

Abstract
Here we report the structural characterization of the product formed from the reaction between hydroethidine (HE) and superoxide (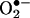 ). By using mass spectral and NMR techniques, the chemical structure of this product was determined as 2-hydroxyethidium (2-OH-E+). By using an authentic standard, we developed an HPLC approach to detect and quantitate the reaction product of HE and
). By using mass spectral and NMR techniques, the chemical structure of this product was determined as 2-hydroxyethidium (2-OH-E+). By using an authentic standard, we developed an HPLC approach to detect and quantitate the reaction product of HE and 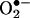 formed in bovine aortic endothelial cells after treatment with menadione or antimycin A to induce intracellular reactive oxygen species. Concomitantly, we used a spin trap, 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO), to detect and identify the structure of reactive oxygen species formed. BMPO trapped the
formed in bovine aortic endothelial cells after treatment with menadione or antimycin A to induce intracellular reactive oxygen species. Concomitantly, we used a spin trap, 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO), to detect and identify the structure of reactive oxygen species formed. BMPO trapped the 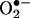 that formed extracellularly and was detected as the BMPO-OH adduct during use of the EPR technique. BMPO, being cell-permeable, inhibited the intracellular formation of 2-OH-E+. However, the intracellular BMPO spin adduct was not detected. The definitive characterization of the reaction product of
that formed extracellularly and was detected as the BMPO-OH adduct during use of the EPR technique. BMPO, being cell-permeable, inhibited the intracellular formation of 2-OH-E+. However, the intracellular BMPO spin adduct was not detected. The definitive characterization of the reaction product of 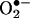 with HE described here forms the basis of an unambiguous assay for intracellular detection and quantitation of
with HE described here forms the basis of an unambiguous assay for intracellular detection and quantitation of 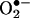 . Analysis of the fluorescence characteristics of ethidium (E+) and 2-OH-E+ strongly suggests that the currently available fluorescence methodology is not suitable for quantitating intracellular
. Analysis of the fluorescence characteristics of ethidium (E+) and 2-OH-E+ strongly suggests that the currently available fluorescence methodology is not suitable for quantitating intracellular 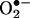 . We conclude that the HPLC/fluorescence assay using HE as a probe is more suitable reactive oxygen species for detecting intracellular
. We conclude that the HPLC/fluorescence assay using HE as a probe is more suitable reactive oxygen species for detecting intracellular 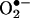 .
.
Keywords: free radical, reactive oxygen species, 2-hydroxyethidium, EPR, spin trapping
One of the most frequently used assays for the detection of superoxide (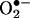 ) that is formed in cells and tissues involves the use of hydroethidine (HE) as an intracellular probe (1–4) (Fig. 1). In previous studies, the red fluorescence (also referred to as the “ethidium fluorescence”) that arises from the oxidation of HE was attributed to
) that is formed in cells and tissues involves the use of hydroethidine (HE) as an intracellular probe (1–4) (Fig. 1). In previous studies, the red fluorescence (also referred to as the “ethidium fluorescence”) that arises from the oxidation of HE was attributed to 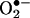 trapping in cells (1–4). Recently, we showed that chemically and enzymatically generated
trapping in cells (1–4). Recently, we showed that chemically and enzymatically generated 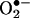 reacts with HE to form a fluorescent product that differs from ethidium (E+), the two-electron oxidation product of HE (5). To our knowledge, this product is formed only from the reaction of
reacts with HE to form a fluorescent product that differs from ethidium (E+), the two-electron oxidation product of HE (5). To our knowledge, this product is formed only from the reaction of 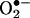 with HE. Other reactive oxygen, nitrogen, and halogen species such as H2O2, hydroxyl radical, peroxynitrite, and hypochlorous acid failed to oxidize HE to the same product (5, 6). These findings essentially nullified the long-held notion that the intracellular
with HE. Other reactive oxygen, nitrogen, and halogen species such as H2O2, hydroxyl radical, peroxynitrite, and hypochlorous acid failed to oxidize HE to the same product (5, 6). These findings essentially nullified the long-held notion that the intracellular 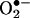 anion reacts with HE to form a red fluorescent E+/DNA complex (1–4).
anion reacts with HE to form a red fluorescent E+/DNA complex (1–4).
