

Abstract
Objective:
To investigate whether creatine administration could slow progressive functional decline in adults with early symptoms of Huntington disease.
Methods:
We conducted a multicenter, randomized, double-blind, placebo-controlled study of up to 40 g daily of creatine monohydrate in participants with stage I and II HD treated for up to 48 months. The primary outcome measure was the rate of change in total functional capacity (TFC) between baseline and end of follow-up. Secondary outcome measures included changes in additional clinical scores, tolerability, and quality of life. Safety was assessed by adverse events and laboratory studies.
Results:
At 46 sites in North America, Australia, and New Zealand, 553 participants were randomized to creatine (275) or placebo (278). The trial was designed to enroll 650 patients, but was halted for futility after the first interim analysis. The estimated rates of decline in the primary outcome measure (TFC) were 0.82 points per year for participants on creatine, 0.70 points per year for participants on placebo, favoring placebo (nominal 95% confidence limits −0.11 to 0.35). Adverse events, mainly gastrointestinal, were significantly more common in participants on creatine. Serious adverse events, including deaths, were more frequent in the placebo group. Subgroup analysis suggested that men and women may respond differently to creatine treatment.
Conclusions:
Our data do not support the use of creatine treatment for delaying functional decline in early manifest HD.
Clinicaltrials.gov identifier:
Classification of evidence:
This study provides Class II evidence that for patients with early symptomatic HD, creatine monohydrate is not beneficial for slowing functional decline.
Huntington disease (HD) is a dominantly inherited neurodegenerative disorder characterized by progressive motor, psychiatric, and cognitive disturbances for which no treatment has been demonstrated to slow clinical decline. HD is caused by the mutant huntingtin protein (mtHtt), which triggers a cascade of molecular perturbations, including impaired energy production, which has been considered a target for disease modification.1 Creatine is a nutritional supplement that is converted to phosphocreatine, which acts as a high-energy phosphate source for restoring adenosine triphosphate (ATP) from adenosine diphosphate.2 In preclinical studies using toxin and transgenic mouse models of HD, creatine delays the onset and slows progression of behavioral and pathologic phenotypes, reverses cerebral ATP deficiency, and extends survival in a dose-dependent manner.3–6 In our initial randomized controlled study in early (stage 1 and II) HD (Creatine Therapy for Huntington's Disease [CREST-HD]), 8 g of creatine daily significantly reduced plasma levels of 8-hydroxy 2-deoxyguanosine (8OH2'dG), a marker of oxidative DNA injury.7 We also completed an 18-month randomized placebo-controlled phase II secondary prevention trial (Creatine Safety and Tolerability in Premanifest HD [PRECREST]) in 70 at-risk and presymptomatic HD participants using 35 g daily, which was well-tolerated and significantly slowed regional brain atrophy in prodromal participants.8 The current study was designed to test the efficacy of high doses of creatine for slowing the progressive functional decline of patients with early manifest HD, in whom a 25% slowing of the total functional capacity (TFC) score is considered clinically significant.
METHODS
Study product.
Active and placebo study drug were manufactured following Current Good Manufacturing Practices under an Investigational New Drug application from the US Food and Drug Administration (FDA). The active pharmaceutical ingredient, creatine monohydrate, was manufactured by AlzChem GmbH (Trostberg, Germany) and shipped to Deseret Laboratories Inc. (St. George, UT) for primary packaging of packets of 7 g each (active packets = 5 g creatine monohydrate and 2 g dextrose; placebo packets = 7 g dextrose). The Clinical Materials Services Unit at the University of Rochester managed packaging, labeling, and distribution to the study sites. Participants were instructed to mix the contents with food or nonclear liquids twice daily with recommendations for avoiding excessive heat and acidity.
Standard protocol approvals, registrations, and patient consents.
Participants were recruited at 46 Huntington Study Group sites in the United States, Canada, Australia, and New Zealand. Protocols and consent procedures for participants and caregivers, consistent with the Declaration of Helsinki, were approved by applicable regulatory agencies, local institutional review boards, or ethics committees. Creatine Safety, Tolerability & Efficacy in Huntington’s Disease (CREST-E) is registered on ClinicalTrials.gov (NCT00712426).
Study design.
We hypothesized that chronic treatment of early manifest HD with high-dose creatine would slow progressive functional decline in early HD (stage I and II) as measured by the TFC scale, a component of the Unified Huntington's Disease Rating Scale (UHDRS).9,10 We designed CREST-E as a multicenter, randomized, double-blind, placebo-controlled, parallel-group study of 40 g daily of creatine monohydrate. The original design included assessing change in study outcome measures after 36 months of treatment. Due to low enrollment and following review and approval by the Data Safety Monitoring Board (DSMB), the design was modified in September 2011 to permit follow-up durations of 18–48 months and annualized assessment of change. Of 151 participants who completed the originally planned 36 months of treatment, 110 consented to 12 months of extended follow-up. Participants titrated to their highest tolerated dose (5–40 g daily). Doses higher than 5 g daily were divided. Unlimited dose adjustments were permitted to help maintain long-term tolerability.
