathic effect.
Virus Purification. Three 10-cm dishes of confluent PK15 cells were infected at a multiplicity of infection of 10. The supernatants were pooled at 17 h postinfection (hpi), and cell debris was removed by centrifugation at 300 × g for 5 min. The cleared supernatant was transferred to a Beckman SW28 centrifugation tube, and 8 ml of 30% sucrose was layered underneath. The sample was spun at 70,000 × g for 3 h at 4°C. The pellet was resuspended in 1 ml of DMEM supplemented with 10% FBS, dispersed by sonication in an ultrasonic processor (VCX-500, Sonics & Materials, Newtown, CT), layered on top of 1 ml of 30% sucrose in a Beckman SW50.1 centrifugation tube, and spun at 28,000 rpm for 1.5 h at 4°C. The pellet was resuspended in 0.1 ml of PBS.
Western Blot Analysis. Equal volumes of purified virions and 2× final sample buffer (10 mM Tris, pH 7.4/150 mM NaCl/1% Triton X-100) were mixed and boiled for 3 min. Fifteen microliters of each sample was electrophoresed through either a 7.5% or a 4–15% (gradient) SDS/polyacrylamide gel (Bio-Rad) then transferred onto a Hybond ECL membrane (Amersham Pharmacia). Western blots were performed as described in ref. 15. The mouse anti-GFP antibody was used at a 1:2,000 dilution, and the M2 anti-FLAG antibody (Sigma) was used at a 1:1,000 dilution. The secondary goat anti-mouse horseradish peroxidase (HRP)-conjugated antibody (Jackson ImmunoResearch) was used at a 1:10,000 dilution. HRP was detected with a luminol–coumaric acid–H2O2 solution, and exposed film was digitized with an EDAS 290 documentation system (Kodak).
Fluorescence Microscopy. The microscope used for these studies was an inverted wide-field Nikon Eclipse TE2000-U equipped with automated fluorescence filter wheels (Sutter Instruments, Novato, CA) and a Cascade:650 camera (Photometrics, Roper Scientific). The microscope was housed in a 37°C environmental box (Life Imaging Services, Reinach, Switzerland), and infected cells were imaged in sealed chambers as described in ref. 9. Image acquisition and processing was done by using the software package metamorph (Universal Imaging, Downingtown, PA). Static images of released viral particles were acquired with a ×100 1.45-numerical aperture (N.A.) oil objective. Time-lapse imaging of mRFP1 and GFP emissions was achieved by automated sequential capture, by using a ×60 1.4-N.A. oil objective with 200-ms exposure times for each channel. Transport to the cell body was imaged in infected DRG explants up to 1 hpi. Transport to axon terminals was imaged in infected disassociated DRG neurons at 7–12 hpi (early egress) and 12–15 hpi (peak egress).
Results
Isolation and Initial Characterization of Dual-Fluorescent Viruses. Five recombinants of PRV, an α-herpesvirus of broad host range, were made by inserting the coding sequence of egfp into a tegument-encoding gene within the full-length viral genome. The parental viral genome was previously engineered to express mRFP1 capsids (7). The five tegument proteins examined in this report were VP1/2, UL37, VP16, VP13/14, and VP22 (see Fig. 7A, which is published as supporting information on the PNAS web site). Single-step viral growth kinetics of these five viruses were not notably affected by the presence of the mRFP1 fused to the VP26 capsid protein or GFP fused to the tegument proteins (Fig. 7B).
Tegument proteins are structural components of the herpesvirus virion (reviewed in ref. 16). We examined, by Western blot analysis, the incorporation of the GFP tegument proteins encoded by each virus into released extracellular viral particles. Three GFP-VP1/2 bands were observed: two prominent bands of ≈250 kDa and one dimmer band of greater mass (Fig. 1A). To confirm that the three bands were expected variants of the VP1/2 tegument protein, a recombinant virus expressing a FLAG-epitope-tagged VP1/2 protein was also examined by Western blot and found to express three similar bands, each of a slightly reduced molecular mass (because of the smaller size of the FLAG epitope relative to GFP). With the remaining four viruses, a single prominent protein band from harvested extracellular viral particles reacted with an anti-GFP antibody, which corresponded to the expected molecular mass of the encoded fusion protein (Fig. 1B).
