

Abstract
Background
Several studies have suggested that global DNA methylation in circulating white blood cells (WBC) is associated with breast cancer risk.
Methods
To address conflicting results and concerns that the findings for WBC DNA methylation in some prior studies may reflect disease effects, we evaluated the relationship between global levels of WBC DNA methylation in white blood cells and breast cancer risk in a case-control study nested within the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO) cohort. A total of 428 invasive breast cancer cases and 419 controls, frequency matched on age at entry (55–59, 60–64, 65–69, ≥70 years), year of entry (on/before September 30, 1997, on/after October 1, 1997) and period of DNA extraction (previously extracted, newly extracted) were included. The ratio of 5-methyl-2’ deoxycytidine [5-mdC] to 2’-deoxyguanine [dG], assuming [dG] = [5-mdC] + [2’-deoxycytidine [dC]] (%5-mdC), was determined by liquid chromatography-electrospray ionization-tandem mass spectrometry, an especially accurate method for assessing total genomic DNA methylation.
Results
Odds ratio (OR) estimates and 95% confidence intervals (CI) for breast cancer risk adjusted for age at entry, year of entry, and period of DNA extraction, were 1.0 (referent), 0.89 (95% CI, 0.6–1.3), 0.88 (95% CI, 0.6–1.3), and 0.84 (95% CI, 0.6–1.2) for women in the highest compared to lowest quartile levels of %5md-C (p for trend = .39). Effects did not meaningfully vary by time elapsed from WBC collection to diagnosis.
Discussion
These results do not support the hypothesis that global DNA hypomethylation in WBC DNA is associated with increased breast cancer risk prior to the appearance of clinical disease.
Keywords: Breast cancer, White blood cells, Global DNA methylation, %5mdC, Cohort
Background
Cytosines can be methylated in the mammalian genome, primarily at CpG sites. CpG sites are located throughout the genome, including gene promoter regions and repetitive DNA sequences. DNA methylation patterns play a key role in gene expression and cell integrity. For example, genome-wide hypomethylation can be associated with chromosomal instability and the expression of oncogenes or repetitive sequences that are normally silenced by methylation [1]. Global loss of DNA methylation is characteristic of cancer tissue [2].
There are several different methods to assess global DNA methylation. Measurement of the ratio of 5-methyl-2’ deoxycytidine [5-mdC] to 2’-deoxyguanine [dG], assuming [dG] = [5-mdC] + [2’-deoxycytidine [dC]] (%5-mdC), by liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) provides a comprehensive measure of genome-wide DNA methylation levels [3]. This method is considered the gold standard as it evaluates the entire genome but is expensive and time-consuming, and requires specialized laboratory equipment. Approximately one-third of the DNA methylation in the genome occurs in the repetitive sequences of the genome, including LINE-1 and Alu [4]. For this reason, DNA methylation levels in LINE-1 or Alu repeats, which can be obtained by high-throughput methods, have often been used as surrogate markers of global DNA methylation [5]. Other high-throughput surrogate methods for estimating global methylation are available. For example, the luminometric methylation assay (LUMA) [6] estimates global DNA methylation levels by using restriction enzymes specific for methylated and unmethylated CCGG, a sequence found throughout the genome. A different approach has been to average methylation levels across the limited set of individual CpG sites represented on the Illumina human methylation bead kits [7, 8]. An advantage of this latter approach, relative to aggregrate methods of assessing global methylation (e.g., %5mdC, LINE-1), is that one can adjust for potential differences in blood cell composition between cases and controls in archived blood specimens [9]. A second advantage is that it is possible to conduct subanalyses to examine methylation in specific locations in the genome (e.g., promoter regions across the genome) [7, 8].
Global DNA hypomethylation of breast tumor tissue is well-established [2]; however, there is some evidence that global hypomethylation in circulating white blood cell (WBC) DNA may also be associated with an increased risk of breast cancer. One possible explanation is that the association represents unidentified environmental and lifestyle determinants that influence both global methylation and breast cancer risk. An alternative possibility is that, in response to very early, preclinical breast cancer, a new clone of circulating leukocytes arises that alters white blood cell DNA methylation [10]. In a relatively small retrospective case-control study, Choi and colleagues [11] observed a nearly threefold increase in breast cancer risk among women in the lowest tertile of %5-mdC in WBC DNA compared to women in the highest tertile. Comparable results were obtained prospectively in the case-cohort study nested in the NIEHS Sister Cohort Study [12], with a nearly twofold increased risk observed among women in the lowest quartile of LINE-1 WBC DNA methylation compared with those in the highest quartile. By contrast, a number of other investigations, including three separate nested case-control studies from Europe, which used pre-diagnostic DNA (and were presented in a single publication) [13], observed no association between LINE-1 methylation in WBC DNA and breast cancer risk [11, 13–18] or between Alu methylation in WBC DNA and breast cancer risk [14, 16, 19]. Findings from the three retrospective case-control studies employing the LUMA assay were inconsistent, with positive, inverse, and null associations [15, 20, 21]. However, three of the four nested case-control studies, which used the Illumina HumanMethylation450 BeadChip on prediagnostic DNA (separate results from three cohorts were reported in a single paper [8]) observed that global hypomethylation was positively associated with increased breast cancer risk [7, 8].
To address these discrepant results, we examined the association between global hypomethylation in WBC DNA and subsequent breast cancer incidence in a study nested in the Prostate, Lung, Colorectal and Ovarian Cancer Screening (PLCO) Trial cohort. We measured WBC DNA %5-mdC levels by LC-ESI-MS/MS because: (1) WBC global hypomethylation as measured by %5-mdC level was found to be strongly and significantly associated with increased breast cancer risk in the one breast cancer study that measured %5-mdC, which was a relatively small restrospective study [11]; (2) global hypomethylation as measured by WBC DNA %5-mdC was reported to be more strongly associated with overall cancer risk than surrogate measures of global methylation (LINE-1, Alu, and LUMA) in a recent meta-analysis [22]; and (3) %5-mdC level measured by LC-ESI-MS/MS is considered the gold standard assay for accurately assessing methylation across the entire genome.
Our study is important because it is the first investigation to examine the association between WBC DNA %5-mdC levels, measured prior to breast cancer diagnosis, and subsequent breast cancer incidence. In this large study of 428 cases and 419 controls, the elapsed time between blood collection and breast cancer development ranged from 1.0 to 9.5 years, enabling us to examine whether risk varied by time elapsed between blood collection and diagnosis.