Fig. 1.

Chemical structures of HE, E+, and 2-OH-E+.
Our previous study (5) demonstrated that the product resulting from the HE reaction with 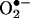 and its DNA complex exhibited fluorescence spectra that differed from those of E+ and its DNA complex. The HPLC and mass spectral parameters of the HE/
and its DNA complex exhibited fluorescence spectra that differed from those of E+ and its DNA complex. The HPLC and mass spectral parameters of the HE/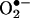 reaction product and E+ also differed (5). Here, by using ion-trap MS and NMR techniques, we characterized the product of the reaction between
reaction product and E+ also differed (5). Here, by using ion-trap MS and NMR techniques, we characterized the product of the reaction between 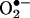 and HE as 2-hydroxyethidium (2-OH-E+) (Fig. 1). By using an authentic standard, we have developed a specific HPLC methodology to quantify the intracellular reaction product of
and HE as 2-hydroxyethidium (2-OH-E+) (Fig. 1). By using an authentic standard, we have developed a specific HPLC methodology to quantify the intracellular reaction product of 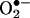 and HE. The limitations of the fluorescence techniques that are currently used to detect
and HE. The limitations of the fluorescence techniques that are currently used to detect 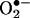 in cells and tissues are also discussed.
in cells and tissues are also discussed.
Materials and Methods
Materials. HE (dihydroethidium) and ethidium bromide were purchased from Molecular Probes. Bovine copper zinc 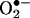 dismutase (SOD) and xanthine oxidase were purchased from Roche Molecular Biochemicals. The spin trap, 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO), was synthesized as described in ref. 7. Stock solutions of HE and E+ (20 mM) were prepared by dissolving in DMSO and stored, protected from light, at –20°C until further use. All other reagents were from Sigma.
dismutase (SOD) and xanthine oxidase were purchased from Roche Molecular Biochemicals. The spin trap, 5-tert-butoxycarbonyl-5-methyl-1-pyrroline N-oxide (BMPO), was synthesized as described in ref. 7. Stock solutions of HE and E+ (20 mM) were prepared by dissolving in DMSO and stored, protected from light, at –20°C until further use. All other reagents were from Sigma.
Cell Culture. Bovine aortic endothelial cells (BAECs) were obtained from Clonetics (San Diego), cultured in a 100-mm culture dish, grown to 80–90% confluence in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS, 4 nmol/liter l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin and incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were passaged as described in ref. 8 and used between passages 4 and 14. On the treatment day, the medium was replaced with DMEM containing 2% FBS.
Preparation of Samples for HPLC. BAECs were treated with menadione for 30 min, subsequently washed with Dulbecco's PBS (DPBS), and treated with 10 μM HE for 20 min in DMEM. The medium was removed, and BAECs were washed with DPBS twice. After centrifugation and removal of supernatant liquid, the resulting cell pellet was stored at –80°C. When using ceramide, manganese (III) tetrakis (5,10,15,20-benzoic acid) porphyrin (MnTBAP), and BMPO, cells were preincubated with these compounds for 2–6 h, washed, and then treated with menadione. Treated BAECs were maintained at –80°C before analysis. After thawing to an ambient temperature, cells were lysed in a 0.25-ml lysis buffer (DPBS with 0.1% Triton X-100, pH 7.4), and cell protein levels were measured. A 10-μl lysis solution was used for measuring protein concentration. The remaining solution was mixed with 0.5 ml of 1-butanol, vortex-mixed for 1 min, and centrifuged. The butanol phase was separated and dried with nitrogen. Dried samples were taken up into the solution by adding 0.1 ml of water for HPLC analysis. The cell number was normalized to protein levels in cell lysates, and HPLC peak areas in individual experiments were normalized to the protein concentration.