Prespecified secondary measures included the UHDRS total motor score (TMS) and a composite of the verbal fluency, symbol digit modalities, and Stroop interference cognitive tests. The primary tolerability measure was study completion on at least 5 g daily. Frequency and severity of adverse events (AEs), changes in vital signs, and laboratory tests served as safety measures. Exploratory measures assessed clinical symptoms (UHDRS subscores), independence, daily function, clinical global impressions, quality of life, caregiver burden, cognition (Hopkins Verbal Learning, Benton Judgment of Line Orientation), biology (plasma creatine, 8OH2'dG,7 and metabolomic markers; huntingtin protein and RNA markers in blood; CAG mutation length [CAGn]), and MRI measures in a subset of 93 participants from 4 sites. Interim safety, futility, and efficacy analyses were planned in coordination with the Steering Committee, DSMB, and FDA.
Eligibility.
Eligible participants were in stage I or II of HD (TFC ≥7), were ≥18 years old, had a confirmatory family history or CAGn ≥36, were capable of providing informed consent and complying with procedures, and were on stable doses of any concomitant psychotropic medications. Women of childbearing potential could not be pregnant or lactating and were required to use adequate contraception methods. Participants were excluded for screening creatinine levels >2.0 mg/dL or other laboratory abnormalities suggesting risk during the study. Exposure to investigational drugs, supplemental creatine at doses >10 g daily or supplemental coenzyme Q10 at doses >600 mg daily within 30 days of randomization, or unstable psychiatric or medical illness were disallowed.
Randomization and blinding.
Randomized treatment assignments (1:1, creatine or matching placebo) were generated by a designated unblinded biostatistics programmer using a SAS (Cary, NC) program and stratified within site with block size 4. Study personnel, participants, caregivers, steering committee members, and NIH program staff were blinded to treatment assignment until study conclusion. Creatinine elevations were expected since creatinine is the primary metabolite of creatine. All creatinine values after screening were redacted and reviewed by the clinical monitor. Serum creatinine levels >2.0 mg/dL led to dose suspensions, close monitoring, and resumption or discontinuation of drug depending on resolution within 12 weeks. Random suspensions generated by an unblinded statistician were instituted so that suspensions for cause would not be unblinding. Participants were encouraged to remain in the study even after permanent drug discontinuation. A questionnaire assessed blinding of participants and investigators and the reasons for their suppositions. A designated unblinded study statistician with access to data by treatment arm prepared closed reports to the DSMB.
Procedures.
Screening, baseline, and randomization.
Prospective participants were evaluated at the screening visit after providing informed consent and randomized within 60 days at the baseline visit. Clinical and laboratory evaluations over the screening and baseline visits included demographics, medical history and concomitant medication review, vitals, weight, height, neurologic and physical examinations, TFC, UHDRS, neuropsychological tests, electrocardiogram, chemistry, hematology and urinalysis, and pregnancy testing, if applicable. Blood and urine were collected for research laboratories. The first dose (5 g) was administered on site followed by 1 hour of observation.
Titration, highest tolerated dose, and maintenance.
Dosage was increased weekly by 5 g increments starting at 10 g (divided) until 40 g daily was reached. Intermittent telephone contacts reviewed AEs, concomitant medications, and dosing issues, and gated titration to the next dose level. If intolerability was experienced, slowed titration and rechallenges were permitted for up to 6 months. Forty grams daily or the dose level immediately below an intolerable dose was considered the participant’s highest tolerated dose for maintenance.
Follow-up visits.
The assessments performed at baseline were repeated periodically during the maintenance phase, which ranged from 12 to 48 months depending on enrollment date. Review of AEs and assessment of compliance was done at every visit. Participants who were enrolled by December 31, 2011, and on study drug at the end of the original 36-month follow-up phase were eligible to continue for an extra year on their assigned treatment. Participants completed a final maintenance phase visit, followed by a posttreatment visit 30 days later.
Dose adjustments, suspensions, and discontinuations.
Dose reductions and suspensions or discontinuations occurred in response to AEs that could be related to treatment. Creatinine levels were monitored in real time by the medical monitor and a nephrology specialist was available for consultation. Elevated creatinine levels (≥2.0) triggered a 2-week suspension. A recheck of creatinine levels was performed every 2 weeks for up to 12 weeks, and study drug was restarted when creatinine returned to the normal range. If, after 12 weeks, creatinine levels had not normalized, participants were referred for nephrology workup.
Statistical analysis.