Methods
Selection of study subjects
Cases and controls for the present analysis were selected from the Etiology and Early Marker Study (EEMS) breast cancer case-control study that was established from the 39,115 women randomized to the intervention arm of the PLCO screening trial [23]. Through June 30, 2005, a total of 1141 eligible cases of breast cancer were identified. A total of 1141 controls were frequency matched to cases by randomly sub-sampling women who had not been diagnosed with breast cancer by June 30, 2005 in eight strata defined by four age categories (55–59, 60–64, 65–69, ≥70 years) and time of entry into the study (on/before September 30, 1997, on/after October 1, 1997).
A total of 732 cases and 928 controls were initially identified as eligible for the present analysis, after further excluding in the following order: subjects who did not give permission for genetic studies (cases = 32, controls = 25), subjects who had a personal history of any cancer prior to the trial (cases = 40, controls = 30), subjects with unconfirmed, erroneous or in situ breast cancer (cases = 229, controls = 13), subjects who developed other types of cancer anytime during follow up (cases = 105, controls = 130), and other reasons (cases = 3, controls = 14).
A total of 649 out of the 726 cases and 787 out of the 928 controls either had: (1) DNA already extracted from buffy coat/whole blood remaining from a prior study; or (2) buffy coat available for extraction. We further excluded 83 breast cancer cases in which diagnosis occurred within one year of the DNA collection, to minimize the likelihood of disease effects, leaving an eligible pool of 566 cases and 787 controls.
For efficiency, our a priori plan was to select 430 cases and 430 controls for this analysis. We first prioritized case selection to include the 151 cases and 147 controls that already had DNA extracted as part of another study [24]. We then supplemented study subject selection to include cases and controls with buffy coat available for DNA extraction for this study. Controls were frequency matched to cases on age, calendar year of entry, and the date of the DNA extraction (already extracted DNA or newly extracted DNA). Matching on date of DNA extraction was done to address the possible concern that DNA methylation patterns may be affected by the timing or method of DNA extraction. We ultimately selected 428 breast cancer cases and 420 controls that had suitable DNA for analysis, slightly less than our goal because some of the subjects we originally selected turned out to have inadequate DNA. One additional control subject was later excluded for an improbable value for %5-mdC (67.4%). Thus, our final analysis consisted of 428 cases and 419 controls. The institutional review boards of the National Cancer Institute, the 10 participating study centers, and the University of Massachusetts Amherst approved this study. Informed consent was obtained from all participants at study enrollment.
DNA extraction
For 298 study subjects, DNA was previously extracted in 2006–2007 from stored buffy coat (98%) or whole blood (2%). About 90% of this DNA was extracted using the Autopure method (Qiagen) and the remaining specimens were extracted using standard phenol/chloroform extraction. In 2014, DNA was extracted for the remaining study subjects using the QIAsymphony SP automated extraction robot (Qiagen). DNA concentrations were quantified using the Picogreen assay and nanodrop technology.
DNA hydrolysis
To provide individual nucleosides for subsequent total methylated cytosine measurements, genomic DNA were hydrolyzed with DNA Degradase Plus (Zymo Research, Cat # E2021) following the manufacturer’s protocol with minor adjustments. Briefly, 400 ng of genomic DNA was incubated with 5 U of DNA Degradase Plus in 25 μl total reaction volumes at 37 °C for 2 hours. Batch control DNA included female genomic DNA (Promega, Cat # G1521), which was considered to be “normally” methylated. Complete DNA hydrolysis of additional control samples were verified by agarose gel electrophoresis. Hydrolyzed DNA samples were stored at −20 °C until %5-mdC analyses.
Measurement of %5-mdC
The %5-mdC levels were determined by LC-ESI-MS/MS after hydrolysis of DNA as described by Song and colleagues [3], with modifications. Both 5-mdC and dG concentrations were quantified with internal standard additions of isotope-labeled 5-mdC and dG (5-mdC-d3 and 15 N5-dG, respectively; obtained from Toronto Research Chemicals). LC separation was performed on an Acquity UPLC system (Waters Corporation) at a flow rate of 400 μL/min. Methanol containing 0.1% formic acid and water containing 0.1% formic acid were used as buffers. The organic buffer ratio was increased at a linear gradient from 0 to 22.5% over 8 min for the elution of nucleosides. The sample injection volume was 20 μL. Detection by ESI-MS/MS was performed on a Quattro Premier XE Mass Spectrometer (Waters Corporation) following LC separation. The following optimized conditions for ESI positive ion mode were used: source temperature, 120 °C; desolvation gas flow, 700 L/h; cone gas flow, 50 L/h; capillary voltage, 4.2 kV; cone voltage, 10 V; extractor voltage, 2 V; entrance potential voltage, 0 V; collision energy, 11 V; and collision cell exit potential, 2.0 V. Multiple reaction monitoring mode was utilized for the quantification of native and labeled nucleosides. The transition pairs of molecular and fragment ions monitored were m/z 242.0/126.0 for 5-mdC, m/z 245.0/129.0 for 5-mdC-d3, m/z 268.1/152.0 for dG, and m/z 273.0/157.0 for 15 N5-dG with a scan time of 150 minutes for each pair. Following QuanLynx (Waters Corporation) analysis for chromatographic peak detection, the resulting peak areas of the native nucleosides were normalized to the labeled internal standards and quantified based on external calibration curves. The 5-mdC and dG nucleosides for external calibration were obtained from Fisher Scientific. We took the average of two injections from each sample vial. Results are reported as the ratio between 5-mdC and dG, assuming that [dG] = [5-mdC] + [dC]. Laboratory personnel were blinded to case status.
Samples were run on twelve plates, with cases and controls distributed on each plate approximately evenly within eight stratum defined by age and time of entry. Each plate had DNA that was either all pre-extracted or all newly extracted. On each plate, we also included three replicate DNA specimens from three women who were in a study site for the PLCO trial that was later dropped (newly extracted DNA). The mean inter-batch coefficient of variation across the twelve plates for each of the three women was 8, 7, and 9%, respectively. The female genomic DNA average inter-batch coefficient of variation was comparable at 9.2%.
In Fig. 1, we show levels of %5-mdC for study subjects in each of the twelve batches and also separately for cases and controls in each batch. Levels of %5-mdC varied by batch and showed limited variation across individuals within batch.
Fig. 1.