Column Chromatography. HE (50 μM) was incubated with xanthine (1 mM) and xanthine oxidase (0.05 units/ml) in a phosphate buffer (pH 7.4, 50 mM) containing diethylenetriaminepentaacetate (100 μM). The mixture was stirred several times in air. After 60 min, the mixture was extracted with chloroform until the chloroform phase became pale red. The organic phase was concentrated, passed through a silica gel column (prewashed with chloroform), and eluted with chloroform. After removing the residual HE, the column was eluted with methanol. Upon evaporation of the excess methanol, a red solid was obtained. After further purification by recrystallization, the sample was analyzed by NMR and MS.
NMR and MS. Mass spectra were run on an LCQ Classic ion-trap instrument (Thermo Electron, Boston). Solutions in 0.1% formic acid in acetonitrile were electrosprayed at 2.5 kV. The capillary temperature was 190°C. NMR spectra were run at 300 MHz on a Varian XL-300 with samples dissolved in deuterated DMSO in a 5-mm tube with an accumulation time of 2 sec and 16,000 data points with DMSO as a reference at 2.5 ppm.
EPR Measurements. EPR spectra were recorded at room temperature on a Bruker EMX spectrometer operating at 9.85 GHz and a cavity equipped with a Bruker AquaX liquid sample cell. BAECs were preincubated with BMPO (25 mM) for 1 h and treated with menadione (10 μM) for 30 min and then with HE (10 μM) for 20 min at 37°C. The medium was collected and analyzed by EPR. In the cell experiments, BAECs were collected and suspended in DPBS and analyzed by EPR. The typical spectrometer parameters were the following: scan range, 100 G; field set, 3,500 G; time constant, 5.12 msec; scan time, 5.12 sec; modulation amplitude, 1.0 G; modulation frequency, 100 kHz; receiver gain, 6.32 × 105; microwave power, 10 mW; and number of scans, 50.
Fluorescence Detection of 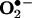 . HE was used to estimate intracellular
. HE was used to estimate intracellular 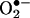 production in BAECs. After treating BAECs with menadione, the medium was removed, and cells were washed with DPBS and incubated in a fresh culture medium with 2% FBS. A final concentration of 10 μM HE was added and incubated for 20 min. Cells were washed twice with DPBS and maintained in a fresh medium. Fluorescence was monitored by using a Nikon Eclipse TE200 fluorescence microscope (excitation and emission at 510 and 590 nm, respectively) equipped with a FITC filter. The intensity values were calculated by using metamorph software (Universal Imaging, Downington, PA). Images were analyzed for intensity of fluorescence within a user-defined region of the cells. Artifactual autofluorescent regions were manually eliminated from the analysis. The relative average fluorescence intensity was normalized for surface area and compared between the control and treated cells.
production in BAECs. After treating BAECs with menadione, the medium was removed, and cells were washed with DPBS and incubated in a fresh culture medium with 2% FBS. A final concentration of 10 μM HE was added and incubated for 20 min. Cells were washed twice with DPBS and maintained in a fresh medium. Fluorescence was monitored by using a Nikon Eclipse TE200 fluorescence microscope (excitation and emission at 510 and 590 nm, respectively) equipped with a FITC filter. The intensity values were calculated by using metamorph software (Universal Imaging, Downington, PA). Images were analyzed for intensity of fluorescence within a user-defined region of the cells. Artifactual autofluorescent regions were manually eliminated from the analysis. The relative average fluorescence intensity was normalized for surface area and compared between the control and treated cells.
