

Abstract
Background:
This study was performed to investigate any association between cellular cholesterol homeostasis and chronic lymphocytic leukemia (CLL). CLL is characterized primarily by an abnormal accumulation of neoplastic B cells in the blood, bone marrow, lymph nodes and spleen.
Methods:
Men aged >50 years participated in this study. Enzyme-based plasma lipid profile estimations, peripheral blood lymphocyte isolation, lysate preparations, SDS-PAGE, western blotting, dil-LDL uptake and ultracentrifugation were employed.
Results:
Our study demonstrated hypocholesterolemia in lymphocytic leukemia in addition to hyper-expression of LDLRs in leukemic lymphocytes. Breakdown of intracellular cholesterol homeostasis and failure to maintain the feedback mechanism normally processed by the transcription factor SREBP-2 in the cytoplasm was apparent. The presence of cholesterol in the nucleus was noted in leukemic lymphocytes. A comparison of cholesterol homeostasis between healthy controls and CLL subjects showed that cholesterol may contribute to lymphocytic leukemia. While plasma cholesterol levels decreased (p < 0.0005), hyper-expression of LDLR (p=0.0001), SREBP-2 (transcription factor of LDLR) (p=0.0001) and PBR (nuclear cholesterol channel protein) (p=0.016) was observed in lymphocytes isolated from CLL subjects in association with a significant increase in intracellular cholesterol in the nuclear (p=0.036) and cytoplasmic (p=0.004) compartments.
Conclusion:
This study provided insights into cholesterol homeostasis in CLL subjects regarding LDLR, SREBP-2 and PBR. Cholesterol may enter the nucleus through highly expressed PBR and may be involved in development of leukemia by influencing cell cycle mechanisms in the lymphocytes of CLL subjects.
Keywords: LDL, LDL receptor, PBR, nuclear cholesterol, chronic lymphocytic leukemia
Introduction
The importance of cellular cholesterol homeostasis has been well documented in modern biology. Cholesterol is needed to maintain the membrane structure and metabolic activities of cells (Brown et al., 1974). Although the literature claims that an over accumulation of cholesterol is toxic to cells (Zhang et al., 2003; Ishikawa et al., 2008), a number of observations also have noted an intimate relationship between intracellular cholesterol homeostasis and its feedback regulation of cellular proliferation and chromosomal maintenance (Brown et al., 1974; Witte et al., 1982; Siperstein MD, 1984; Dessi et al., 1997). Intracellular cholesterol homeostasis is maintained by the joint performance of intracellular cholesterol biosynthesis and receptor-mediated LDL cholesterol endocytosis (Brown et al., 1986).
Intracellular saturation of sterols (e.g., cholesterol) down-regulates the expression of LDL receptor (LDLR) on surface membranes, which has been shown in various cell types including fibroblasts (Brown et al., 1975), lymphocytes (Ho et al., 1976; Yen et al., 1995), and hepatocytes (Havekes et al., 1986). LDLR expression is regulated by sterols via sterol regulatory element (SRE) (Smith et al., 1990). This DNA element binds to Sterol Regulatory Element Binding Proteins (SREBPs), a family of endoplasmic reticulum-generated transcription factors that are processed through the Golgi apparatus and activate ldlr-gene expression (Brown et al., 1997) after entering into the nucleus. The intracellular saturation of cholesterol down-regulates LDLR and SREBP2 transcription. SREBP2 is a potent regulator of the ldlr promoter in normal cells but not in cancer cells (Chen et al., 2001).
Several studies have provided the evidence for low plasma cholesterol in patients diagnosed with carcinoma (Feinleib M., 1983; Kreger et al., 1992). Hypocholesterolemia is a common finding in several malignant disorders, including acute myelogenous leukemia, chronic myeloproliferative disorders, and colon cancer (Vitols et al., 1985; Vitols, 1991). It is unclear whether cholesterol is a risk factor for the development of malignancy or is secondary to the cancer (Vitols, 1991). Human leukemic cells and certain tumor tissues, as compared to the corresponding normal cells or tissues, display greater receptor-mediated uptake of LDL cholesterol(Peterson et al., 1985).
A relationship between the expression of PBR (peripheral type benzodiazepine receptor) and its nuclear localization and its role in mediating the transport of cholesterol into the cell nucleus has been reported in proliferating breast cancer cells of an aggressive phenotype (Hardwick et al., 2002). Cancer cells have increased metabolic requirements that are due, in part, to increased cell proliferation and mitochondrial biogenesis. PBR functions alter mitochondrial lipid metabolism (Ravagnan et al., 1999). It is not yet clear whether PBR is a cholesterol channel on the nuclear membrane in all types of malignancy or only an incidence in breast carcinoma, as reported previously (Hardwick et al., 2002). Cholesterol is now found in chromosomes within the nucleus (Albi et al., 2004). This evidence regarding LDLR, SREBP2, and PBR has been targeted in this study to determine the plausibility of any direct link between their intra-cellular regulation and intra-nuclear cholesterol deposition or homeostasis, as compared to control subjects.