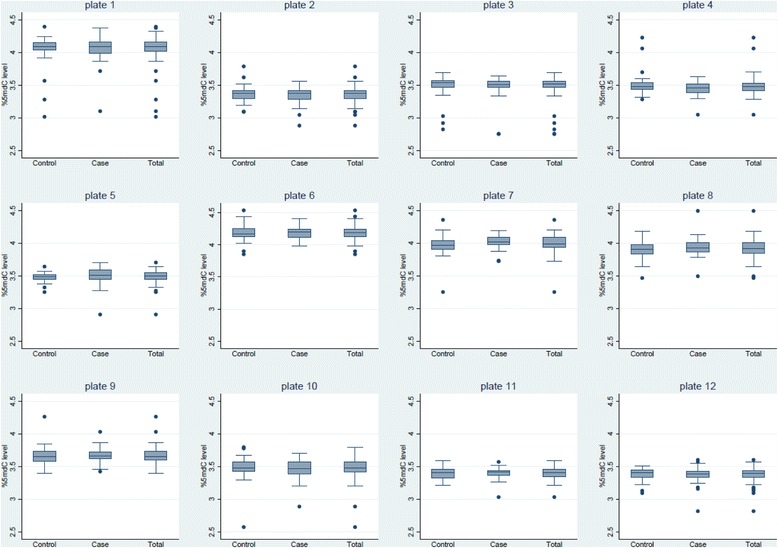
Boxplot distribution of the ratio of 5-methyl-2’ deoxycytidine (5-mdC) to 2’-deoxyguanine (dG) (%5-mdC) levels by case-control status in each of twelve laboratory plates. The ends of the boxes are the 25th and 75th percentiles and the center line is the median. The lengths of the whiskers are defined as 1.5*(75th percentile – 25th percentile). The circles are values outside the whisker length
Breast cancer ascertainment
Breast and other cancers were primarily identified through an annual study update mailed to participants, which established cancer diagnosis in the previous year, including type and date [25]. Non-respondents were contacted by mail and telephone. In order to confirm the self-reported cancers, medical records (for standardized medical record abstraction of pathology reports) were retrieved and usually obtained within 2 years of self-report. Cancers were also identified through death certificates, data obtained from state cancer registries, and information from next-of-kin for deceased participants.
Ascertainment of other variables
Demographic information, medical history, and health-related behavior were obtained through baseline questionnaires completed by study participants at or around the time of randomization.
Statistical analysis
Logistic regression was used to estimate odds ratios and 95% CI, for breast cancer with case/control status as the outcome and a categorical variable denoting quartiles of the %5-mdC levels as the primary exposure of interest. Because of the evidence of inter-batch variation, we created %5-mdc quartile levels separately for each batch and then created a new summary variable with four levels based on the batch-specific cut points. For example, individuals in the lowest category of the summary variable included individuals from each of the twelve batches who were placed in the lowest batch-specific quartile ranking. Models were adjusted for matching variables, including age in four categories (55–59, 60–64, 65–69, ≥70 years), time of entry into the study (on/before September 30, 1997, on/after October 1, 1997), and period of DNA extraction (previously extracted, newly extracted). Additional models considered adjustment for established or suspected breast cancer risk factors, including race, body mass index, age at menarche, age at first live birth, number of children, type of menopause, age at natural menopause, personal history of benign breast disease, cigarette smoking, recent alcohol intake, family history of breast cancer, and menopausal hormone use. Statistical significance was assessed for each level of the primary exposure variable using two-sided Wald hypothesis tests.
Results
Relative risks for established breast cancer risk factors were generally comparable in our analytic subgroup of cases and controls to those reported previously in the literature [25] (Table 1). Nulliparity, later age at first birth, late age at natural menopause, a personal history of benign breast disease, a family history of breast cancer, and alcohol consumption were associated with increased breast cancer risk. Late age at menarche, three or more live births and surgical menopause were associated with reduced breast cancer risk.
Table 1.
Associations between demographic and lifestyle characteristics and risk of breast cancer, adjusted for age and time of entry into the cohort
| Controls | % | Cases | % | Odds Ratio, 95% CI | |||
|---|---|---|---|---|---|---|---|
| Age (years) | |||||||
| <59 | 150 | 35.8 | 151 | 35.3 | 1.0 (referent) | ||
| 60–64 | 140 | 33.4 | 141 | 32.9 | 1.00 | 0.72 | 1.39 |
| 65–69 | 93 | 22.2 | 102 | 23.8 | 1.09 | 0.76 | 1.56 |
| > =70 | 36 | 8.6 | 34 | 7.9 | 0.94 | 0.56 | 1.58 |
| Time of entry | |||||||
| > =October 10, 1997 | 168 | 40.1 | 174 | 40.7 | 1.0 (referent) | ||
| < =September 30, 1997 | 251 | 59.9 | 254 | 59.4 | 1.02 | 0.78 | 1.35 |
| Race | |||||||
| White, non-Hispanic | 380 | 90.7 | 395 | 92.3 | 1.0 (referent) | ||
| Other | 39 | 9.3 | 33 | 7.7 | 0.81 | 0.50 | 1.32 |
| Body mass index (kg/m2) | |||||||
| Underweight-normal (15–24.99) | 175 | 41.9 | 176 | 41.2 | 1.0 (referent) | ||
| Overweight (25.0–29.99) | 141 | 33.7 | 152 | 35.6 | 1.07 | 0.79 | 1.46 |