The primary hypothesis was that participants assigned to creatine would have a reduced rate of decline in TFC compared with controls. The study was originally designed to have 96% power to detect a 25% reduction in the overall rate of TFC decline, approximately 80% to detect a 20% reduction. The revised statistical analysis plan specified power of 84% to detect a 25% reduction and 66% power to detect a 20% reduction. Analyses were conducted in accordance to the intent-to-treat (ITT) principle. The original protocol specified that the primary analysis use an analysis of covariance model for the changes in TFC from baseline to 3 years including terms for baseline TFC, enrolling site, and treatment. To incorporate the protocol modification for variable lengths of follow-up, the primary analysis was changed to compare the mean annualized rates of change in TFC between the 2 groups. These were estimated by a mixed model incorporating adjustments for baseline values and site. Analyses of secondary outcome variables were conducted similarly. Data on the TMS score are also provided since it has been the primary outcome in trials examining progressive motor decline.
Interim analyses were prespecified for efficacy and futility at 50%, and for efficacy at 75%, of projected study information. Efficacy analyses were based on an O'Brien–Fleming α-spending function and the futility analysis used a predictive power criterion. Supportive analyses included changes from baseline to the 1-, 2-, and 3-year time points conducted according to the original analysis of covariance model for those participants with the requisite data. Prespecified subgroup analyses, based on age, sex, and disease severity, were also conducted. Safety and tolerability data were tabulated by treatment group and blindedness questionnaires by participant and investigator. Premature withdrawal rates and times to discontinuation of study medication were compared by the log-rank test. Data from one site (16 participants) were excluded due to Good Clinical Practice concerns identified prior to the interim analysis. This study provides Class II evidence for assessing whether creatine monohydrate is beneficial for slowing functional decline in patients with early symptomatic HD.
RESULTS
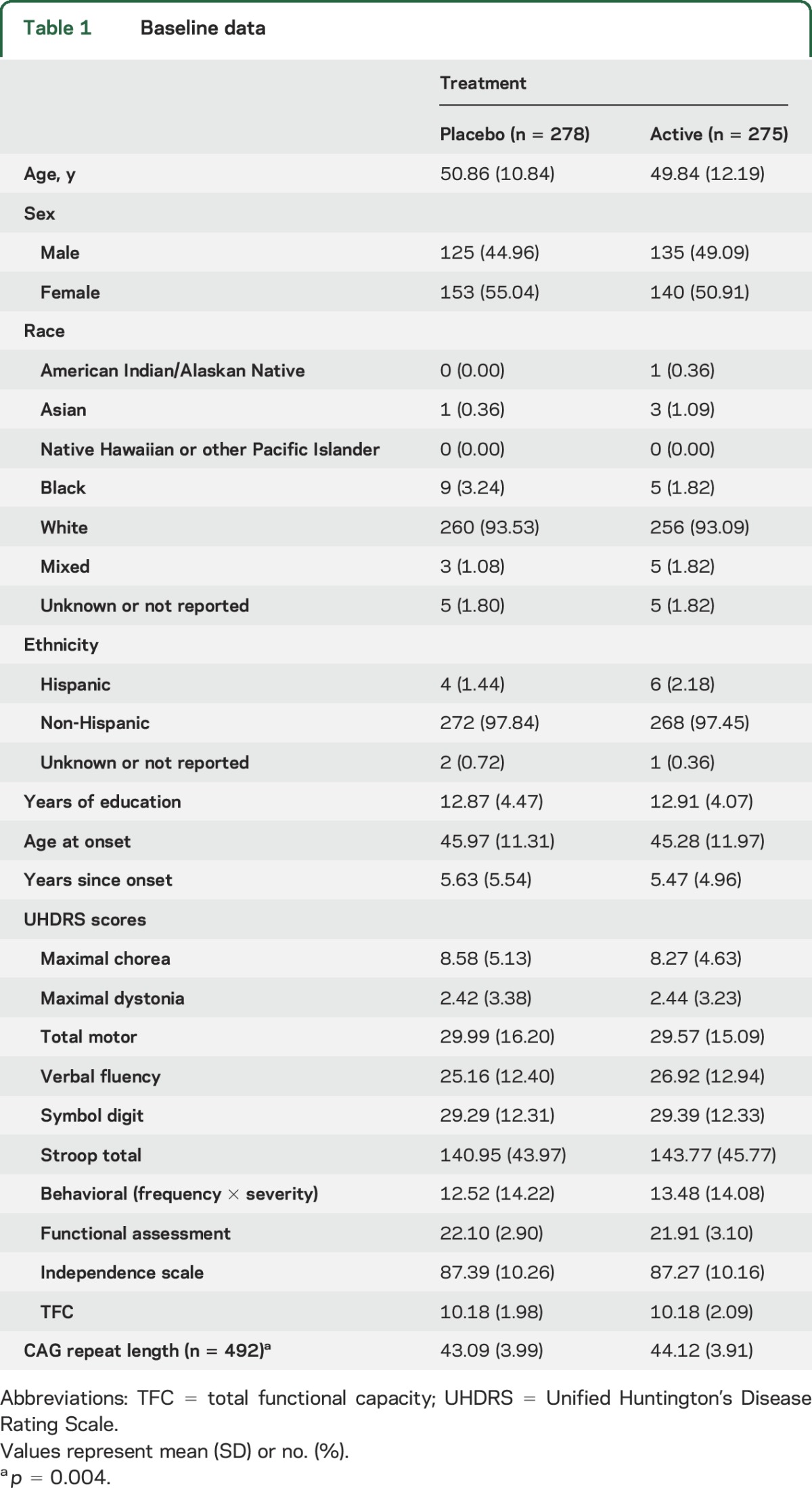
Enrollment began in September 2009 and follow-up ceased in December 2014. After 553 participants had been randomized to the creatine (275) and placebo (278) groups (figure 1), results from the first prespecified interim analysis revealed the futility stopping criterion had been reached, indicating a high likelihood that the study, if completed, would not demonstrate a beneficial effect of creatine. The DSMB recommended early termination of the trial. No safety concerns were noted. Baseline demographic and clinical characteristics were well-balanced between the 2 groups (table 1) except the mean CAG length in the creatine group was 1 unit greater than in controls. Participants had a mean (SD) age of 50 (11.5) years, with a mean (SD) age at onset of 46 (12) years and were predominantly white (93%) and female (53%). Mean baseline TFC scores in the 2 groups were similar (10.2).