Materials and methods
Materials and reagents
Enzyme-based plasma lipid profile estimation kits (total cholesterol, LDL- and HDL cholesterol) were purchased from ERBA Diagnostics Gmbh, Mannheim, Germany. Lymphoprep solution for the isolation of peripheral blood lymphocytes was obtained from Axis-shield, Oslo, Norway. Enhanced chemiluminescence reagent and primary antibodies for LDLR, SREBP-2, and PBR were bought from Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA. Secondary antibodies with HRP conjugates were from Bangalore Genei, Bangalore India. Nitrocellulose membranes were obtained from Genotech Inc. CA, USA. Dil (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) was purchased from Sigma, St. Louis, MO, USA. All other chemicals used were of analytical-reagent grade. Fresh human plasma for the LDL isolation was obtained from the Blood Bank, All India Institute of Medical Sciences, New Delhi, India, maintaining all the guidelines and ethical regulations of the institute. LDL was isolated according to the procedure of Havel et al. (Havel et al., 1955).
Study design
Participants
In this case-control study, seventy-five male individuals (twenty-five in each of the following categories) older than 50 yr of age were included as study population according to the stipulated guidelines of and clearance from the Ethical Clearance Committee, AIIMS, New Delhi, India. There was approximately 90% overlap between untreated and treated subjects, i.e., the same subject participated in the untreated and treated groups. Approximately 10% of participants with chronic lymphocytic leukemia (CLL) discontinued themselves from the longr treatment protocol and left the clinic for their own reasons. All the participants were selected randomly. The normal subjects were recruited for the study after undergoing a routine clinical screening procedure; being aware about the importance of the study, and acknowledging their consent. Similarly, the patients in the second group were included based on prefixed criteria determined by the clinics, and written informed consent was obtained prior to their involvement in the study. Each patient’s disease history, blood lymphocyte counts, immune-phenotyping, and other investigations, as decided by the hospital clinic of the outpatient unit, was obtained, in addition to. Subjects were also screened for any drug intake expected to affect lipid metabolism.
Sample collection
Blood samples (10 –12 ml) were aseptically drawn from the superficial veins of each of the study subjects. Whole blood was used for the lymphocytes isolation. Plasma was used for the lipid profile measurement. Plasma was separated from whole blood using a routine laboratory protocol.
Isolation of peripheral blood lymphocytes (PBLs)
Peripheral blood lymphocytes were isolated according to the modified method of Böyum (Böyum, 1968). In brief, whole blood (10 ml) with anticoagulant was mixed with 10 ml of 0.15 M NaCl. Then, the blood-NaCl mixture was layered onto 10 ml of Lymphoprep in a centrifuge tube (50 ml Falcon). The mixture was then centrifuged at 1400 rpm at 25°C for 40 min. The white interphase layer (predominantly lymphocytes) was collected and mixed with 30 ml of Hank’s balanced salt solution. The mixture was then centrifuged at 1,400 rpm for 10 min at 25°C to collect the lymphocyte pellet. The lymphocytes were resuspended in RPMI 1640 medium and centrifuged at 1,000 rpm for 10 min at room temperature; this step was repeated twice. The pure lymphocyte pellet (PBL) was stored for the following experiments.
Preparation of lysate from isolated PBLs
PBLs were isolated from blood and the cells were washed twice in ice-cold PBS. The cells were pelleted by centrifugation at 2500 rpm for 5 minutes at 4°C. The supernatant was discarded, and the pellet was resuspended in lysis buffer containing 50 mM Tris–HCl (pH=7.4), 300 mM NaCl, 0.5% (v/v) Triton X-100, 5 mM EDTA with 2 mM PMSF, 10 mg/mL leupeptin and 10 U/mL aprotinin and then lysed by strong vortexing. The volume of the used lysis buffer was approximately 5–10 times the volume of the pellet. This lysed suspension was kept on ice for 30 minutes and then centrifuged at 12,000 rpm for 15 minutes at 4°C. The supernatant was collected and stored at 4°C. Protein content was estimated using the Bradford method (Bradford MM., 1976) before performing SDS-PAGE on the lysate.