| Obese (> = 30) | 102 | 24.4 | 99 | 23.2 | 0.96 | 0.68 | 1.36 |
| Age at menarche (years) | |||||||
| <12 | 70 | 16.8 | 88 | 20.6 | 1.0 (referent) | ||
| 12–13 | 238 | 56.9 | 219 | 51.2 | 0.73 | 0.51 | 1.05 |
| > =14 | 110 | 26.3 | 121 | 28.3 | 0.87 | 0.58 | 1.31 |
| Number of live births | |||||||
| None | 31 | 7.4 | 48 | 11.2 | 1.0 (referent) | ||
| 1–2 births | 123 | 29.4 | 147 | 34.4 | 0.77 | 0.46 | 1.29 |
| 3+ births | 265 | 63.3 | 233 | 54.4 | 0.57 | 0.35 | 0.92 |
| Age at first birth (years) | |||||||
| <20 | 72 | 17.2 | 55 | 12.9 | 1.0 (referent) | ||
| 20–24 | 209 | 49.9 | 181 | 42.5 | 1.14 | 0.76 | 1.70 |
| 25–29 | 80 | 19.1 | 108 | 25.4 | 1.77 | 1.12 | 2.79 |
| > =30 | 27 | 6.4 | 34 | 8.0 | 1.66 | 0.89 | 3.07 |
| Nulliparous | 31 | 7.4 | 48 | 11.3 | 2.03 | 1.14 | 3.59 |
| Type of menopause | |||||||
| Natural | 253 | 60.4 | 283 | 66.1 | 1.0 (referent) | ||
| Bilateral oophorectomy | 49 | 11.7 | 29 | 6.8 | 0.52 | 0.31 | 0.85 |
| Other | 117 | 27.9 | 116 | 27.1 | 0.88 | 0.65 | 1.20 |
| Age at natural menopause (years) | |||||||
| <50 | 91 | 36.0 | 95 | 33.7 | 1.0 (referent) | ||
| 50–54 | 122 | 48.2 | 141 | 50.0 | 1.15 | 0.79 | 1.69 |
| 55+ | 40 | 15.8 | 46 | 16.3 | 1.15 | 0.69 | 1.93 |
| History of benign breast disease | |||||||
| No | 285 | 69.9 | 261 | 61.9 | 1.0 (referent) | ||
| Yes | 123 | 30.2 | 161 | 38.2 | 1.43 | 1.07 | 1.91 |
| Smoking status | |||||||
| Never | 257 | 61.3 | 229 | 53.5 | 1.0 (referent) | ||
| Former | 135 | 32.2 | 163 | 38.1 | 1.36 | 1.02 | 1.82 |
| Current | 27 | 6.4 | 36 | 8.4 | 1.51 | 0.89 | 2.56 |
| Recent alcohol intake | |||||||
| No | 112 | 29.2 | 99 | 24.9 | 1.0 (referent) | ||
| Yes | 272 | 70.8 | 298 | 75.1 | 1.25 | 0.91 | 1.72 |
| Family history of breast cancer | |||||||
| No | 353 | 85.9 | 340 | 81.0 | 1.0 (referent) | ||
| Yes | 58 | 14.1 | 80 | 19.1 | 1.43 | 0.99 | 2.07 |
| Hormone user | |||||||
| Never | 111 | 26.7 | 102 | 23.9 | 1.0 (referent) | ||
| Former | 70 | 16.8 | 55 | 12.9 | 0.86 | 0.55 | 1.34 |
| Current | 235 | 56.5 | 269 | 63.2 | 1.26 | 0.91 | 1.75 |
The batch-standardized quartile distribution of %5-mdC was unrelated to the distribution of breast cancer risk factors or study design matching factors (Table 2).
Table 2.
Baseline distribution of study design and lifestyle risk factors by quartile of DNA methylationa
| Q4 (high) | Q3 | Q2 | Q1 (low) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | N | % | p value | |
| Age at entry (years) | |||||||||
| <59 | 85 | 39.5 | 67 | 31.5 | 76 | 37.3 | 73 | 34.0 | |
| 60–64 | 66 | 30.7 | 79 | 37.1 | 60 | 29.4 | 76 | 35.4 | |
| 65–69 | 46 | 21.4 | 48 | 22.5 | 52 | 25.5 | 49 | 22.9 | |
| > =70 | 18 | 8.4 | 19 | 8.9 | 16 | 7.8 | 17 | 7.9 | |
| 0.75 | |||||||||
| Time at entry | |||||||||
| < =September 30,1997 | 134 | 62.3 | 125 | 58.7 | 114 | 55.9 | 132 | 61.4 | |
| > =October 1, 1997 | 81 | 37.7 | 88 | 41.3 | 90 | 44.1 | 83 | 38.6 | |
| 0.53 | |||||||||
| Date of DNA extraction | |||||||||
| Pre-extracted DNA | 69 | 32.1 | 69 | 32.4 | 70 | 34.3 | 90 | 41.9 | |
| Newly-extracted DNA | 146 | 67.9 | 144 | 67.6 | 134 | 65.7 | 125 | 58.1 | |
| 0.12 | |||||||||
| Race/ethnicity | |||||||||
| White, non-Hispanic | 194 | 90.2 | 197 | 92.5 | 190 | 93.1 | 194 | 90.2 | |
| Other | 21 | 9.8 | 16 | 7.5 | 14 | 6.9 | 21 | 9.8 | |
| 0.60 | |||||||||
| Body mass index (kg/m2) | |||||||||
| Underweight-normal (15–24.9) | 77 | 35.8 | 84 | 39.4 | 98 | 48.3 | 92 | 43.0 | |
| Overweight (25.0 − 29.9) | 91 | 42.3 | 72 | 33.8 | 60 | 29.6 | 70 | 32.7 | |
| Obese (> = 30) | 47 | 21.9 | 57 | 26.8 | 45 | 22.2 | 52 | 24.3 | |
| 0.09 | |||||||||
| Age at menarche (years) | |||||||||
| <12 | 37 | 17.3 | 44 | 20.7 | 31 | 15.2 | 46 | 21.4 | |
| 13 | 116 | 54.2 | 122 | 57.3 | 107 | 52.5 | 112 | 52.1 | |
| > =14 | 61 | 28.5 | 47 | 22.1 | 66 | 32.5 | 57 | 26.5 | |
| 0.26 | |||||||||
| Number of live births | |||||||||
| None | 18 | 8.4 | 20 | 9.4 | 16 | 7.8 | 25 | 11.6 | |
| 1–2 | 66 | 30.7 | 65 | 30.5 | 66 | 32.4 | 73 | 34.0 | |
| > =3 | 131 | 60.9 | 128 | 60.1 | 122 | 59.8 | 117 | 54.4 | |
| 0.75 | |||||||||
| Age at first birth (years) | |||||||||
| <20 | 36 | 16.8 | 32 | 15.0 | 24 | 11.8 | 35 | 16.3 | |
| 20–24 | 101 | 47.2 | 94 | 44.1 | 100 | 49.3 | 95 | 44.2 | |