Figure 1. Patient flow.

At the time of study closure, 533 (278 + 275) participants had been randomized, of whom 169 (72 + 97) had withdrawn from the study, 110 (55 + 55) had completed the study, and 274 (151 + 123) were still active in the study. The total numbers of participants with at least 2, 3, and 4 years primary outcome data at the time of study conclusion were 296, 152, and 36, respectively. Sixteen participants at one site were excluded from the analysis due to Good Clinical Practice issues identified prior to the interim analysis. Data from one participant enrolled after study closure were also excluded.
Table 1.
Baseline data
Premature withdrawals included 97 participants (35%) on creatine and 72 (26%) controls (p < 0.001). Participants who withdrew were slightly more impaired at baseline than those who did not withdraw (mean TFC 9.9 vs 10.3). The most common reasons for withdrawal were study burden and side effects. Permanent discontinuations and temporary suspensions were more common in the creatine group, the latter being primarily due to creatinine elevations. The mean dosage at 2 years was 31.6 g on creatine and 38.3 g on placebo due to a lower proportion reaching the 40 g/d dosage of creatine (58%) than on placebo (91%). Mean plasma levels of creatine at 12 months in a subset of participants were 31.9 µg/mL (SD 232.4) on placebo (n = 128) and 224.8 μg/mL (SD 268.7) on creatine (n = 118). Mean 8-OH2DG levels were 18.87 pg/mL (SD 9.26) at baseline and 16.49 pg/mL (SD 5.85) in participants taking creatine compared to 17.05 pg/mL (SD 7.80) in controls at 12 months. Questionnaires addressing study blinding revealed some association between the actual and guessed treatments, with investigators guessing assignment to placebo 55% correctly and 45% incorrectly, creatine 60% correctly and 40% incorrectly. Participants guessed placebo correctly 43% and incorrectly 57%, creatine correctly 73% and incorrectly 27%.
Primary efficacy outcome.
Primary outcome assessment was performed on an ITT population, excluding 16 participants at one site and one further participant without follow-up data. The estimated rates of decline in the primary outcome measure (TFC) were 0.82 points per year for participants on creatine, 0.70 points per year for controls, a difference of 0.12 points per year (nominal 95% confidence limits −0.11 to 0.35) favoring placebo (figure 2). The decline of TFC over time was nearly linear in both groups except for the longest follow-up times where the data were sparse. Imputation of a zero TFC score for visits following a death did not materially change the numbers, nor did inclusion of the data from the site excluded due to Good Clinical Practice violations. A supportive analysis of the primary outcome was conducted omitting data collected after a participant was permanently withdrawn from study drug. The results for the entire population and for men and women separately were similar to those for the full follow-up period.
Figure 2. Changes from baseline to each visit in total functional capacity (TFC) and total motor score (TMS), mean ± standard error.

(A) TFC for all patients assigned to active creatine and to placebo. (B) TMS for all patients assigned to active creatine and to placebo. In A (primary analysis) and B (supporting analysis), the 2 track curves track closely except at the final point where the data are sparse (see text for details).
Prespecified subgroup analyses.
Additional analyses of the primary outcome variable in subgroups defined by sex, age, TFC at baseline, and CAG revealed no significant interaction of treatment with age, baseline TFC, or CAG length. However, there was a statistically significant interaction of treatment by sex (table e-1 at Neurology.org). Among women, the rate of decline of TFC was 0.42 points per year greater for those on creatine vs placebo (p = 0.0028), whereas among men, the rate of decline was 0.25 points per year less for those on creatine than placebo (p = 0.12). A negative effect of creatine treatment among women (+1.52, p = 0.0799) and a possible benefit among men (−1.30, p = 0.1699) was also observed for the TMS.