HPLC Analysis. HE, E+, and the 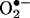 -derived oxidation product were separated on an HPLC system equipped with fluorescence and UV detectors. The mobile phase was H2O/CH3CN. The stationary phase was a C18 reverse-phase column (Partisil ODS-3 250 × 4.5 mm, Alltech Associates Deerfield, IL). Fluorescence detection at 510 nm (excitation) and 595 nm (emission) was used to monitor these compounds. Typically, 50 μl of sample was injected into the HPLC system (HP 1100, Agilent Technologies, Palo Alto, CA) with a C18 column (250 × 4.5 mm) equilibrated with 10% CH3CN in 0.1% trifluoroacetic acid. HE, E+, and the
-derived oxidation product were separated on an HPLC system equipped with fluorescence and UV detectors. The mobile phase was H2O/CH3CN. The stationary phase was a C18 reverse-phase column (Partisil ODS-3 250 × 4.5 mm, Alltech Associates Deerfield, IL). Fluorescence detection at 510 nm (excitation) and 595 nm (emission) was used to monitor these compounds. Typically, 50 μl of sample was injected into the HPLC system (HP 1100, Agilent Technologies, Palo Alto, CA) with a C18 column (250 × 4.5 mm) equilibrated with 10% CH3CN in 0.1% trifluoroacetic acid. HE, E+, and the 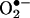 -derived product were by a linear increase separated in CH3CN concentration from 10% to 70% in 46 min at a flow rate of 0.5 ml/min. The elution was monitored by a variable UV detector at 210 and 350 nm and a fluorescence detector with excitation and emission at 510 and 595 nm, respectively.
-derived product were by a linear increase separated in CH3CN concentration from 10% to 70% in 46 min at a flow rate of 0.5 ml/min. The elution was monitored by a variable UV detector at 210 and 350 nm and a fluorescence detector with excitation and emission at 510 and 595 nm, respectively.
Statistical Analysis. All data are reported as mean ± SEM. The significance was determined with the instat program (GraphPad, San Diego) for Student's unpaired t tests or ANOVA, where appropriate. A value of P < 0.05 was considered to be significant.
Results
Identification of the Reaction Product Between 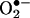 and HE as 2-OH-E+. In a previous study (5), we indicated that the proton NMR of the
and HE as 2-OH-E+. In a previous study (5), we indicated that the proton NMR of the 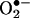 /HE reaction product did not differ significantly from E+. This finding raised the possibility that an oxygen atom was attached to one of the amino groups of HE (Fig. 1). In an effort to locate the oxygen atom, the ion-trap MS spectra of ethidium bromide, its dihydro derivative, the
/HE reaction product did not differ significantly from E+. This finding raised the possibility that an oxygen atom was attached to one of the amino groups of HE (Fig. 1). In an effort to locate the oxygen atom, the ion-trap MS spectra of ethidium bromide, its dihydro derivative, the 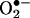 oxidation product, and its sodium borohydride reduction product were examined. Basically, the main fragmentation reactions of the (M+H)+ ion of E+ were the loss of ethyl and ethylene from the N-ethyl group, –NH2 from the aromatic ring, and phenyl from the 6 position (data not shown). The oxyethidium (M+H)+ ion was fragmented in a similar fashion (Fig. 2A), ultimately losing a molecule of ammonia, after a loss of ethylene. Surprisingly, there was no loss of water or –NHOH (32 Da) as might have been expected if a hydroxylamino group (–NHOH) had indeed been present on one of the rings. This result led us to believe that an oxygen atom was in fact attached to the ring, and therefore we reexamined the NMR spectrum of the
oxidation product, and its sodium borohydride reduction product were examined. Basically, the main fragmentation reactions of the (M+H)+ ion of E+ were the loss of ethyl and ethylene from the N-ethyl group, –NH2 from the aromatic ring, and phenyl from the 6 position (data not shown). The oxyethidium (M+H)+ ion was fragmented in a similar fashion (Fig. 2A), ultimately losing a molecule of ammonia, after a loss of ethylene. Surprisingly, there was no loss of water or –NHOH (32 Da) as might have been expected if a hydroxylamino group (–NHOH) had indeed been present on one of the rings. This result led us to believe that an oxygen atom was in fact attached to the ring, and therefore we reexamined the NMR spectrum of the 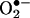 product (Fig. 2B).
product (Fig. 2B).
Fig. 2.