| 25–29 | 47 | 22.0 | 54 | 25.4 | 48 | 23.7 | 39 | 18.1 | |
| > =30 | 12 | 5.6 | 13 | 6.1 | 15 | 7.4 | 21 | 9.8 | |
| Nulliparous | 18 | 8.4 | 20 | 9.4 | 16 | 7.9 | 25 | 11.6 | |
| 0.56 | |||||||||
| Type of menopause | |||||||||
| Natural | 129 | 60.0 | 136 | 63.9 | 126 | 61.8 | 145 | 67.4 | |
| Bilateral oophorectomy | 20 | 9.3 | 21 | 9.9 | 17 | 8.3 | 20 | 9.3 | |
| Other | 66 | 30.7 | 56 | 26.3 | 61 | 29.9 | 50 | 23.3 | |
| 0.67 | |||||||||
| Age at natural menopause (years) | |||||||||
| <50 | 53 | 41.1 | 46 | 33.8 | 41 | 32.8 | 46 | 31.7 | |
| 50–54 | 56 | 43.4 | 69 | 50.7 | 66 | 52.8 | 72 | 49.7 | |
| 55+ | 20 | 15.5 | 21 | 15.4 | 18 | 14.4 | 27 | 18.6 | |
| 0.65 | |||||||||
| History of benign breast disease | |||||||||
| No | 133 | 62.7 | 143 | 69.4 | 130 | 64.4 | 140 | 66.7 | |
| Yes | 79 | 37.3 | 63 | 30.6 | 72 | 35.6 | 70 | 33.3 | |
| 0.51 | |||||||||
| Smoking status | |||||||||
| Never | 115 | 53.4 | 118 | 55.4 | 115 | 56.4 | 138 | 64.2 | |
| Former | 81 | 37.7 | 78 | 36.6 | 72 | 35.3 | 67 | 31.2 | |
| Current | 19 | 8.8 | 17 | 8.0 | 17 | 8.3 | 10 | 4.7 | |
| 0.31 | |||||||||
| Recent alcohol intake | |||||||||
| No | 45 | 22.6 | 54 | 28.0 | 56 | 29.6 | 56 | 28.0 | |
| Yes | 154 | 77.4 | 139 | 72.0 | 133 | 70.4 | 144 | 72.0 | |
| Family history of breast cancer | |||||||||
| No | 169 | 81.3 | 180 | 86.1 | 168 | 83.2 | 176 | 83.0 | |
| Yes | 39 | 18.8 | 29 | 13.9 | 34 | 16.8 | 36 | 17.0 | |
| 0.60 | |||||||||
| Hormone use | |||||||||
| Never | 61 | 28.6 | 51 | 23.9 | 55 | 27.1 | 46 | 21.6 | |
| Former | 32 | 15.0 | 33 | 15.5 | 24 | 11.8 | 36 | 16.9 | |
| Current | 120 | 56.3 | 129 | 60.6 | 124 | 61.1 | 131 | 61.5 | |
| 0.54 | |||||||||
aMissing values for cases and controls, respectively, were as follows: body mass index (1, 1), age at menarche (1, 0), number of livebirths (0, 2), age at first birth (0, 3), personal history of benign breast disease (11, 6), alcohol intake (35,31), family history of breast cancer (8, 8), and hormone use (2, 3). Q quartile
As shown in Table 3, odds ratios and 95% CI, adjusted for age and time of entry and period of DNA extraction, were 1.0 (referent), 0.89 (95% CI, 0.6–1.3), 0.88 (95% CI, 0.6–1.3), and 0.84 (95% CI, 0.6–1.2) comparing women in the highest to the lowest batch-specific quartile levels of %5-mdC (p for trend = .39). Results were essentially unchanged after additional adjustment for breast cancer risk factors (p for trend = .35). When we restricted analyses to 298 cases who were hormone-receptor-positive (estrogen-receptor-positive and progesterone-receptor-positive) and 419 controls, comparable fully-adjusted odds ratios and 95% CI were similar: 1.0, 0.82 (0.5–1.3), 0.98 (0.6–1.5), and 0.85 (0.5–1.3) (data not shown).
Table 3.
Quartile level of WBC DNA methylation and risk of breast cancer
| Quartiles of DNA methylation | ||||
|---|---|---|---|---|
| Q4 (high) | Q3 | Q2 | Q1 (low) | |
| Adjusted ORa | 1.0 (referent) | 0.89 | 0.88 | 0.84 |
| 95% CI | (0.6–1.3) | (0.6–1.3) | (0.6–1.2) | |
| Cases, controls | 114, 101 | 107, 106 | 102, 102 | 105, 110 |
| Fully adjusted ORb | 1.0 (referent) | 0.90 | 0.87 | 0.83 |
| 95% CI | (0.6–1.3) | (0.6–1.3) | (0.6–1.2) | |
| Cases, controls | 114, 101 | 107, 106 | 102, 102 | 105, 110 |
WBC white blood cells, Q quartile, OR Odds Ratio
aAdjusted for age, time of entry and date of DNA extraction (previously extracted, newly extracted)
bAdditionally adjusted for race, body mass index, age at menarche, age at first live birth, number of children, type of menopause, age at natural menopause, personal history of benign breast disease, cigarette smoking, alcohol intake, family history of breast cancer, and hormone use
In further analyses, we stratified on period of DNA extraction (Table 4). Among the group with previously extracted DNA, we observed a nonsignificant increase (OR = 1.44, 95% CI, 0.8–2.7) in risk in the lowest quartile of %5-mdC compared to those in highest quartile. In the group with newly extracted DNA, however, there was an unexpected decreasing trend in the ORs from the highest to lowest quartile of %5-mdC (p for trend = .05). Risk was also significantly decreased in the lowest quartile of %5-mdC compared to those in the highest quartile (OR = 0.61, 95% CI, 0.40–1.0).
Table 4.