Fig. 1.

Incorporation of fluorescent fusion proteins into extracellular viral particles. Western blots of proteins from purified extracellular viral particles electrophoresed through 7.5% SDS/polyacrylamide (A) or 4–15% gradient SDS/polyacrylamide gels (B) are shown. Blots were probed with an anti-GFP antibody and, in one case, reprobed with an anti-FLAG antibody (A; FLAG-VP1/2 lane in image at right resulted from reprobing indicated lane in left image). Viruses expressing GFP-VP1/2 alone or in combination with mRFP1-VP26 showed similar GFP-VP1/2 incorporation. (C) Illustration depicting method used to image newly released fluorescent viral particles from cells transfected with recombinant herpesvirus DNA. (D) Imaging of individual diffraction-limited fluorescent virions, as illustrated in C, at 2 d posttransfection. Labels at the left indicate the GFP fusion present in each sample, and the percentages at the right indicate the fraction of mRFP1 particles that also emit GFP fluorescence. All images are 10 μm × 10 μm.
Incorporation of the GFP tegument proteins into released viral particles was further assessed by fluorescence microscopy. Newly released viral particles from infected PK15 cells were imaged on regions of a coverslip devoid of cells (Fig. 1C). Infection of PK15 cells was initiated by transfection of a full-length infectious clone encoding each virus; therefore, fluorescence emissions were restricted to de novo synthesized proteins and not input inoculum. Red fluorescent punctae, consistent with emission from single viral particles, were also observed to emit green fluorescence for all viruses (Fig. 1D) (7, 9). Occasionally, punctae of green or red fluorescence alone were observed. The former were observed with each virus and are likely “light particles,” which consist of tegument and envelope but lack capsids (17). Particles observed to emit red fluorescence alone were generally limited to the VP16-GFP (8% of the total), GFP-VP13/14 (38% of the total), and GFP-VP22 (34% of the total) viruses and appeared to result from highly variable emission intensities from particle to particle, with the low range either beyond detection or, possibly, lacking incorporation altogether. Consistent with these findings, the GFP emissions from the GFP-VP13/14 and GFP-VP22 viruses were ≈10-fold lower than those of the other three dual-fluorescent viruses, which likely resulted in the larger proportions of viral particles lacking detectable GFP fluorescence (the GFP images of the latter two viruses were scaled to bring out the dim emissions in Fig. 1C).
Viral Particle Transport and Composition After Entry into Sensory Neurons. Although the gross subcellular localization of tegument proteins after infection has been examined in fixed cells, whether tegument proteins remain associated with capsids during transport to and docking at nuclear pores is not known (18). To address this question, translocation of mRFP1 and GFP emissions from individual dual-fluorescent viral particles were tracked in axons after infection of primary sensory neurons. A correspondence in motion of the two emission profiles was used as an indication of cotransport of the two components as a complex, a procedure we refer to as correlative-motion analysis.
Within the first hpi, mRFP1 punctae were frequently observed transporting in axons toward sensory neuron cell bodies in the explant center. The fluorescent signals were consistent with the emission profile previously observed for individual capsids (7, 9). mRFP1 capsids traveled predominantly in the retrograde direction, and the dynamics of transport were as expected based on previous studies (data not shown) (7). Detection of cotransport of mRFP1 and GFP signals was achieved by sequentially imaging mRFP1 followed by GFP continuously with 200-ms exposure times for each channel. Because imaging of the fluorophores was staggered, a motile dual-fluorescent particle was expected to emit a displaced mRFP1 and GFP signal, with the amount of displacement proportional to the velocity of the particle. Based on this consideration, both the VP1/2- and UL37-tagged viruses had GFP and mRFP1 signals transported in axons as single units (see Movie 1, which is published as supporting information on the PNAS web site). In contrast, mRFP1 capsids lacked associated GFP emissions in neurons infected with the VP16-, VP13/14-, or VP22-tagged viruses (Figs. 2 and 3A). The 3.3% of mRFP1 capsids in cells infected with UL37-tagged virus that lacked detectable GFP emissions may have resulted from particles moving slightly out of the focal plane (the high-contrast mRFP1 emissions required less optimal focus to detect than did the dimmer GFP emissions) (Fig. 3A).