SDS-PAGE and western blotting
The protein samples (lysate) were mixed with 5x protein loading buffer containing 1% SDS, 5% β-mercaptoethanol, 50 mM Tris-HCl (pH= 6.8), 10% glycerol, and 0.02% bromo phenol blue. Then, they were boiled for 5 minutes. Next, 10% SDS-PAGE was carried out on the samples and the proteins were transferred to nitrocellulose sheets. The transfer was performed at 35 V for 12 hours at 4°C in a transfer buffer containing 48 mM Tris Base, 39 mM glycine, and 20% methanol, pre-chilled at 4°C. The sheet was washed with blocking buffer (5% Blot-Quick blocking power in 0.01 M PBS containing 0.05% Tween-20) for 2 to 4 hours at room temperature on a shaker. Anti-LDLR mouse monoclonal antibody, anti-SREBP-2 mouse monoclonal antibody, anti-PBR goat polyclonal antibody, and β-actin mouse monoclonal antibody diluted in 0.01 M PBS–0.1% Tween-20 were added to the appropriate blots at dilutions of 1:2,000, 1:500, 1:1000, and 1: 2000, respectively. The membrane was incubated at room temperature for 2 hours. The membrane was then washed three times in 0.01 M PBS –0.1% Tween-20 for 10 min each. Anti-mouse and anti-goat HRP conjugates were added at a dilution of 1: 8,000 (anti-mouse) for LDLR, 1:3,000 (anti-mouse) for SREBP-2 and 1:10,000 (anti-goat) for PBR and β Actin, followed by incubation for 2 hours at room temperature. Following three washes in PBS–Tween buffer, the blots were developed using enhanced chemiluminescence in the dark, and the bands were imaged using Kodak XK-5 X-Ray film. An alpha tech imager was used for the analysis of the intensity/density of the respective bands.
Dil-LDL preparation
Dil-LDL was prepared in the laboratory as described in our earlier reports (Ramakrishnan et al., 2012; Arjuman et al., 2013).
Dil-LDL uptake studies
LDL uptake by lymphocytic cells was carried out on samples from patients and healthy subjects. Fluorescently labeled Dil-LDL was used as a substrate to determine the uptake rate of lymphocyte cells. Isolated lymphocytic cells were seeded in 12-well plates at 2 x 106 cells/well. The cells were serum starved for 12-16 hr in incomplete RPMI medium. Dil-LDL was then added to the wells at different concentrations, i.e., 0, 25, 50, 75, 100 and 125 μg/ml. The cells were incubated for 5 hr at 37°C. The remaining cell-surface-bound LDL (i.e., the LDL that did not internalize into the cells during the incubation) was removed by treating the cells with dextran sulphate buffer (50 mM NaCl, 10 mM HEPES, 10 mg/ml dextran sulphate) for 1 hour at 4°C. The medium was removed, 1 ml of isopropanol was added to each well, and the mixture was mixed thoroughly for 15 minutes at room temperature in the dark. The isopropanol was then collected and centrifuged at 5,000 g for 10 minutes at room temperature. The supernatant was applied for Dil quantification (Ramakrishnan et al., 2012). The readings were utlized to construct a graph of the added Dil-LDL and to compare the amount of Dil-LDL taken up by the cells.
Isolation of nucleus and cytoplasm of lymphocytes
Isolated lymphocytes were pelleted by centrifugation at 2,000 rpm at 4°C for 5 minutes. Then, 2 x 106 cells were washed in ice-cold PBS. The cell pellet was re-suspended in 300 µl of ice-cold nuclei preparation buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% Nonidet P-40, 0.15 mM spermine and 0.5 mM spermidine) and incubated on ice for 5 minutes. The cell suspension was centrifuged at 3,000 rpm for 5 minutes to pellet the nuclei. The remaining supernatant was ultracentrifuged at a speed of 100,000g for 60 min to obtain the cytoplasm.
Cholesterol measurement in nuclear and cytoplasmic fractions of lymphocytes
Total cell cholesterol was extracted from the cells using 1 ml of hexane:isopropanol (3:2, v:v). The extracts were transferred to microcentrifuge tubes and were air dried. The cholesterol standard, the samples, and the blank were dissolved in 1 ml of isopropanol:Nonidet P-40 (9:1, v:v). The cholesterol content in the nucleus and cytoplasm of lymphocytes was estimated using an enzymatic cholesterol assay based on a fluorometric method (Robinet et al., 2010). Fluorescence was read at an excitation wavelength of 530 nm and an emission wavelength of 580 nm.
Statistical analysis
Results from experimental repeats have been expressed as the mean ± SD. All expression data have been normalized against the respective internal control as indicated in the Figures. Statistical analysis was carried out using a 2-tailed unpaired Student’s t-test and a one-way analysis of variance (ANOVA) test. The significance level was set at p<0.05.
Results
Biochemical parameters across different study groups
With respect to Table 1, a significant decrease in the plasma LDL-cholesterol concentrations can be seen in the leukemic group as compared to the normal subjects. Only a partial recovery was noticed in treated patients. While LDL levels decreased in leukemic subjects, on average, the HDL concentration remained unchanged. Considering the results, it can be concluded that a malignancy such as leukemia can affect LDL cholesterol homeostasis. Low plasma LDL concentration in leukemic patients can be attributed to clearance of LDL particles from blood plasma by highly expressing LDLRs. Eventually, leukemia represented a metabolically opposite scenario to that of dyslipidemia affected atherosclerosis.
Table 1.