Structural determination of the product formed from the reaction between 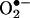 and HE. (A) The ion-trap MS (MS2) spectrum of the red product isolated from the reaction mixture containing HE and xanthine/xanthine oxidase in 100 mM phosphate buffer, pH 7.4, containing 100 μM diethylenetriaminepentaacetate. (B) High-resolution 1H-NMR spectrum of the product formed from the reaction between
and HE. (A) The ion-trap MS (MS2) spectrum of the red product isolated from the reaction mixture containing HE and xanthine/xanthine oxidase in 100 mM phosphate buffer, pH 7.4, containing 100 μM diethylenetriaminepentaacetate. (B) High-resolution 1H-NMR spectrum of the product formed from the reaction between 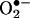 and HE.
and HE.
The 1H-NMR spectrum of the product isolated from the reaction between 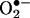 and HE was obtained in deuterated DMSO. This procedure enabled us to avoid the complications discussed by Thomas and Roques (9). An expansion of the aromatic rings A and B is shown in Fig. 2B Inset. The chemical shifts (in ppm) of the reaction product of HE and
and HE was obtained in deuterated DMSO. This procedure enabled us to avoid the complications discussed by Thomas and Roques (9). An expansion of the aromatic rings A and B is shown in Fig. 2B Inset. The chemical shifts (in ppm) of the reaction product of HE and 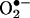 and E are shown in Table 1. Decoupling experiments established the relationships among the resonances (Table 1). A three-spin system consisting of H7, H9, and H10 establishes that the A ring (Fig. 2B Inset), with its H7 strongly shifted upfield by the aromatic ring π -orbital effect of the phenyl group, is virtually unchanged. However, the B ring protons are now a two-spin paracoupled system [coupling constant (J) <1 Hz] with H1 shifted strongly upfield from 8.58 to 7.88 ppm. H4 is shifted slightly downfield from 7.32 to 7.42 ppm. These effects are consistent with the addition of an oxygen atom in the 2 position, and, consequently, the H2 at 7.48 ppm detected for E+ is absent in 2-OH-E+ (Table 1). These data are consistent with the proposed structure in Fig. 2B.
and E are shown in Table 1. Decoupling experiments established the relationships among the resonances (Table 1). A three-spin system consisting of H7, H9, and H10 establishes that the A ring (Fig. 2B Inset), with its H7 strongly shifted upfield by the aromatic ring π -orbital effect of the phenyl group, is virtually unchanged. However, the B ring protons are now a two-spin paracoupled system [coupling constant (J) <1 Hz] with H1 shifted strongly upfield from 8.58 to 7.88 ppm. H4 is shifted slightly downfield from 7.32 to 7.42 ppm. These effects are consistent with the addition of an oxygen atom in the 2 position, and, consequently, the H2 at 7.48 ppm detected for E+ is absent in 2-OH-E+ (Table 1). These data are consistent with the proposed structure in Fig. 2B.
Table 1. Chemical shifts (ppm) and couplings (Hz).
| Compound | Chemical shifts | Couplings | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | H2 | H4 | H7 | H9 | H10 | J1,2 | J2,4 | J7,9 | J9,10 | |
| E+ | 8.58 | 7.48 | 7.32 | 6.18 | 7.29 | 8.52 | 9.5 | 1.75 | 2.5 | 9.5 |
| 2-OH-E+ | 7.88 | — | 7.42 | 6.24 | 7.48 | 8.32 | — | — | 2.4 | 8.99 |
The reference was set with DMSO at 2.52 ppm. —, Not applicable.
Intracellular Detection and Quantitation of 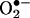 with 2-OH-E+ as the Standard. BAECs were treated with 5–20 μM menadione, a redox-cycling quinone known to generate intracellular
with 2-OH-E+ as the Standard. BAECs were treated with 5–20 μM menadione, a redox-cycling quinone known to generate intracellular 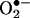 . After a 30-min exposure to menadione, BAECs were washed free of extracellular menadione and treated with 10 μM HE for 20 min. Cells were washed again, and fluorescent images were obtained by using a fluorescence microscope. As shown in Fig. 3A, menadione caused a dose-dependent increase in red fluorescence. The densitometric analysis shows the dose-dependent increase in fluorescence intensity (Fig. 3B). Before treatment with menadione, when cells were pretreated with MnTBAP (10), a SOD mimetic, the red fluorescence was greatly diminished (Fig. 3). To probe whether the increase in red fluorescence originated from E+ or 2-OH-E+, we used the HPLC/fluorescence technique.