Fig. 2.

Transport of capsids and tegument proteins in axons of sensory neurons after viral entry. Correlative motion analysis of viral-particle transport in axons immediately after exposure of cultured DRG explants to dual-fluorescent viruses is shown. Frames of alternating mRFP1 (R) and GFP (G) emissions are shown for each virus from left to right. All viruses express mRFP1-VP26 (capsid) and the indicated tegument protein fused to GFP. Frames from the recordings are oriented with the explant above the field of view (i.e., retrograde transport is from bottom to top). Capsids are ≈125 nm in diameter, making the emissions diffraction-limited. As such, the apparent size of the particles is proportional to their brightness and not to their physical dimensions. All frames are 2.5 μm × 14.1 μm.
Fig. 3.

Summary of herpesvirus axonal transport. Quantitation of capsid transport in axons is plotted with the proportion of capsids moving anterograde (to the axon terminal) above and retrograde (to the cell body) below. The proportion of transported capsids alone and capsids complexed with tegument is presented as the percentage of the total number of transported capsids (n) observed for each virus. (A) Capsid transport to the soma during the first hpi. (B) Capsid transport to axon terminals from 12 to 15 hpi.
The fate of capsid–tegument complexes posttransport was examined by imaging the nuclear rims of neuronal cell bodies within the DRG explant 2 hpi (Fig. 4). Infection with each of the five viruses resulted in the accumulation of individual mRFP1 capsid emissions at nuclear rims, presumably as a result of capsid docking at nuclear pore complexes (19–22). The GFP emissions of the VP1/2- and UL37-tagged viruses were coincident with the mRFP1 signals at the nuclear rims, whereas the remaining viruses consistently lacked nuclear-rim-associated GFP signals.
Fig. 4.

Accumulation of capsids and tegument proteins at the nuclear rims of infected sensory neurons after retrograde transport. (A) Differential interference-contrast image of the center of a DRG explant infected with the mRFP1-VP26–GFP-VP1/2 virus (2 hpi). The fluorescence signals associated with the nucleus of a sensory neuron (boxed) are shown below. (B) Nuclear rim images of the remaining dual-fluorescent viruses, carried out as in A. Labels at the left indicate the GFP fusion present in each infection.
Viral Particle Transport and Composition During Egress. To determine whether tegument proteins associate with capsids during transport from the cell body to axon terminals, correlative motion analysis was used to monitor particle transport and assembly in axons during the egress phase of infection. mRFP1 capsids were transported primarily in the anterograde direction (toward axon terminals), although processive retrograde motion was also observed at a lower frequency due to spontaneous reversals in transport direction, as described in ref. 9. Infected neurons were imaged from 12 to 15 hpi, when egressing capsids were observed transporting to axon terminals most frequently (9). Similar to transport after entry, GFP signals were detected associated with nearly all mRFP1 capsids from the VP1/2- and UL37-tagged viruses. In contrast to entry transport to the nucleus, cotransport of mRFP1 and GFP signals was observed with the remaining three viruses as well, indicating a difference in composition of the capsid-transport complex when targeted to the nucleus or distal axon (Figs. 3B and 5). Association of mRFP1 capsids with detectable GFP tegument emissions was not absolute. This was most notable with the GFP-VP13/14 and GFP-VP22 viruses and, to a lesser extent, with the viruses encoding UL37-GFP and VP16-GFP. This heterogeneity in GFP fluorescence paralleled that observed for released extracellular viral particles (Fig. 1D), consistent with viral particles transporting to axon terminals having an equivalent tegument composition as released extracellular virions.
Fig. 5.

Transport of capsids and tegument proteins in axons of sensory neurons during viral egress. Correlative motion analysis of de novo assembled viral particles during egress is shown. Images were captured between 7 and 15 hpi in axons of dissociated DRG sensory neurons. Frames of alternating mRFP1 (R) and GFP (G) emissions are shown for each virus from left to right. All viruses express mRFP1-VP26 (capsid) and the indicated tegument protein fused to GFP. Frames from the recordings are oriented with the neuronal cell body above the field of view (i.e., anterograde transport is from top to bottom). All frames are 2.1 μm × 15.2 μm.