Biochemical Parameters (Concentrations in Blood Plasma) of the Different Study Groups: A significant decrease in plasma cholesterol concentrations (total, LDL and HDL) was observed in the untreated leukemic group as compared to that in the normal subjects. The variation of HDL concentrations remained within normal limits, whereas LDL concentrations decreased to a significantly low value. Only a partial recovery was noted in treated patients. An ANOVA was applied for the statistical evaluation. The data are presented as the mean ± SD for 25 samples from each respective category
| Variables in plasma | Normal range (Mean±SD) | 1. Control subjects (Mean±SD) (n=25) | 2. Untreated patients (Mean±SD) (n=25) | 3. Treated patients (Mean±SD) (n=25) | p-value |
|---|---|---|---|---|---|
| Total cholesterol | 140-250 mg/dl | 182.0 ± 21.8 | 136.5 ± 21.5 | 168.3 ± 32.1 | 0.0001 |
| 1 vs. 2 <0.0005 | |||||
| 1 vs. 3 ns | |||||
| LDL | <130 mg/dl | 116.6 ± 21.3 | 69.0 ± 14.7 | 109.1 ± 36.0 | 0.0001 |
| 1 vs. 2 <0.0005 | |||||
| 1 vs. 3 ns | |||||
| HDL | 30-65 mg/dl | 39.9 ± 9.0 | 31.2 ± 6.8 | 36.4 ± 7.7 | 0.0043 |
| 1 vs. 2 <0.005 | |||||
| 1 vs. 3 ns |
LDL, low-density lipoprotein; HDL, high-density lipoprotein
Expression of LDLR, SREBP-2, and nuclear PBR
To find out the reason behind low plasma cholesterol concentrations in leukemic patients, the expression of three proteins involved in intracellular cholesterol homeostasis-LDLR, SREBP-2, and PBR-were studied. LDLR on the cell surface is involved in cholesterol clearance from the periphery; SREBP2 is a transcription factor that regulates LDLR and comes from a precursor in the endoplasmic reticulum, and PBR is a receptor-associated cholesterol channel on the nuclear membrane and is involved in cholesterol transport from the cytoplasm to the nucleoplasm. The immunoblots in Figure 1 (panels A, B, and C) show that LDLR, SREBP-2, and PBR were highly expressed in untreated leukemic lymphocytes compared to normal lymphocytes in the control group. Treatment caused a partial recovery of LDLR and SREBP-2 to their normal profile. Treatment did not show a consistent change at the level of PBR expression.
Figure 1.
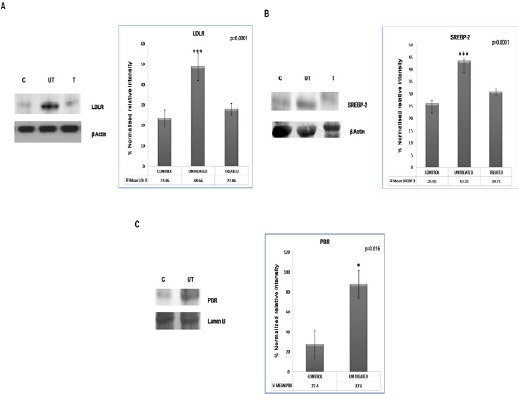
Expression of LDLR, SREBP2 and Nuclear PBR: Respective antibodies were used to visualize the expression of LDLR, SREBP2 and PBR by western blotting. Three groups of subjects-control (C), untreated (UT) and treated (T)-were included in the study. A 2-tailed unpaired Student’s t-test was employed to determine statistical significance. Each bar represents the mean ± SD for 25 samples from each respective category. Significantly greater LDL receptor (panel A; p=0.0001), SREBP2 (panel B; p=0.0001) and PBR (panel C; p=0.016) expression was observed in the untreated leukemic group than in lymphocytes from the control subjects. The values here depict the densitometric measurements of the blots using an alpha image analyzer. β-actin and lamin B were used as internal markers. Treatment showed (data not shown for PBR) partial recovery
Functional activity of LDLR by Dil-LDL uptake assay
The functional activity of LDLR was measured estimating the uptake of Dil-labeled LDL by the cells (see method section). The amount of Dil internalized by the cells (proportional to internalized LDL) was used as a measure to determine the LDL taken up by the cells. Figure 2 shows a higher rate of LDL uptake by leukemic lymphocytes in comparison with normal subjects. It was also evident that leukemic lymphocytes needed more LDL particles than normal lymphocytes to reach the saturation point. The delayed saturation in leukemic lymphocytes suggested a comparatively more rapid utilization of the cytoplasmic cholesterol pool than its normal counterparts. The differential time to reach the equilibrium between incoming and outgoing concentrations of cholesterol within the cytoplasm determined the time delay in reaching the saturation point of the cytoplasmic cholesterol pool. Since the leukemic lymphocytes differed from normal lymphocytes due to having highly expressed PBR and leukemia led to cell density augmentation, increased cholesterol utilization was expected in new membrane formation and/or in chromatin structures.
Figure 2.