Quartile level of WBC DNA methylation and risk of breast cancer by date of DNA extraction, and by time between blood collection and diagnosis, stratified by DNA extraction method
| Quartiles of DNA methylation | ||||
|---|---|---|---|---|
| Q4 (high) | Q3 | Q2 | Q1 (low) | |
| Period of DNA extraction | ||||
| Previously extracted DNA | ||||
| Adjusted ORa | 1. 0 (referent) | 0.99 | 1.01 | 1.44 |
| 95% CI | (0.5–1.9) | (0.5–2.0) | (0.8–2.7) | |
| Cases, controls | 33, 36 | 33, 36 | 34, 36 | 51, 39 |
| Newly extracted DNA | ||||
| Adjusted ORa | 1.0 (referent) | 0.84 | 0.82 | 0.61 |
| 95% CI | (0.5–1.3) | (0.5–1.3) | (0.4–1.0) | |
| Cases, controls | 81, 65 | 74, 70 | 68, 66 | 54, 71 |
| Time from blood collection to diagnosis | ||||
| Previously extracted DNA | ||||
| 1 to <2 years | ||||
| Adjusted ORa,b | 1.0 (referent) | 0.99 | 1.16 | 1.41 |
| 95% CI | (0.4–2.7) | (0.4–3.0) | (0.6–3.6) | |
| Cases, controls | 11, 36 | 10, 36 | 13, 36 | 16, 39 |
| 2 to <4 years | ||||
| Adjusted ORa,b | 1.0 (referent) | 1.15 | 1.00 | 1.39 |
| 95% CI | (0.5–2.7) | (0.4–2.4) | (0.6–3.2) | |
| Cases, controls | 14, 36 | 16, 36 | 14, 36 | 21, 39 |
| ≥ 4 years | ||||
| Adjusted ORa,b | 1.0 (referent) | 0.89 | 0.97 | 1.53 |
| 95% CI | (0.3–2.8) | (0.3–3.1) | (0.6–4.3) | |
| Cases, controls | 8, 36 | 7, 36 | 7, 36 | 14, 39 |
| Newly extracted DNA | ||||
| 2 to <4 years | ||||
| Adjusted ORa,b | 1. 0 (referent) | 0.88 | 0.73 | 0.68 |
| 95% CI | (0.5–1.5) | (0.4–1.3) | (0.4–1.2) | |
| Cases, controls | 43, 65 | 43, 70 | 32, 66 | 33, 71 |
| ≥4 years | ||||
| Adjusted ORa,b | 1.0 (referent) | 0.80 | 1.00 | 0.51 |
| 95% CI | (0.4–1.5) | (0.6–1.8) | (0.3–1.0) | |
| Cases, controls | 38, 65 | 31, 70 | 36, 66 | 21, 71 |
aAdjusted for age and time at entry
bSame controls are used for each category of elapsed time from blood collection to diagnosis, within period of DNA extraction (previously extracted, newly extracted); OR Odds Ratio
We further stratified on both period of DNA extraction and elapsed time from blood collection to diagnosis. Inherent to the study design, all of the breast cancer cases diagnosed within 1- < 2 years of blood collection had previously extracted DNA. When we restricted the analysis to study subjects with previously extracted DNA, we observed nonsignificant slight increases in risk in the lowest quartile level of %5-mdC in all three categories of years since blood collection (i.e. 1- < 2, 2- < 4, ≥4 years). When we restricted analysis to study subjects with newly extracted DNA, however, we found no evidence of any increased risk in women in either the 2- < 4, or ≥4 years since blood collection.
Discussion
Overall, we found no evidence that lower levels of %5-mdC in white blood cell DNA were associated with increased breast cancer risk in a case-control study nested in the PLCO cohort. The %5mdC assay provides a comprehensive measure of genome-wide DNA methylation and is considered the gold standard for accuracy. Odds ratio (OR) estimates and 95% confidence intervals (CI) for breast cancer risk adjusted for age at entry, year of entry, and period of DNA extraction, were 1.0 (referent), 0.89 (95% CI, 0.6–1.3), 0.88 (95% CI, 0.6–1.3), and 0.84 (95% CI, 0.6–1.2) for women in the highest compared to lowest quartile levels of %5md-C (p for trend = .39). There was some variability in our results depending on whether the DNA was previously or newly extracted. We observed a nonsignificant increased risk in the lowest quartile of %5mdC in the subset of women with previously extracted DNA, whereas risk was significantly decreased in the lowest quartile of %5mdC in the subset of women with newly extracted DNA. One possibility is that this difference in the results by period of DNA extraction is the result of technical issues or sample degradation. Conceivably, the earlier method of extraction or shorter buffy coat storage of the previously extracted samples may have resulted in less non-differential misclassification in our methylation measure. In a recent reliability study of methylation measures from mononuclear cells using the HumanMethylation450K Bead Array, differences in DNA extraction methods (and possible differences in cell composition resulting from them) were suggested to have contributed to lower observed reliability for repeated samples across studies than for technical replicates within a study [26]. Given that our findings that previously and newly extracted DNA were in opposing directions and that there was a larger sample size of newly extracted DNA, chance due to small numbers after stratification is a likely explanation for the observed variability.
Our results are not consistent with those of the one other breast cancer study to measure global DNA hypomethylation with %5-mdC, a small case-control study in which WBC DNA was collected after breast cancer diagnosis [11]. In that study, risk of breast cancer was nearly three times higher among those with in the lowest tertile of %5-mdC compared to those in the highest tertile. A major strength of our study is that we purposely included only breast cancer cases in which DNA was collected at least 12 months prior to diagnosis. We found no evidence that risk varied according to time elapsed between blood collection and breast cancer diagnosis.
As well-summarized in a recent systematic review [27], the weight of evidence also does not support an association between breast cancer risk and WBC DNA methylation, measured in other studies by surrogate marker methods, such as Alu, LINE-1, or LUMA. Of seven retrospective case-control studies [11, 14–19] and four studies with prospectively collected pre-diagnostic WBC DNA (results from three cohorts were presented in a single publication) [12, 13] that measured global methylation by Alu and LINE-1 methylation, only one [12] observed a significantly higher risk of breast cancer among those who had lower LINE-1 methylation levels. Three retrospective case-control studies have examined the relation between WBC DNA global methylation levels and breast cancer risk using LUMA, with inconsistent results [15, 20, 21]. As suggested by Brennan and colleages [22], LINE-1 and other surrogate assays are likely not sufficiently sensitive to detect slight inter-individual differences in WBC DNA methylation. Indeed, Brennan and colleagues reported that the population variability in WBC DNA LINE-1 methylation measured by pyrosequencing in prospectively collected blood did not statistically exceed that of technical duplicates [13]. Further, Tang and colleagues [27] noted that the findings from WBC DNA methylation studies that have evaluated LINE-1 and breast cancer have been null with one exception, despite using different methods of detection (e.g., combined bisulfite restriction analysis, pyrosequencing, MethylLight). Interestingly, a particular strength noted of the prospective study that detected a statistical association between WBC DNA LINE-1 hypomethylation and breast cancer risk was that it employed three independent bisulfite conversions, PCR, and pyrosequencing reactions on each sample [12]. In our prospectively collected blood specimens, we also observed limited population variability in %5mdC levels in WBC DNA, adding to concern that even a small amount of laboratory error is problematic in studies that involve quantification of global WBC DNA methylation from healthy individuals.