The VP16-GFP, GFP-VP13/14, and GFP-VP22 fusion proteins were frequently observed transporting in axons independently of capsids (Fig. 5 and Movie 2, which is published as supporting information on the PNAS web site). These GFP emissions could often be detected in axon terminals before accumulation of mRFP1 capsids (see below). The emissions from capsid-independent VP16-GFP often ceased to be diffraction-limited, appearing instead as slightly elongated structures (Fig. 5). Independent transport of GFP-VP1/2 or UL37-GFP was never observed.
The fate of capsid and tegument transport during egress was examined by imaging axon terminals. With each of the dual-fluorescent viruses, mRFP1 capsids were often accumulated throughout the axon terminal to varying degrees. Nearly all mRFP1 and GFP signals colocalized at the axon terminals of neurons infected with the VP1/2-, or UL37-tagged viruses. mRFP1 emissions were also associated with GFP in neurons infected with the VP16-, VP13/14-, and VP22-tagged viruses; however, with these three viruses, GFP punctae lacking mRFP1 emissions typically outnumbered punctae emitting fluorescence from both fusion proteins, often >10-fold (Fig. 6). The large proportion of GFP monofluorescent punctae resulted from accumulation of these particles in advance of dual-fluorescent capsid-transport complexes and could be seen as early as 7–12 hpi (Fig. 6). Therefore, anterograde movement of the VP16, VP13/14, and VP22 tegument proteins occurs both independently of, and in association with, the core VP1/2–UL37 capsid-transport complex.
Fig. 6.

Accumulation of capsids and tegument proteins at axon terminals during viral egress. Axon terminals of dissociated DRG sensory neurons infected with the dual-fluorescent viruses were imaged 12–15 or 7–12 hpi (VP13/14 early) for accumulation of capsid and tegument proteins. Images of mRFP1 capsids, GFP tegument, and an overlay are shown for each virus. Labels at left indicate the GFP fusion expressed in each infection. All images are 33 μm × 39 μm. Axon terminals had varied morphologies typical of growth cones.
Discussion
Herpesvirus capsids possess a targeting mechanism that directs their transport in neurons to either the cell body or the distal axon at distinct stages of infection (7). However, the structure of the capsid is not known to vary, and whether additional viral factors associate with capsids to regulate intracellular capsid trafficking has remained unclear. Although more than a dozen interactions between herpesvirus proteins and components of host microtubulemotor complexes have been detected by in vitro and two-hybrid studies, interactions required for capsid transport have yet to be found (23–29). For this reason, determining the composition of the actively transported capsid complex was a necessary prerequisite to understanding the mechanism of transport and its regulation. We show here that herpesvirus tegument proteins are constituents of capsid-transport complexes. Furthermore, the composition of these complexes is dynamic, such that alterations in tegument makeup are coupled with trafficking of capsids to opposing locations inside neurons.
To examine the composition of the capsid-transport complex, capsid and tegument proteins were simultaneously tracked in living neurons. A collection of recombinant viruses was isolated that express mRFP1 fused to the capsid and GFP fused to one of five different viral tegument proteins. The mRFP1 protein was a boon for these studies because, unlike commercial red fluorescent proteins (i.e., DsRed and HcRed), mRFP1 is monomeric and is therefore appropriate for fusion studies of protein localization in cells (10). Additionally, the sequences of the mRFP1 and egfp ORFs are sufficiently diverged as to remove concern of recombination between the two in the context of the viral genome during infection. Each of the recombinant viruses grew with wild-type kinetics, and the fusion proteins were incorporated into released virions. From these results, we conclude that the presence of the two fusions was not deleterious to the infection process in cell culture. However, other GFP fusions (GFP fused to the N terminus of VP16 or UL37) were found to compromise viral growth and were not further pursued (data not shown).