. After a 30-min exposure to menadione, BAECs were washed free of extracellular menadione and treated with 10 μM HE for 20 min. Cells were washed again, and fluorescent images were obtained by using a fluorescence microscope. As shown in Fig. 3A, menadione caused a dose-dependent increase in red fluorescence. The densitometric analysis shows the dose-dependent increase in fluorescence intensity (Fig. 3B). Before treatment with menadione, when cells were pretreated with MnTBAP (10), a SOD mimetic, the red fluorescence was greatly diminished (Fig. 3). To probe whether the increase in red fluorescence originated from E+ or 2-OH-E+, we used the HPLC/fluorescence technique.
Fig. 3.

Menadione-induced HE fluorescence in BAECs. (A) Phase-contrast and fluorescence images of BAECs treated with different concentrations of menadione for 30 min. After menadione treatment, BAECs were washed with DPBS and incubated with HE (10 μM) for 20 min. Cells were washed twice with DPBS and kept in the culture medium. The red fluorescence generated from HE was monitored by using fluorescence microscopy. BAECs were preincubated with MnTBAP (150 μM) for 2 h. After washing the cells free of extracellular MnTBAP, they were treated with menadione (10 μM) for 30 min, and HE-derived fluorescence was measured. (B) Densitometric analysis of data shown in A.
As shown in Fig. 4A, the HPLC profiles of authentic 2-OH-E+ and E+ were well resolved and clearly different. Fig. 4A Inset shows the calibration of the HPLC peak intensity of 2-OH-E+ and E+ as a function of concentrations. The HPLC peak height of 2-OH-E+ was nearly 3-fold greater than the peak height of E+ over a range of concentrations (0–20 μM). Cells treated with various concentrations of menadione (Fig. 3A) and subsequently treated with HE were lysed, and 1-butanol extracts of lysates were analyzed by HPLC as described in Materials and Methods. Fig. 4B shows the typical HPLC traces of 1-butanol extracts. HPLC showed the presence of two major fluorescent products, one corresponding to 2-OH-E+ and the other to E+. With the increase in menadione concentration, the peak intensity corresponding to 2-OH-E+ and E+ also increased. Fig. 4C shows the quantitation of intracellular 2-OH-E+ and E+ levels. The detection limit of 2-OH-E+ and E+ in cells was 3–5 nmol/μg protein. Pretreatment with MnTBAP significantly decreased the formation of 2-OH-E+ and E+ in cells.
Fig. 4.

HPLC identification of 2-OH-E+ from the 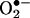 reaction in BAECs. (A) HPLC/fluorescence chromatograms of authentic 2-OH-E+ (labeled 1) and E+ (labeled 2). (Inset) HPLC peak intensity of 2-OH-E+ and E+ at different concentrations. (B) BAECs were treated with various concentrations of menadione as described in the Fig. 3 legend. HPLC samples were prepared and analyzed as described in Materials and Methods. (C) The actual concentrations of 2-OH-E+ and E+ generated under the conditions described above were calculated by using the calibration data shown in A Inset.(Inset) The effect of antimycin A (10 μM) on 2-OH-E+ and E+ formation in BAECs. Cells were treated with antimycin A for 30 min.
reaction in BAECs. (A) HPLC/fluorescence chromatograms of authentic 2-OH-E+ (labeled 1) and E+ (labeled 2). (Inset) HPLC peak intensity of 2-OH-E+ and E+ at different concentrations. (B) BAECs were treated with various concentrations of menadione as described in the Fig. 3 legend. HPLC samples were prepared and analyzed as described in Materials and Methods. (C) The actual concentrations of 2-OH-E+ and E+ generated under the conditions described above were calculated by using the calibration data shown in A Inset.(Inset) The effect of antimycin A (10 μM) on 2-OH-E+ and E+ formation in BAECs. Cells were treated with antimycin A for 30 min.