Secondary and exploratory outcomes.
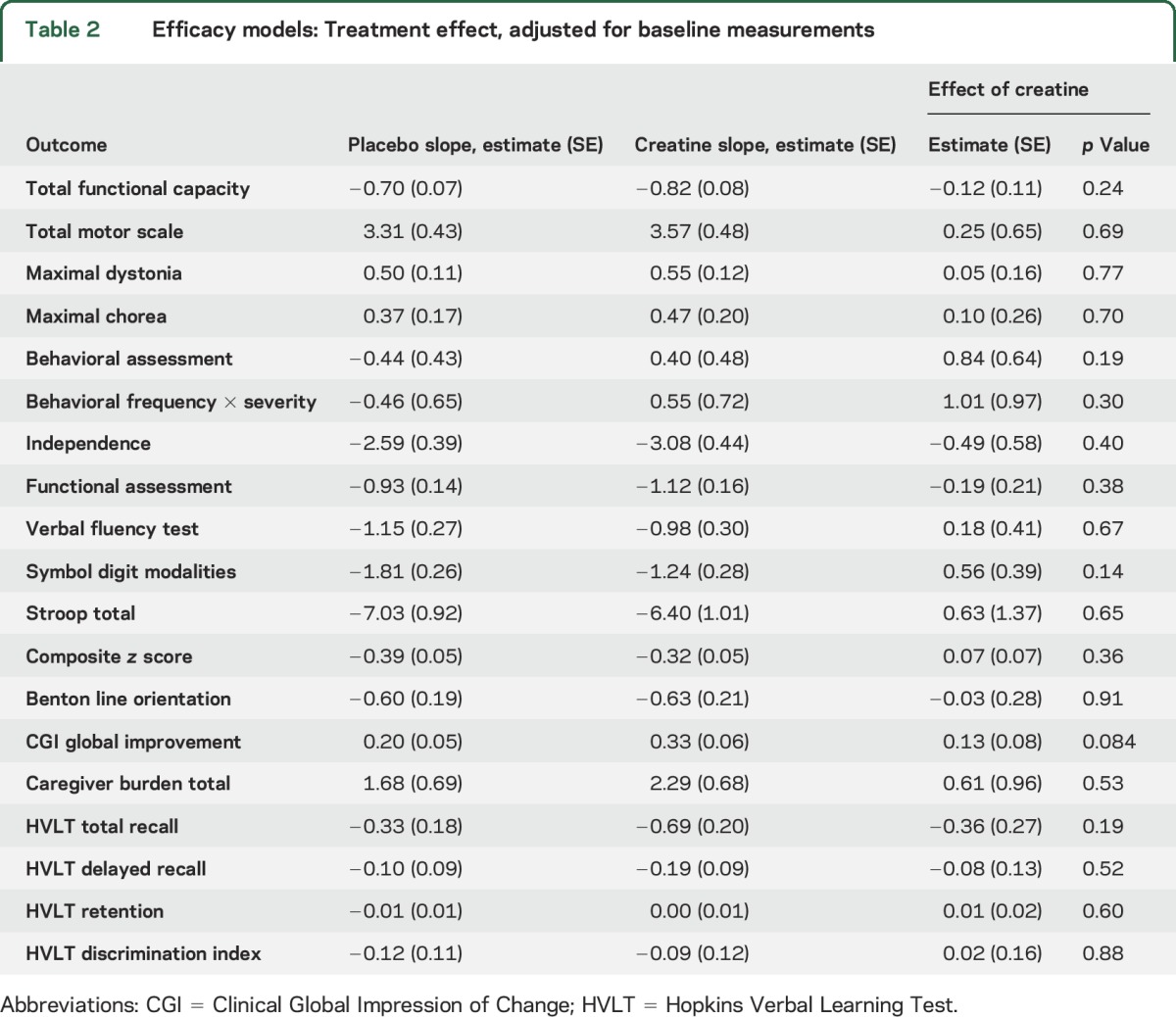
Among secondary and exploratory response variables (table 2), motor assessments (e.g., TMS and total maximal chorea score) generally followed a similar pattern to the TFC, with slightly greater worsening for participants on creatine than for controls. An exception was the rigidity component of the UHDRS. The pattern was also reversed for most cognitive measures; for example, the composite z score declined at a rate of 0.39 units/year in controls but only 0.32 units in the creatine group. Among 41 measures analyzed, none showed even nominally significant differences between treatment groups.
Table 2.
Efficacy models: Treatment effect, adjusted for baseline measurements
Safety and tolerability outcomes.