Several recent studies have estimated global methylation by averaging individual CpG site-specific methylation levels over the hundreds of thousands of CpG sites on the Illumina HumanMethylation450 BeadChip [7, 8]. This alternative approach measures less than 5% of the 28 million CpG sites in the genome [28]. In two prospective cohort analyses that used this approach to measure WBC DNA methylation, women in the highest quartile of methylation had about a 50% decrease in risk of breast cancer compared to women in the lowest quartile of methylation [7, 8]. Another cohort analysis, which used pooled samples and next-generation sequencing of the overlapping CpG sites from the Illumina HumanMethylation450 BeadChip, also found higher levels of genome-wide methylation in controls than in cases [7]. However, findings from a fourth cohort study were null [7]. Subanalyses based on genomic region have been inconsistent. One study found that WBC epigenome-wide methylation in the promoter region was associated with an increase in breast cancer risk whereas epigenome-wide methylation outside the promoter region was associated with a decreased risk of breast cancer [8]. A second study confirmed that WBC epigenome-wide methylation in gene bodies was associated with decreased risk but was unable to replicate the increase in risk with promoter region methylation [7]. These findings may need to be interpreted with caution given a recent report that measurement error and limited variability in DNA methylation measures from mononuclear cells is problematic for a substantial proportion of of CpG sites on the HumanMethylation450 BeadChip [26].
Another potential limitation of our study is that we studied %5-mdC in a composite of DNA from different types of white blood cells. As previously noted by others [29, 30], the WBC distribution can vary across individuals and the level of global DNA methylation can vary by cell type. The study by Choi and colleagues also used composite DNA [11] as have nearly all other prior WBC methylation studies because it is simple and yields the most DNA [30]. Another potential limitation of our study is the batch-to-batch variation in %5mdC that we observed. This issue necessitated creating quartile cut points for %5mdC separately for individuals within each laboratory batch, a method also employed in the case-control study by Choi and colleagues [11].
Conclusions
In summary, our study, as well as the published literature, which has used a range of methods for assessing global methylation, including Alu, LINE-1, LUMA, and %5-mdC assays, does not support a link between WBC DNA methylation and breast cancer risk. It is conceivable a more accurate and precise measure of global methylation is needed. However, if there is an association between WBC DNA methylation patterns and breast cancer risk, it is likely that the at-risk methylation pattern is more complex and restricted to specific regions and will need to be identified through measures of sequence-specific or gene-specific DNA methylation [13, 31].
Acknowledgements
Not applicable.
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number R15CA170111 and the intramural research program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Availability of data and materials
Data are available for request through the PLCO Cancer Data Access Site (CDAS): (https://biometry.nci.nih.gov/cdas/plco/).
Abbreviations
- %5-mdC
Ratio of 5-methyl-2’ deoxycytidine (5-mdC) to 2’-deoxyguanine (dG)
- CCGG
Cytosine-cytosine-guanine-guanine sequence
- CI
Confidence interval
- CpG
Cytosine-phosphate-guanine dinucleotide
- LC-ESI-MS/MS
Liquid chromatography-electrospray ionization-tandem mass spectrometry
- LINE-1
Long interspersed nuclear element 1
- LUMA
Luminometric methylation assay
- m/z
Mass-to-charge ratio
- OR
Odds ratio
- PCR
Polymerase chain reaction
- PLCO
Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial
- WBC
White blood cell
Authors’ contributions
SRS and KFA conceived and designed the study, with additional guidance from JRP, ARK, CS, RGZ, RB, and DAR. KI, SM, NCT, and HW performed the laboratory analyses, supervised by DAR and JRP. SRS and RB analyzed the data. SRS wrote the manuscript with input and critical review from JRP, KFA, CS, RGZ, KI, SK, NCT, HW, RB, ARK, and DAR. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Ethics approval and consent to participate
The University of Massachusetts Amherst Institutional Review Board, the Special Studies Institutional Review Board at the National Cancer Institute, and institutional review boards at the 10 participating study centers (Colorado Multiple Institutional Review Board, CB F490, MedStar Health Research Institute-Georgetown University Oncology Institutional Review Board, Pacific Health Research and Education Institute Institutional Review Board, Henry Ford Health System Institutional Review Board, University of Minnesota Twin Cities Institutional Review Board, Washington University in St. Louis Institutional Review Board, University of Pittsburgh Institutional Review Board, The University of Utah Institutional Review Board, Marshfield Clinic Research Foundation Institutional Review Board, and University of Alabama at Birmingham Institutional Review Board) approved this study. Informed consent was obtained from all study participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Susan R. Sturgeon, Phone: 413-577-1364, Email: ssturgeon@schoolph.umass.edu
J. Richard Pilsner, Email: rpilsner@schoolph.umass.edu.
Kathleen F. Arcaro, Email: karcaro@vasci.umass.edu
Kaoru Ikuma, Email: kikuma@lastate.edu.
Haotian Wu, Email: haotianwu@schoolph.umass.edu.
Soon-Mi Kim, Email: soonmi@umass.edu.
Nayha Chopra-Tandon, Email: nchoprat@umass.edu.
Adam R. Karpf, Email: adam.karpf@unmc.edu
Regina G. Ziegler, Email: regina.ziegler@nih.gov
Catherine Schairer, Email: schairerc@exchange.nih.gov.
Raji Balasubramanian, Email: rbalasub@schoolph.umass.edu.