As shown in the “untreated” control (Fig. 4B), E+ was consistently present as a minor impurity in HE. In commercial samples, the amount of this impurity was unpredictable. With prolonged storage and continued exposure of HE stock solutions to room light, the relative amount of E+ increased. In addition, E+ was also formed intracellularly in cells treated HE under conditions with in which 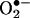 was generated. The mechanism of E+ formation under these conditions is not fully understood. However, the present HPLC results clearly demonstrate that the increase in red fluorescence from HE in cells cannot be solely attributed to E+.
was generated. The mechanism of E+ formation under these conditions is not fully understood. However, the present HPLC results clearly demonstrate that the increase in red fluorescence from HE in cells cannot be solely attributed to E+.
Intracellular Vs. Extracellular Formation of 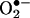 : Comparison Between Spin Trapping and HPLC. Fig. 5 shows the HPLC profiles of intracellular and extracellular levels of 2-OH-E+ and E+ in BAECs treated with 20 μM menadione and HE as described in Fig. 3B. Clearly, menadione stimulated 2-OH-E+ and E+ in cells (Fig. 5A) considerably more than in the extracellular medium (Fig. 5B). SOD (100 units/ml) totally blocked the formation of 2-OH-E+ in the medium but had very little effect on intracellular formation of 2-OH-E+.
: Comparison Between Spin Trapping and HPLC. Fig. 5 shows the HPLC profiles of intracellular and extracellular levels of 2-OH-E+ and E+ in BAECs treated with 20 μM menadione and HE as described in Fig. 3B. Clearly, menadione stimulated 2-OH-E+ and E+ in cells (Fig. 5A) considerably more than in the extracellular medium (Fig. 5B). SOD (100 units/ml) totally blocked the formation of 2-OH-E+ in the medium but had very little effect on intracellular formation of 2-OH-E+.
Fig. 5.

The effect of SOD on 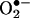 product formation in BAECs treated with menadione. (A) HPLC traces of HE-derived products in cell lysates were obtained from control, menadione-treated, and menadione/SOD-treated cells. BAECs were preincubated with 100 units/ml SOD for 1 h and then treated with 40 μmol/liter menadione and HE as described in the Fig. 3 legend. (B) Experimental conditions were the same as in A except that HE-derived products were analyzed in the cell culture medium.
product formation in BAECs treated with menadione. (A) HPLC traces of HE-derived products in cell lysates were obtained from control, menadione-treated, and menadione/SOD-treated cells. BAECs were preincubated with 100 units/ml SOD for 1 h and then treated with 40 μmol/liter menadione and HE as described in the Fig. 3 legend. (B) Experimental conditions were the same as in A except that HE-derived products were analyzed in the cell culture medium.
Next, we compared the HPLC with the spin-trapping method. Cells were treated with 20 μM menadione in the presence and absence of the BMPO spin trap for 30 min and were subsequently treated with HE as described in Fig. 3. Fig. 6 A and B Upper show both HPLC and EPR traces of cell lysates and media. BMPO significantly diminished both the intracellular and extracellular formation of 2-OH-E+, suggesting that BMPO could effectively scavenge 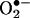 generated in the intracellular and extracellular compartments. BMPO has previously been shown to be cell-permeable (11). Concomitantly, we monitored the cell lysates and media by EPR. Fig. 6A Lower shows no discernible BMPO adduct in cell lysates, whereas an intense spectrum due to BMPO-OH was detected in the cell culture media. The BMPO-OH formation in the cell culture media was inhibited when SOD was present. These results clearly demonstrate that BMPO or a similar nitrone trap is useful only for detecting extracellularly formed
generated in the intracellular and extracellular compartments. BMPO has previously been shown to be cell-permeable (11). Concomitantly, we monitored the cell lysates and media by EPR. Fig. 6A Lower shows no discernible BMPO adduct in cell lysates, whereas an intense spectrum due to BMPO-OH was detected in the cell culture media. The BMPO-OH formation in the cell culture media was inhibited when SOD was present. These results clearly demonstrate that BMPO or a similar nitrone trap is useful only for detecting extracellularly formed 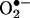 anion and that the HPLC/fluorescence assay technique using HE as a probe is suitable for detecting the intracellular formation of
anion and that the HPLC/fluorescence assay technique using HE as a probe is suitable for detecting the intracellular formation of 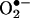 .