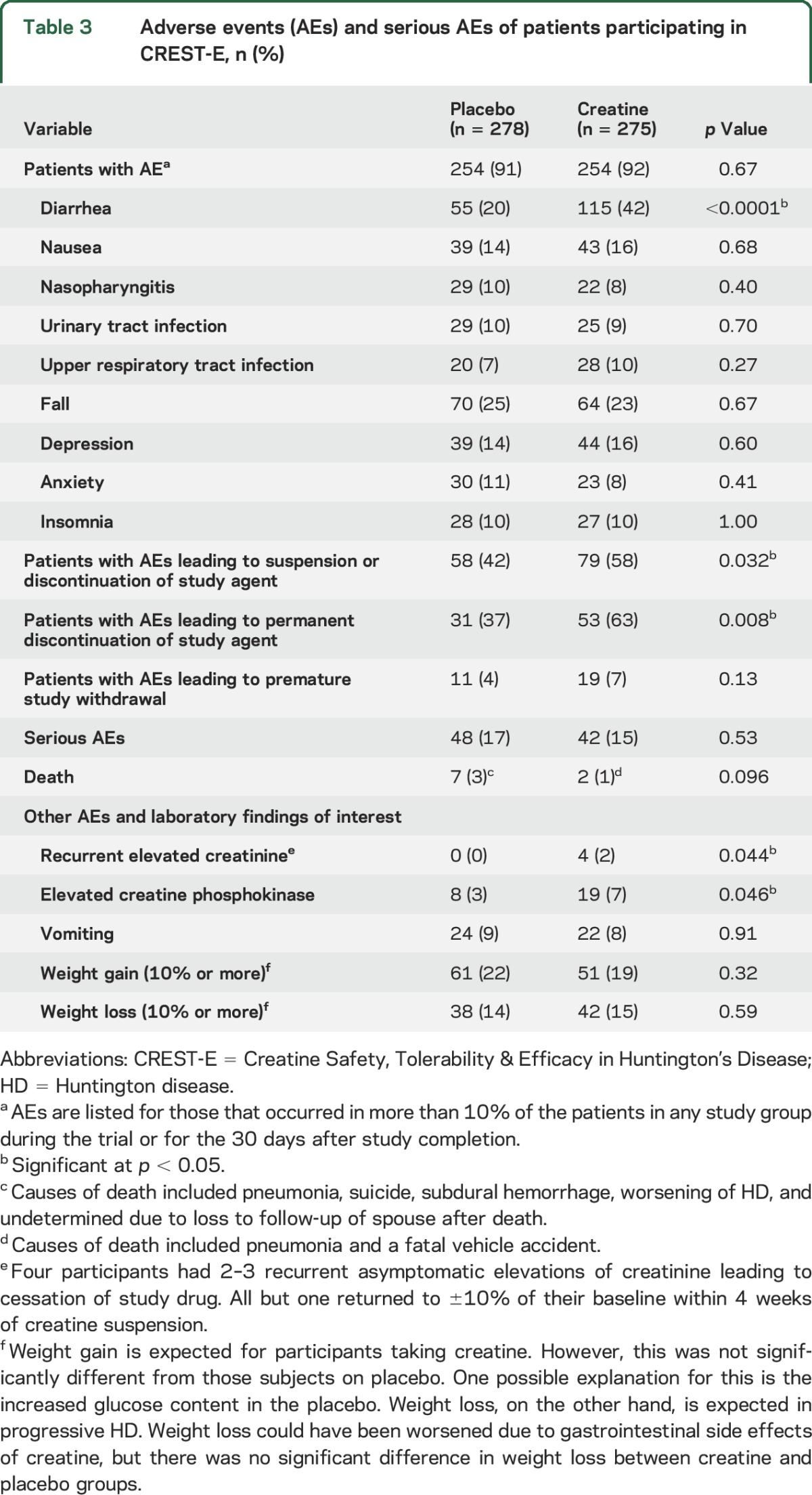
AEs, mainly gastrointestinal, were significantly more common in participants on creatine (table 3). Nevertheless, neither weight gain nor weight loss was observed on creatine. Eleven controls and 19 creatine-treated participants listed AEs as the reason for withdrawal. Transient increases in creatinine levels were noted in participants taking creatine. Mild, unexplained, asymptomatic elevations of creatine phosphokinase were more frequent in creatine-treated participants throughout the study. Serious AEs were more frequent in controls (78 among 48 participants) than in the creatine group (62 among 42 participants). Of 9 deaths reported (pneumonia [30], vehicle accident [2], suicide [1], subdural hemorrhage [1], worsening of HD [1], and unknown [1]), 7 occurred among controls.
Table 3.