Viral transport was tracked in axons of primary sensory neurons as diffraction-limited fluorescent emissions (the herpesvirus capsid is ≈125 nm in diameter). Correlated motion between red and green fluorescence provided a readout for the presence of specific tegument proteins in the capsid-transport complex. This method, which we refer to as “correlative motion analysis,” was facilitated by the spectral separation of GFP from mRFP1 and an electron-multiplying charge-coupled device.
Neurotropic herpesviruses encode ≈15 tegument proteins, at least two of which, VP1/2 and UL37, remained associated with capsids as they traveled toward the nucleus and ultimately accumulated at the nuclear rim after entry into neurons. However, three other tegument proteins examined, VP16, VP13/14, and VP22, were removed from the capsid surface before retrograde transport. The absence of VP16, VP13/14, and VP22 from transported capsids is consistent with this transport occurring independently of endosomal trafficking, because viral particles potentially entering axons by endocytosis would remain intact until fusion of viral and cellular membranes occurred (4, 7). The fate of the VP16, VP13/14, and VP22 proteins was unclear because GFP emissions could not be detected after viral entry into axons, indicating that the proteins may have diffused in the cytosol.
During the egress of progeny capsids from infected neurons, all five tegument proteins examined (VP1/2, UL37, VP16, VP13/14, and VP22) transported with capsids. Cotransport was most evident with the GFP-VP1/2, UL37-GFP, and VP16-GFP viruses, each of which emitted bright GFP fluorescence. Although observation of GFP-VP13/14 and GFP-VP22 cotransport with capsids was less consistent, we expect that this lack of consistency was an imaging limitation because these viruses had highly variable GFP emission levels that were, on average, ≈10-fold lower than those of the other three viruses (Fig. 1D). Furthermore, detection of GFP emissions from moving particles in neurons was more challenging than imaging static extracellular viral particles. We note that the variability in GFP emissions indicates that the composition of fully assembled viral particles is highly heterogeneous, which is consistent with a recent observation of VP22 incorporation levels in virions (40).
VP16, VP13/14, and VP22 frequently underwent capsid-independent anterograde transport in axons during the egress phase of infection. In some neurons, as much as 90% of fluorescent viral punctae moving to distal axons consisted of VP16-GFP that lacked mRFP1 capsid fluorescence (Movie 2). This transport resulted in a subpopulation of these proteins at axon terminals that were spatially distinct from accumulated capsids. Capsid-independent transport of tegument proteins began in advance of capsid transport, and the occasional appearance of VP16-GFP as dim tubular structures may indicate that this transport occurs as part of the vesicular secretory pathway (for example, see ref. 30). Viral membrane protein transport has also been noted to occur in advance of capsids and, by definition, is membrane-bound (31, 32). Whether viral tegument and membrane proteins transport as a complex will require further studies, but transport as a complex seems likely.
Our findings are consistent with capsid acquisition of a final complement of tegument proteins before axonal transport to the periphery (33). Because several tegument proteins bind to viral membrane proteins, including the VP16 and VP22 proteins examined in this report, the egressing capsid-transport complex may be a fully assembled infectious particle (34–37). If true, the independent transport of tegument proteins may represent a constitutive process into which capsids can be fed. Unfortunately, examining whether egressing viral particles in axons are mature enveloped structures has produced contradictory results (6, 8, 38). It remains possible that capsids egress in axons as protein complexes in the cytosol or as fully assembled virions within the secretory pathway. Because the tegument proteins are cytosolic, they are well positioned to direct trafficking of the viral particle in either scenario (either on the capsid or around the cytosolic surface of a transport vesicle).
In summary, we find that capsids transport in axons while complexed to tegument proteins, and the composition of the capsid-transport complex is distinct during the entry and egress phases of infection (see Fig. 8, which is published as supporting information on the PNAS web site). Because VP1/2 and UL37 are basal components of the capsid-transport complex, they are prime candidates for viral factors that may interact with host transport machinery, such as the dynein–dynactin complex, resulting in net retrograde transport to the soma and nucleus of neurons (20). Additional tegument proteins, including VP16, VP13/14, and VP22, undergo anterograde transport apart from capsids, and their association with progeny capsids may direct viral egress to the periphery late in infection. Determining the mechanism of capsid trafficking in axons is essential to understanding herpesvirus pathogenesis. Furthermore, the ease of manipulating the virus provides a model for studying the fundamental cellular process of coordinated bidirectional transport.