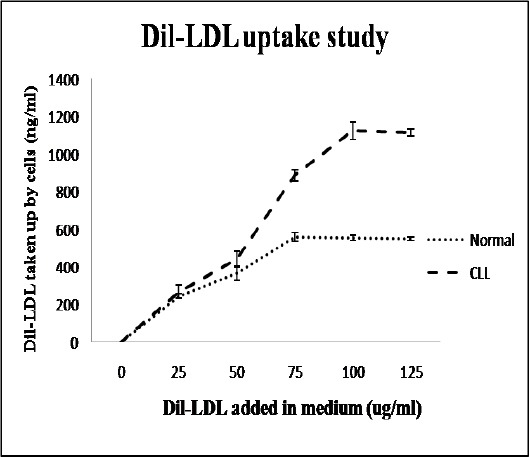
Functional Activity of LDLR by Dil-LDL Uptake Assay: For this assay, 2 x 106 cells/ml were incubated with different concentrations of Dil-LDL (0, 25, 50, 75, 100, 125 µg/ml) for 5 hr in a multi-well plate. Amount of Dil internalized into the cells was proportional to the quantity of LDL taken up by the cells. The graphs show a higher rate of LDL uptake by leukemic lymphocytes as compared to normal counterparts. It was also evident that leukemic lymphocytes required more LDL particles than normal lymphocytes to reach the saturation point. A 2-tailed unpaired Student’s t-test was employed to determine statistical significance. The data show the mean ± SD for 25 samples from each respective category
In vitro feedback regulation of LDLR and SREBP-2 expression by saturated dose of LDL cholesterol in normal and leukemic lymphocytes
In normal lymphocytes (control cells in Figure 3), LDLR and SREBP2 expressions decreased with cholesterol saturation (100 µg/ml LDL). However, in patient’s cells (leukemic lymphocytes in Figure 4), no significant decrease was found in the expression of both LDLR and SREBP2 with equivalent cholesterol saturation by LDL. Earlier reports have shown that the net intracellular cholesterol concentration is the regulator for net concentration of activated SREBP2 (Brown et al., 1997) in normal cells. Since SREBP2 is the transcription factor for the ldlr gene, the cholesterol-mediated regulation of SREBP2 (Brown et al., 1997) can also regulate the expression of LDLR on the cell surface. The rule worked in normal lymphocytes (Figure 3) but not in leukemic lymphocytes (Figure 4). This result apparently demonstrated that the cholesterol mediated feedback regulation on LDLR transcription fails in leukemic cells. This result corroborates the findings of Chen et al., hold that human prostate cancer cells lack feedback regulation of LDLR and its regulator, SREBP-2 (Chen et al., 2001).
Figure 3.
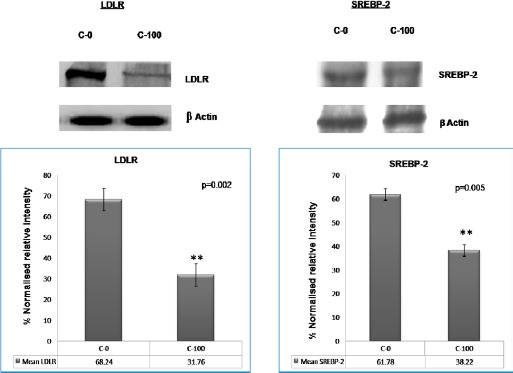
In Vitro Feedback Regulation of LDLR and SREBP-2 Expression Caused by a Saturated Dose of LDL Cholesterol in Lymphocytes from Control Subjects:
Figure 4.
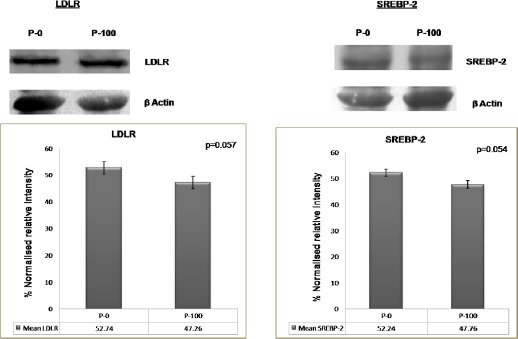
In Vitro Feedback Regulation of LDLR and SREBP-2 Expression Caused by a Saturated Dose of LDL Cholesterol in Lymphocytes from Leukemic Subjects: LDLR and SREBP-2 expression after LDL treatment (lymphocytes from leukemic subjects): No significant decrease in the expression level of either LDLR or SREBP-2 was observed in cells from patients (leukemic lymphocytes) with cholesterol saturation (P-100). p = 0.057 (LDLR), p = 0.054 (SREBP-2). P-0: Cells incubated without LDL; P-100: Cells incubated with 100 µg/ml LDL. A 2-tailed paired Student’s t-test was applied for these parameters, and a p value was obtained. Each bar shows the mean ± SD for 25 samples from each respective category.
LDLR and SREBP-2 expression after LDL treatment (control): LDLR and SREBP-2 expression in normal lymphocytes from control subjects went down with cholesterol saturation (C-100). p = 0.002 (LDLR), p = 0.005 (SREBP-2). C-0: cells incubated without LDL; C-100: cells incubated with 100 µg/ml LDL. A 2-tailed paired Student’s t-test was applied for these parameters, and a p value was obtained. Each bar shows the mean ± SD for 25 samples from each respective category.