Adverse events (AEs) and serious AEs of patients participating in CREST-E, n (%)
DISCUSSION
CREST-E represents the largest, longest duration, and highest dose study of creatine to date. Previous trials in HD11–13 and other neurologic disorders14–22 tested doses up to 10 g daily for less than a year. The CREST-E cohort was highly representative at baseline for the demographic and symptom characteristics of the individuals with early HD for whom CREST-E was designed. CREST-E did not address the possibility that creatine could be beneficial in prodromal HD. Although CREST-E was well-powered to detect a clinically meaningful beneficial effect of creatine, it failed to demonstrate such an effect in the primary and secondary clinical outcomes. However, a prespecified subgroup analysis of sex revealed a positive trend in male participants and a negative effect in female participants. Sex differences have not been reported in previous clinical studies of HD but have been described in relation to ergogenic and lean body mass effects of creatine and to exercise and sex hormone modulation of creatine kinase.23–25
Challenges arose in the conduct of CREST-E, which reinforce the need for biomarkers predictive of efficacy, clinical outcome measures that could support more efficient phase III studies, and reducing the burden of study participation. Despite having 46 sites and extensive marketing and outreach, competition with other trials, the wide availability of over-the-counter forms of creatine, and unwillingness to risk placebo assignment inhibited enrollment. This prompted redesign and primary endpoint revision 2 years into the trial and reduced the potential power to detect a small reduction in the rate of TFC decline. Participant and caregiver burden due to the length and complexity of CREST-E contributed to the substantial withdrawal rate. The higher withdrawal rate in the creatine group compared to controls (35% vs 26%) suggested that treatment-related gastrointestinal side effects may have been a factor despite the high proportion of participants reaching the targeted dose range, the allowance of unlimited dose adjustments, and stable body weight.
Maintaining a blinded study was a concern from the outset and the imbalance in the blindedness questionnaire data at the end of the study indicates that we were not completely successful. Risk of unblinding was mitigated by effective matching of creatine and placebo compounds, by instituting random suspensions to help mask suspensions due to true elevations caused by creatine metabolism, and by redacting potentially unblinding creatinine results, which were only visible to the clinical monitor. Nevertheless, AEs or other factors may have enabled some correct guessing about treatment assignment. Participants concerned about being assigned to placebo could have purchased creatine; however, heightened blood levels in 5/178 placebo participants suggested that such supplementation was rare. 8-OH2DG levels were not useful as a biomarker, largely because baseline levels were only minimally elevated. Expanding community use of antioxidant supplements could have been a factor.
Finally, while CREST-E proceeded based on earlier human data, the results suggest that animal model experiments may be more valuable for addressing biological aspects of a possible treatment than for predicting clinical efficacy.
Supplementary Material
GLOSSARY
- AE
adverse event
- ATP
adenosine triphosphate
- CREST-E
Creatine Safety, Tolerability & Efficacy in Huntington's Disease
- DSMB
Data Safety Monitoring Board
- FDA
Food and Drug Administration
- HD
Huntington disease
- ITT
intent-to-treat
- TFC
total functional capacity
- TMS
total motor score
- UHDRS
Unified Huntington's Disease Rating Scale
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Hersch had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Drs. Hersch, Schifitto, Oakes, and Rosas were responsible for the study concept and design. Drs. Hersch, Schifitto, Oakes, Rosas, and Bredlau were involved in data acquisition, analysis, and interpretation. Drs. Hersch, Schifitto, Oakes, Rosas, and Bredlau drafted the manuscript. Drs. Hersch, Schifitto, Oakes, Rosas, Bredlau, Meyers, and Nahin critically revised the manuscript for important intellectual content. Dr. Oakes led the biostatistical analyses. Drs. Hersch, Schifitto, and Rosas obtained study funding. Drs. Hersch, Schifitto, Oakes, Rosas, Bredlau, Meyers, and Nahin were responsible for providing administrative, technical, and material support and supervision.
STUDY FUNDING
This work was primarily supported by cooperative agreements from the NIH/National Center for Complementary and Integrative Health (NIH/NCCIH) to Dr. Hersch (U01AT000613) and to Drs. Hersch and Schifitto (U01AT008197). NIH/NCCIH staff reviewed the study design, provided oversight and assistance for study conduct, served as an interface between the study and the DSMB, and critically reviewed the manuscript. Additional funding was provided by the NIH Office of Dietary Supplements and by the FDA Orphan Products Division (R01FD003359) to Dr. Hersch. Support for biomarkers was provided by NIH/NINDS grants P01NS058793 and U01NS071789 to Dr. Hersch and R01NS042861 to Dr. Rosas.