Supplementary Material
Acknowledgments
We thank Joy Lee for critical evaluation of the manuscript, Gary Borisy and Anjen Chenn for additional suggestions, Tony Del Rio for assistance in virus purification, and David Smith and the Fryer Company (Huntley, IL) for microscopy support. We also thank Roger Tsien (University of California at San Diego, La Jolla) for development and sharing of the mRFP1 fluorescent protein, and Dane Chetkovich (Northwestern University) for anti-GFP antibody. This work was supported by National Institutes of Health Grant 1R01AI056346 and a Schweppe Foundation award (to G.A.S.). G.W.G.L. was supported, in part, by a travel award from the Center for Genetic Medicine of Northwestern University.
Author contributions: G.W.G.L. and G.A.S. designed research; G.W.G.L., S.H., K.E.C., S.E.A., A.P., and G.A.S. performed research; G.W.G.L. and G.A.S. analyzed data; G.W.G.L. and G.A.S. wrote the paper; and S.H., K.E.C., S.E.A., and A.P. contributed new reagents/analytic tools.
Abbreviations: DRG, dorsal root ganglion; hpi, h postinfection; mRFP1, monomeric red fluorescent protein 1; PRV, pseudorabies virus.
See Commentary on page 5639.
References
- 1.Rixon, F. J. (1993) Semin. Virol. 4, 135–144. [Google Scholar]
- 2.Zhou, Z. H., Chen, D. H., Jakana, J., Rixon, F. J. & Chiu, W. (1999) J. Virol. 73, 3210–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grunewald, K., Desai, P., Winkler, D. C., Heymann, J. B., Belnap, D. M., Baumeister, W. & Steven, A. C. (2003) Science 302, 1396–1398. [DOI] [PubMed] [Google Scholar]
- 4.Lycke, E., Hamark, B., Johansson, M., Krotochwil, A., Lycke, J. & Svennerholm, B. (1988) Arch. Virol. 101, 87–104. [DOI] [PubMed] [Google Scholar]
- 5.Marchand, C. F. & Schwab, M. E. (1986) Brain Res. 383, 262–270. [DOI] [PubMed] [Google Scholar]
- 6.Lycke, E., Kristensson, K., Svennerholm, B., Vahlne, A. & Ziegler, R. (1984) J. Gen. Virol. 65, 55–64. [DOI] [PubMed] [Google Scholar]
- 7.Smith, G. A., Pomeranz, L., Gross, S. P. & Enquist, L. W. (2004) Proc. Natl. Acad. Sci. USA 101, 16034–16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penfold, M. E. T., Armati, P. & Cunningham, A. L. (1994) Proc. Natl. Acad. Sci. USA 91, 6529–6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith, G. A., Gross, S. P. & Enquist, L. W. (2001) Proc. Natl. Acad. Sci. USA 98, 3466–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A. & Tsien, R. Y. (2002) Proc. Natl. Acad. Sci. USA 99, 7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Card, J. P., Rinaman, L., Schwaber, J. S., Miselis, R. R., Whealy, M. E., Robbins, A. K. & Enquist, L. W. (1990) J. Neurosci. 10, 1974–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith, G. A. & Enquist, L. W. (1999) J. Virol. 73, 6405–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith, C. L. (1998) in Culturing Nerve Cells, eds. Banker, G. & Goslin, K. (MIT Press, Cambridge, MA), pp. 261–287.