Relative cholesterol content in cell nucleus and cytoplasm of lymphocytes across control and leukemic groups
There was a significant increase in the cholesterol content in the nucleus and cytoplasm of leukemic lymphocytes (Figure 5). The nuclear cholesterol concentration per lymphocyte in leukemic patients amplified almost twofold and the cytoplasmic cholesterol augmented approximately threefold over the control lymphocyte cholesterol concentration.
Figure 5.
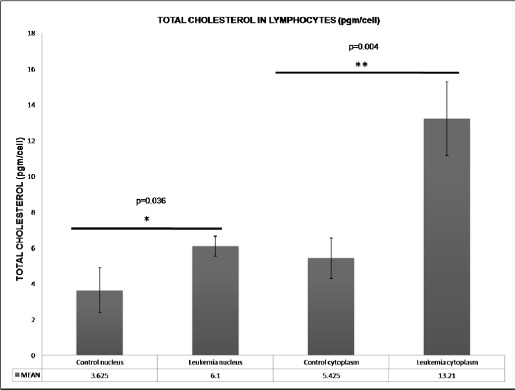
Relative Cholesterol Content in the Cell Nucleus and Cytoplasm of Lymphocytes from Control and Leukemic Subjects: A significant increase in the cholesterol concentration was observed in the nucleus (p=0.036) and cytoplasm (p=0.004) of leukemic cells as compared to normal lymphocytes. Each bar shows the mean ± SD for 25 samples from each respective category
Discussion
As with other cancers; namely, acute myelogenous leukemia (AML), chronic myeloproliferative disorders, and colon cancer (Vitols et al., 1985; Vitols, 1991), lipid abnormality was observed in CLL patients of our study. The major observation in this study was the correlation between the significant changes in plasma lipid profile (cholesterol, LDL, and HDL), particularly LDL, and the expression of cholesterol-processing molecules (LDLR, SREBP-2, and PBR) in lymphocytes of CLL participants. The altered cholesterol level in CLL patients can be attributed to the increased utilization of cholesterol by the hyper expressed LDLRs in neoplastic cells. This type of observation is in line with in vitro findings revealed by various mononuclear cells studies on patients and animals with leukemia (Budd et al., 1986). Accordingly, prolonged low LDL levels may provide a clue for a predisposition to malignant tumors and can serve as a screening tool to provide the basis for preventive measures.
Our findings showed that in a disease such as leukemia, cholesterol metabolism is affected. The internalization of exogenous cholesterol (blood cholesterol) is mainly achieved through LDLR-mediated endocytosis (Brown et al., 1986). Considering the results of the current study, both the LDLR and its transcription factor SREBP-2 were more highly expressed in the lymphocytes of CLL participants than in those of the control group. This result corroborates the findings of Peterson et al., demonstrating an elevated activity of highly expressed LDLR in malignant cells (Peterson et al., 1985). In this study, the Dil-LDL uptake assay was used as a measure of the LDLR functional activity and depicted a high rate of LDL utilization by leukemic lymphocytes. Moreover, a delayed saturation curve for cholesterol (more time was taken to reach the plateau of the cholesterol saturation curve in leukemic cells) was found indicating faster utilization of the cytoplasmic cholesterol pool in leukemic lymphocytes in comparison with the normal counterparts. Utilization of more cholesterol is expected by the greater mass of cancer cells as well as its use in chromatin structures (Albi et al., 2003). A newly growing consensus on the role of cholesterol in the cell nucleus has been emerged by discovering cholesterol within the chromosomal DNA (Albi et al., 2003), sphingomyelin-bound cholesterol (esterified cholesterol) in the cell nucleus (Albi et al., 2003), and the liberation of more free cholesterol by liver regeneration processes (Rao, 1986; Albi et al., 2003).
Although several studies have investigated the aspects of LDLR expression, including the feedback regulation loss of intracellular cholesterol homeostasis and the presence of a highly expressed cholesterol transporter on nuclear membrane, the reports are mostly fragmented. No intercorrelation was attempted among these parameters in the same cell. In the present study, we attempted to correlate these parameters in cancer cells, i.e., lymphocytic leukemia ones.
Weaker suppression of LDLR activity and inactivation of transcription factor SREBP-2 by the intracellular accumulation of LDL cholesterol was found in lymphocytes from CLL patients as compared to those from healthy controls.
In other words, these leukemic cells overrode the feedback regulatory mechanism for LDLR expression. This phenomenon could explain the extra demand of cholesterol by leukemic cells, i.e., the need for cholesterol in the cell nucleus, as shown by the reports on the presence of cholesterol in the nuclear chromosome (Albi et al., 2004).
With respect to increase of intracellular (both cytoplasm and nucleus) cholesterol levels and loss of feedback regulation on the transcription of LDLR in leukemic lymphocytes compared to normal lymphocytes, this study suggests the passage of cytoplasmic cholesterol into the cell nucleus through proper channels or transporters found on the nuclear membrane such as PBR.