DISCLOSURE
S. Hersch reports receiving grant support from NIH, FDA-Orphan Products Development (OPD) Program, Huntington's Disease Society of America, Hereditary Disease Foundation, PRANA, the Novartis Institutes for Biomedical Research, California Institute for Regenerative Medicine, and the Dake Foundation and consulting fees from Azevan, Link Medicine, Mitochon Pharmaceuticals, Pfizer, PRANA, Wave Life Sciences, and Voyager Therapeutics. G. Schifitto reports receiving grants from NIH. D. Oakes reports grant support from NIH, DOD, Michael J. Fox Foundation, TEVA Pharmaceuticals, Prana Biotechnology, and Vaccinex Inc. He has received consulting fees from Raptor Pharmaceuticals and Voyager Therapeutics. A. Bredlau reports receiving grant support from NIH/NCATS and consulting fees from Azevan and Borvo-Lenus Pharmaceuticals. C. Meyers and R. Nahin report no disclosures relevant to the manuscript. H. Rosas reports receiving grant support from NIH, the Huntington Study Group, the Dake Foundation, and PRANA and consulting fees from Pfizer. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10:204–216. [DOI] [PubMed] [Google Scholar]
- 2.Andres RH, Ducray AD, Schlattner U, Wallimann T, Widmer HR. Functions and effects of creatine in the central nervous system. Brain Res Bull 2008;76:329–343. [DOI] [PubMed] [Google Scholar]
- 3.Matthews RT, Yang L, Jenkins BG, et al. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington's disease. J Neurosci 1998;18:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andreassen OA, Dedeoglu A, Ferrante RJ, et al. Creatine increases survival and delays motor symptoms in a transgenic animal model of Huntington's disease. Neurobiol Dis 2001;8:479–491. [DOI] [PubMed] [Google Scholar]
- 5.Ferrante RJ, Andreassen OA, Jenkins BG, et al. Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J Neurosci 2000;20:4389–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dedeoglu A, Kubilus JK, Yang L, et al. Creatine therapy provides neuroprotection after onset of clinical symptoms in Huntington's disease transgenic mice. J Neurochem 2003;85:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hersch SM, Gevorkian S, Marder K, et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2'dG. Neurology 2006;66:250–252. [DOI] [PubMed] [Google Scholar]
- 8.Rosas HD, Doros G, Gevorkian S, et al. PRECREST: a phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology 2014;82:850–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unified Huntington's Disease Rating Scale: reliability and consistency. Huntington Study Group. Mov Dis 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 10.Marder K, Zhao H, Myers RH, et al. Rate of functional decline in Huntington's disease: Huntington Study Group. Neurology 2000;54:452–458. [DOI] [PubMed] [Google Scholar]
- 11.Verbessem P, Lemiere J, Eijnde BO, et al. Creatine supplementation in Huntington's disease: a placebo-controlled pilot trial. Neurology 2003;61:925–930. [DOI] [PubMed] [Google Scholar]
- 12.Tabrizi SJ, Blamire AM, Manners DN, et al. Creatine therapy for Huntington's disease: clinical and MRS findings in a 1-year pilot study. Neurology 2003;61:141–142. [DOI] [PubMed] [Google Scholar]
- 13.Tabrizi SJ, Blamire AM, Manners DN, et al. High-dose creatine therapy for Huntington disease: a 2-year clinical and MRS study. Neurology 2005;64:1655–1656. [DOI] [PubMed] [Google Scholar]
- 14.Writing Group for the NETiPDI, Kieburtz K, Tilley BC, Elm JJ, et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: a randomized clinical trial. JAMA 2015;313:584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Investigators NN-P. A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin Neuropharmacol 2008;31:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Investigators NN-P. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 2006;66:664–671. [DOI] [PubMed] [Google Scholar]
- 17.Atassi N, Ratai EM, Greenblatt DJ, et al. A phase I, pharmacokinetic, dosage escalation study of creatine monohydrate in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2010;11:508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenfeld J, King RM, Jackson CE, et al. Creatine monohydrate in ALS: effects on strength, fatigue, respiratory status and ALSFRS. Amyotroph Lateral Scler 2008;9:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shefner JM, Cudkowicz ME, Schoenfeld D, et al. A clinical trial of creatine in ALS. Neurology 2004;63:1656–1661. [DOI] [PubMed] [Google Scholar]
- 20.Groeneveld GJ, Veldink JH, van der Tweel I, et al. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann Neurol 2003;53:437–445. [DOI] [PubMed] [Google Scholar]
- 21.Kley RA, Tarnopolsky MA, Vorgerd M. Creatine for treating muscle disorders. Cochrane Database Syst Rev 2013;6:CD004760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee B, Sharma U, Balasubramanian K, Kalaivani M, Kalra V, Jagannathan NR. Effect of creatine monohydrate in improving cellular energetics and muscle strength in ambulatory Duchenne muscular dystrophy patients: a randomized, placebo-controlled 31P MRS study. Magn Reson Imaging 2010;28:698–707. [DOI] [PubMed] [Google Scholar]
- 23.Ellery SJ, Walker DW, Dickinson H. Creatine for women: a review of the relationship between creatine and the reproductive cycle and female-specific benefits of creatine therapy. Amino Acids 2016;48:1807–1817. [DOI] [PubMed] [Google Scholar]
- 24.Koch AJ, Pereira R, Machado M. The creatine kinase response to resistance exercise. J Musculoskelet Neuronal Interact 2014;14:68–77. [PubMed] [Google Scholar]
- 25.Fukuda DH, Smith AE, Kendall KL, et al. The effects of creatine loading and gender on anaerobic running capacity. J Strength Cond Res 2010;24:1826–1833. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.