- 14.Lee, E. C., Yu, D., Martinez de Velasco, J., Tessarollo, L., Swing, D. A., Court, D. L., Jenkins, N. A. & Copeland, N. G. (2001) Genomics 73, 56–65. [DOI] [PubMed] [Google Scholar]
- 15.Smith, G. A. & Enquist, L. W. (2000) Proc. Natl. Acad. Sci. USA 97, 4873–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mettenleiter, T. C. (2002) J. Virol. 76, 1537–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szilagyi, J. F. & Cunningham, C. (1991) J. Gen. Virol. 72, 661–668. [DOI] [PubMed] [Google Scholar]
- 18.Morrison, E. E., Stevenson, A. J., Wang, Y. F. & Meredith, D. M. (1998) J. Gen. Virol. 79, 2517–2528. [DOI] [PubMed] [Google Scholar]
- 19.Sodeik, B., Ebersold, M. W. & Helenius, A. (1997) J. Cell Biol. 136, 1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dohner, K., Wolfstein, A., Prank, U., Echeverri, C., Dujardin, D., Vallee, R. & Sodeik, B. (2002) Mol. Biol. Cell 13, 2795–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ojala, P. M., Sodeik, B., Ebersold, M. W., Kutay, U. & Helenius, A. (2000) Mol. Cell. Biol. 20, 4922–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Batterson, W., Furlong, D. & Roizman, B. (1983) J. Virol. 45, 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye, G. J., Vaughan, K. T., Vallee, R. B. & Roizman, B. (2000) J. Virol. 74, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diefenbach, R. J., Miranda-Saksena, M., Diefenbach, E., Holland, D. J., Boadle, R. A., Armati, P. J. & Cunningham, A. L. (2002) J. Virol. 76, 3282–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Douglas, M. W., Diefenbach, R. J., Homa, F. L., Miranda-Saksena, M., Rixon, F. J., Vittone, V., Byth, K. & Cunningham, A. L. (2004) J. Biol. Chem. 279, 28522–28530. [DOI] [PubMed] [Google Scholar]
- 26.Martinez-Moreno, M., Navarro-Lerida, I., Roncal, F., Albar, J. P., Alonso, C., Gavilanes, F. & Rodriguez-Crespo, I. (2003) FEBS Lett. 544, 262–267. [DOI] [PubMed] [Google Scholar]
- 27.Ogawa-Goto, K., Irie, S., Omori, A., Miura, Y., Katano, H., Hasegawa, H., Kurata, T., Sata, T. & Arao, Y. (2002) J. Virol. 76, 2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diefenbach, R. J., Diefenbach, E., Douglas, M. W. & Cunningham, A. L. (2004) Biochem. Biophys. Res. Commun. 319, 987–992. [DOI] [PubMed] [Google Scholar]
- 29.Nishiyama, Y. (2004) Rev. Med. Virol. 14, 33–46. [DOI] [PubMed] [Google Scholar]
- 30.Nakata, T., Terada, S. & Hirokawa, N. (1998) J. Cell Biol. 140, 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miranda-Saksena, M., Armati, P., Boadle, R. A., Holland, D. J. & Cunningham, A. L. (2000) J. Virol. 74, 1827–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holland, D. J., Miranda-Saksena, M., Boadle, R. A., Armati, P. & Cunningham, A. L. (1999) J. Virol. 73, 8503–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miranda-Saksena, M., Boadle, R. A., Armati, P. & Cunningham, A. L. (2002) J. Virol. 76, 9934–9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs, W., Klupp, B. G., Granzow, H., Hengartner, C., Brack, A., Mundt, A., Enquist, L. W. & Mettenleiter, T. C. (2002) J. Virol. 76, 8208–8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross, S. T., Harley, C. A. & Wilson, D. W. (2003) Virology 317, 1–12. [DOI] [PubMed] [Google Scholar]
- 36.Ng, T. I., Ogle, W. O. & Roizman, B. (1998) Virology 241, 37–48. [DOI] [PubMed] [Google Scholar]
- 37.Chi, J. H., Harley, C. A., Mukhopadhyay, A. & Wilson, D. W. (2005) J. Gen. Virol. 86, 253–261. [DOI] [PubMed] [Google Scholar]
- 38.Card, J. P., Rinaman, L., Lynn, R. B., Lee, B. H., Meade, R. P., Miselis, R. R. & Enquist, L. W. (1993) J. Neurosci. 13, 2515–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klupp, B. G., Fuchs, W., Granzow, H., Nixdorf, R. & Mettenleiter, T. C. (2002) J. Virol. 76, 3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.del Rio, T., Ch'ng, T. H., Flood, E. A., Gross, S. P. & Enquist, L. W. (2005) J. Virol., in press. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}