Furthermore, high expression of PBR in leukemic lymphocytes was discovered in this study. As mentioned above, numerous studies have also indicated a role for nuclear cholesterol in the mechanism of cell proliferation and cancer progression (Hardwick et al., 1999). Normally, PBR is located on outer mitochondrial membrane in endocrine tissues, where it transports cholesterol and helps in steroidogenesis. A role for nuclear PBR in the regulation of tumor cell proliferation along with their aggressive phenotypic expression have also been shown by Hardwick et al., (1999). We observed a higher expression of the 32 kDa PBR protein (channel protein) in untreated leukemic lymphocytes than in lymphocytes from the control group indicating the plausibility of higher cholesterol transport from the cytoplasmic compartment to the nucleus in leukemic cells. These results thus have shed some light on the utilization of cholesterol in the nucleus of highly proliferated cancer/leukemic cells. Therefore, the previously reported hypocholesterolemia in cancer patients might be the result of such a link where more cholesterol is transported into the cell nucleus or to nuclear chromosomes in order to support the existence of tumor cells. It is noteworthy to mention that a relationship between cholesterol metabolism and tumor growth has been shown in previous investigations (Brown et al., 1974; Witte et al., 1982) with a plausible association between cholesterol esterification and the control of cell cycle progression (Dessi et al., 1997; Mulas et al., 2011).
This study provided new insight into cholesterol homeostasis in CLL participants in relation to LDLR, SREBP-2, and PBR. Cholesterol may enter the cell nucleus through highly expressed PBR and may be involved in developing leukemia by manipulating cell cycle progression in lymphocytes of CLL patients. Further studies are needed to explain the mechanism by which cholesterol is involved within the nucleus for cell growth. In addition to its participation in membrane formation, the role of cholesterol in DNA replication following cell proliferation is another challenging perspective for future research on cholesterol homeostasis in cellular metabolism.
Author contribution
SS collected most of the data, performed the experiments, contributed to the discussion, and conducted statistical analyses when needed. GS shared the biochemical analysis shown in Table 1. MM and LK reviewed the manuscript. NCC analyzed all the data, contributed to the discussion, wrote the manuscript and reviewed/edited the manuscript. All authors read and approved the final version of the manuscript.
Statement conflict of Interest
All authors declare no competing or conflicts of interests
Acknowledgments
The authors are thankful to the Institute Intramural grant committee AIIMS, New Delhi, India, for financial support.
References
- Albi E, Cataldi S, Rossi G, et al. A possible role of cholesterol-sphingomyelin/phosphatidylcholine in nuclear matrix during rat liver regeneration. J Hepatol. 2003;38:623–28. doi: 10.1016/s0168-8278(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Albi E, Pieroni S, Viola Magni MP, et al. Chromatin sphingomyelin changes in cell proliferation and/or apoptosis induced by ciprofibrate. J Cell Physiol. 2003;196:354–61. doi: 10.1002/jcp.10314. [DOI] [PubMed] [Google Scholar]
- Albi E, Viola Magni MP. The role of intranuclear lipids. Biol Cell Auspices Eur Cell Biol Organ. 2004;96:657–67. doi: 10.1016/j.biolcel.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Arjuman A, Chandra NC. Effect of IL-10 on LOX-1 expression, signalling and functional activity:an atheroprotective response. Diabetes Vasc Dis Res. 2013;10:442–51. doi: 10.1177/1479164113489042. [DOI] [PubMed] [Google Scholar]
- Böyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Investig Suppl. 1968;97:77–89. [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brown MS, Dana SE, Siperstein MD. Properties of 3-hydroxy-3-methylglutaryl coenzyme A reductase solubilized from rat liver and hepatoma. J Biol Chem. 1974;249:6585–89. [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and inhibition of growth of human fibroblasts by 7-ketocholesterol. J Biol Chem. 1974;249:7306–14. [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Regulation of the activity of the low density lipoprotein receptor in human fibroblasts. Cell. 1975;6:307–16. doi: 10.1016/0092-8674(75)90182-8. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway:regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Budd D, Ginsberg H. Hypocholesterolemia and acute myelogenous leukemia. Association between disease activity and plasma low-density lipoprotein cholesterol concentrations. Cancer. 1986;58:1361–65. doi: 10.1002/1097-0142(19860915)58:6<1361::aid-cncr2820580630>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Chen Y, Hughes-Fulford M. Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2. Int J Cancer. 2001;91:41–5. doi: 10.1002/1097-0215(20010101)91:1<41::aid-ijc1009>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Dessi S, Batetta B, Pani A, et al. Role of cholesterol synthesis and esterification in the growth of CEM and MOLT4 lymphoblastic cells. Biochem J. 1997;321:603–08. doi: 10.1042/bj3210603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinleib M. Review of the epidemiological evidence for a possible relationship between hypocholesterolemia and cancer. Cancer Res. 1983;43:2503s–7s. [PubMed] [Google Scholar]