David A. Reckhow, Email: reckhow@ecs.umass.edu
References
- 1.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 2.Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, Ehrlich M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11:6883–94. doi: 10.1093/nar/11.19.6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song L, James SR, Kazim L, Karpf AR. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:504–10. doi: 10.1021/ac0489420. [DOI] [PubMed] [Google Scholar]
- 4.Bestor TH. The host defence function of genomic methylation patterns. Novartis Found Symp. 1998;214:187–95. doi: 10.1002/9780470515501.ch11. [DOI] [PubMed] [Google Scholar]
- 5.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karimi M, Johansson S, Ekstrom TJ. Using LUMA: a luminometric-based assay for global DNA-methylation. Epigenetics. 2006;1:45–8. doi: 10.4161/epi.1.1.2587. [DOI] [PubMed] [Google Scholar]
- 7.van Veldhoven K, Polidoro S, Baglietto L, Severi G, Sacerdote C, Panico S, Mattiello A, Palli D, Masala G, Krogh V, et al. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin Epigenetics. 2015;7:67. doi: 10.1186/s13148-015-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Severi G, Southey MC, English DR, Jung CH, Lonie A, McLean C, Tsimiklis H, Hopper JL, Giles GG, Baglietto L. Epigenome-wide methylation in DNA from peripheral blood as a marker of risk for breast cancer. Breast Cancer Res Treat. 2014;148:665–73. doi: 10.1007/s10549-014-3209-y. [DOI] [PubMed] [Google Scholar]
- 9.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Closas M, Gail MH, Kelsey KT, Ziegler RG. Searching for blood DNA methylation markers of breast cancer risk and early detection. J Natl Cancer Inst. 2013;105:678–80. doi: 10.1093/jnci/djt090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi JY, James SR, Link PA, McCann SE, Hong CC, Davis W, Nesline MK, Ambrosone CB, Karpf AR. Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis. 2009. doi:10.1093/carcin/bgp143. PMID: 1958413 [DOI] [PMC free article] [PubMed]
- 12.Deroo LA, Bolick SC, Xu Z, Umbach DM, Shore D, Weinberg CR, Sandler DP, Taylor JA. Global DNA methylation and one-carbon metabolism gene polymorphisms and the risk of breast cancer in the sister study. Carcinogenesis. 2014;35:333–8. doi: 10.1093/carcin/bgt342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brennan K, Garcia-Closas M, Orr N, Fletcher O, Jones M, Ashworth A, Swerdlow A, Thorne H, KConFab Investigators. Riboli E, et al. Intragenic ATM methylation in peripheral blood DNA as a biomarker of breast cancer risk. Cancer Res. 2012;72:2304–13. doi: 10.1158/0008-5472.CAN-11-3157. [DOI] [PubMed] [Google Scholar]
- 14.Wu HC, Delgado-Cruzata L, Flom JD, Perrin M, Liao Y, Ferris J, Santella RM, Terry MB. Repetitive element DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the BCFR. Carcinogenesis. 2012. doi:10.1093/carcin/bgs201. [DOI] [PMC free article] [PubMed]
- 15.Xu X, Gammon MD, Hernandez-Vargas H, Herceg Z, Wetmur JG, Teitelbaum SL, Bradshaw PT, Neugut AI, Santella RM, Chen J. DNA methylation in peripheral blood measured by LUMA is associated with breast cancer in a population-based study. FASEB J. 2012;26:2657–66. doi: 10.1096/fj.11-197251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho YH, Yazici H, Wu HC, Terry MB, Gonzalez K, Qu M, Dalay N, Santella RM. Aberrant promoter hypermethylation and genomic hypomethylation in tumor, adjacent normal tissues and blood from breast cancer patients. Anticancer Res. 2010;30:2489–96. [PMC free article] [PubMed] [Google Scholar]
- 17.Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, Fletcher O, Dos Santos Silva I, Peto J, Boshoff C, et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum Mol Genet. 2009;18:1332–42. doi: 10.1093/hmg/ddp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitkumthorn N, Tuangsintanakul T, Rattanatanyong P, Tiwawech D, Mutirangura A. LINE-1 methylation in the peripheral blood mononuclear cells of cancer patients. Clin Chim Acta. 2012;413:869–74. doi: 10.1016/j.cca.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 19.Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, Stegmaier C, Jacobs IJ, Brenner H. Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS One. 2008;3:e2656. doi: 10.1371/journal.pone.0002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delgado-Cruzata L, Wu HC, Perrin M, Liao Y, Kappil MA, Ferris JS, Flom JD, Yazici H, Santella RM, Terry MB. Global DNA methylation levels in white blood cell DNA from sisters discordant for breast cancer from the New York site of the breast cancer family registry. Epigenetics. 2012;7:868-74. doi:10.4161/epi.20830. Epub 2012 Jun 18. [DOI] [PMC free article] [PubMed]
- 21.Kuchiba A, Iwasaki M, Ono H, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Tsugane S, Yoshida T. Global methylation levels in peripheral blood leukocyte DNA by LUMA and breast cancer: a case-control study in Japanese women. Br J Cancer. 2014;110:2765–71. doi: 10.1038/bjc.2014.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brennan K, Flanagan JM. Is there a link between genome-wide hypomethylation in blood and cancer risk? Cancer Prev Res (Phila) 2012;5:1345–57. doi: 10.1158/1940-6207.CAPR-12-0316. [DOI] [PubMed] [Google Scholar]
- 23.Hayes RB, Sigurdson A, Moore L, Peters U, Huang WY, Pinsky P, Reding D, Gelmann EP, Rothman N, Pfeiffer RM, et al. Methods for etiologic and early marker investigations in the PLCO trial. Mutat Res. 2005;592:147–54. PMID: 16054167. [DOI] [PubMed]
- 24.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, et al. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–4. PMID: 17529973. [DOI] [PMC free article] [PubMed]
- 25.Lacey JV, Jr, Kreimer AR, Buys SS, Marcus PM, Chang SC, Leitzmann MF, Hoover RN, Prorok PC, Berg CD, Hartge P, et al. Breast cancer epidemiology according to recognized breast cancer risk factors in the prostate, lung, colorectal and ovarian (PLCO) cancer screening trial cohort. BMC Cancer. 2009;9:84. doi: 10.1186/1471-2407-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dugue PA, English DR, MacInnis RJ, Jung CH, Bassett JK, FitzGerald LM, Wong EM, Joo JE, Hopper JL, Southey MC, et al. Reliability of DNA methylation measures from dried blood spots and mononuclear cells using the HumanMethylation450k BeadArray. Sci Rep. 2016;6:30317. doi:10.1038/srep30317. [DOI] [PMC free article] [PubMed]
- 27.Tang Q, Cheng J, Cao X, Surowy H, Burwinkel B. Blood-based DNA methylation as biomarker for breast cancer: a systematic review.Clin Epigenetics. 2016;8:115. doi:10.1186/s13148-016-0282-6. [DOI] [PMC free article] [PubMed]
- 28.Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet. 2014;30:75–84. doi:10.1016/j.tig.2013.11.004. [DOI] [PubMed]
- 29.Wu HC, Delgado-Cruzata L, Flom JD, Kappil M, Ferris JS, Liao Y, Santella RM, Terry MB. Global methylation profiles in DNA from different blood cell types. Epigenetics. 2011;6. doi:10.4161/epi.6.1.13391. [DOI] [PMC free article] [PubMed]
- 30.Terry MB, Delgado-Cruzata L, Vin-Raviv N, Wu HC, Santella RM. DNA methylation in white blood cells: association with risk factors in epidemiologic studies. Epigenetics. 2011;6:828-37. doi:10.4161/epi.6.7.16500. Epub 2011 July 1. [DOI] [PMC free article] [PubMed]
- 31.Xu Z, Bolick SC, Deroo LA, Weinberg CR, Sandler DP, Taylor JA. Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst. 2013;105:694–700. doi: 10.1093/jnci/djt045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available for request through the PLCO Cancer Data Access Site (CDAS): (https://biometry.nci.nih.gov/cdas/plco/).