- Hardwick M, Fertikh D, Culty M, et al. Peripheral-type benzodiazepine receptor (PBR) in human breast cancer:correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res. 1999;59:831–42. [PubMed] [Google Scholar]
- Hardwick M, Cavalli LR, Barlow KD, et al. Peripheral-type benzodiazepine receptor (PBR) gene amplification in MDA-MB-231 aggressive breast cancer cells. Cancer Genet Cytogenet. 2002;139:48–51. doi: 10.1016/s0165-4608(02)00604-0. [DOI] [PubMed] [Google Scholar]
- Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–53. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havekes LM, Verboom H, de Wit E, et al. Regulation of low density lipoprotein receptor activity in primary cultures of human hepatocytes by serum lipoproteins. Hepatol Baltim Md. 1986;6:1356–60. doi: 10.1002/hep.1840060623. [DOI] [PubMed] [Google Scholar]
- Ho YK, Brown S, Bilheimer DW, et al. Regulation of low density lipoprotein receptor activity in freshly isolated human lymphocytes. J Clin Invest. 1976;58:1465–74. doi: 10.1172/JCI108603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Iwasaki Y, Yatoh S, et al. Cholesterol accumulation and diabetes in pancreatic beta-cell-specific SREBP-2 transgenic mice:a new model for lipotoxicity. J Lipid Res. 2008;49:2524–34. doi: 10.1194/jlr.M800238-JLR200. [DOI] [PubMed] [Google Scholar]
- Kreger BE, Anderson KM, Schatzkin A, et al. Serum cholesterol level, body mass index, and the risk of colon cancer. The Framingham Study. Cancer. 1992;70:1038–43. doi: 10.1002/1097-0142(19920901)70:5<1038::aid-cncr2820700505>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Mulas MF, Abete C, Pulisci D, et al. Cholesterol esters as growth regulators of lymphocytic leukaemia cells. Cell Prolif. 2011;44:360–71. doi: 10.1111/j.1365-2184.2011.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C, Vitols S, Rudling M, et al. Hypocholesterolemia in cancer patients may be caused by elevated LDL receptor activities in malignant cells. Med Oncol Tumor Pharmacother. 1985;2:143–47. doi: 10.1007/BF02934541. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan G, Arjuman A, Suneja S, et al. The association between insulin and low-density lipoprotein receptors. Diabetes Vasc Dis Res. 2012;9:196–204. doi: 10.1177/1479164111430243. [DOI] [PubMed] [Google Scholar]
- Rao KN. Regulatory aspects of cholesterol metabolism in cells with different degrees of replication. Toxicol Pathol. 1986;14:430–37. doi: 10.1177/019262338601400408. [DOI] [PubMed] [Google Scholar]
- Ravagnan L, Marzo I, Costantini P, et al. Lonidamine triggers apoptosis via a direct, Bcl-2-inhibited effect on the mitochondrial permeability transition pore. Oncogene. 1999;18:2537–46. doi: 10.1038/sj.onc.1202625. [DOI] [PubMed] [Google Scholar]
- Robinet P, Wang Z, Hazen SL, et al. A simple and sensitive enzymatic method for cholesterol quantification in macrophages and foam cells. J Lipid Res. 2010;51:3364–69. doi: 10.1194/jlr.D007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siperstein MD. Role of cholesterogenesis and isoprenoid synthesis in DNA replication and cell growth. J Lipid Res. 1984;25:1462–68. [PubMed] [Google Scholar]
- Smith JR, Osborne TF, Goldstein JL, et al. Identification of nucleotides responsible for enhancer activity of sterol regulatory element in low density lipoprotein receptor gene. J Biol Chem. 1990;265:2306–10. [PubMed] [Google Scholar]
- Vitols S, Gahrton G, Björkholm M, et al. Hypocholesterolaemia in malignancy due to elevated low-density-lipoprotein-receptor activity in tumour cells:evidence from studies in patients with leukaemia. Lancet. 1985;2:1150–54. doi: 10.1016/s0140-6736(85)92679-0. [DOI] [PubMed] [Google Scholar]
- Vitols S. Uptake of low-density lipoprotein by malignant cells-possible therapeutic applications. Cancer Cells Cold Spring Harb N. 1991;3:488–95. [PubMed] [Google Scholar]
- Witte LD, Cornicelli JA, Miller RW, et al. Effect of platelet-derived and endothelial cell-derived growth factors on the low density lipoprotein receptor pathway in cultured human fibroblasts. J Biol Chem. 1982;257:5392–5401. [PubMed] [Google Scholar]
- Yen CF, Kalunta CI, Chen FS, et al. Regulation of low-density lipoprotein receptors and assessment of their functional role in Burkitt’s lymphoma cells. Biochim Biophys Acta. 1995;1257:47–57. doi: 10.1016/0005-2760(95)00051-d. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Unfolding the toxicity of cholesterol. Nat Cell Biol. 2003;5:769–70. doi: 10.1038/ncb0903-769. [DOI] [PubMed] [Google Scholar]