.
Fig. 6.

Comparison between HPLC and EPR spin-trapping detection of 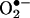 in BAECs treated with menadione. (A Upper) BAECs were treated with menadione (10 μM) for 1 h, then washed with DPBS and incubated with HE (10 μM) for 20 min. (Lower) BAECs were preincubated with BMPO (25 mM) and then treated with menadione (10 μM) and HE (10 μM). HE-derived oxidation products were obtained in cell lysates as described earlier. (B) Experimental conditions were the same as in A except that the HE-derived products were analyzed in the cell culture medium. (C Upper) BAECs were treated with menadione (10 μM) and BMPO (25 mM) for 1 h. EPR spectra of cell lysates were recorded. (Lower) Experimental conditions were the same as in Upper except for the presence of SOD (100 units/ml). (D) Experimental conditions were the same as in C except that EPR spectra of media were analyzed.
in BAECs treated with menadione. (A Upper) BAECs were treated with menadione (10 μM) for 1 h, then washed with DPBS and incubated with HE (10 μM) for 20 min. (Lower) BAECs were preincubated with BMPO (25 mM) and then treated with menadione (10 μM) and HE (10 μM). HE-derived oxidation products were obtained in cell lysates as described earlier. (B) Experimental conditions were the same as in A except that the HE-derived products were analyzed in the cell culture medium. (C Upper) BAECs were treated with menadione (10 μM) and BMPO (25 mM) for 1 h. EPR spectra of cell lysates were recorded. (Lower) Experimental conditions were the same as in Upper except for the presence of SOD (100 units/ml). (D) Experimental conditions were the same as in C except that EPR spectra of media were analyzed.
Quantitation of Ceramide-Induced Intracellular 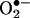 . Ceramide has been shown to induce intracellular reactive oxygen species (12) and synergistically enhance the quinone-mediated oxidative stress in neuronal cells (13). We investigated whether the pretreatment of endothelial cells with menadione would enhance ceramide-induced intracellular
. Ceramide has been shown to induce intracellular reactive oxygen species (12) and synergistically enhance the quinone-mediated oxidative stress in neuronal cells (13). We investigated whether the pretreatment of endothelial cells with menadione would enhance ceramide-induced intracellular 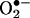 generation. Fig. 7 shows that 2-OH-E+ formation was the greatest in cells pretreated with ceramide followed by menadione (5 μM) and HE treatment, compared with cells treated with either ceramide or menadione (Fig. 7). As shown earlier (Fig. 4), menadione and ceramide also increased the formation of E+. However, the intracellular mechanism of E+ formation remains unclear. These results indicate that the HPLC technique could detect and quantitate 2-OH-E+ formed from
generation. Fig. 7 shows that 2-OH-E+ formation was the greatest in cells pretreated with ceramide followed by menadione (5 μM) and HE treatment, compared with cells treated with either ceramide or menadione (Fig. 7). As shown earlier (Fig. 4), menadione and ceramide also increased the formation of E+. However, the intracellular mechanism of E+ formation remains unclear. These results indicate that the HPLC technique could detect and quantitate 2-OH-E+ formed from 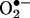 generated from a redox-signaling mechanism.
generated from a redox-signaling mechanism.
Fig. 7.

HPLC analysis of 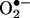 production in BAECs treated with menadione and ceramide. (A) HPLC traces of cell lysates obtained from the control cells and from cells preincubated with ceramide (20 μM) for 6 h followed by menadione (5 μM) treatment. Cells were washed and treated with HE (10 μM) as described in the Fig. 3 legend. (B) The actual concentrations of 2-OH-E+ and E+ peaks calculated by using the calibration shown in Fig. 4.
production in BAECs treated with menadione and ceramide. (A) HPLC traces of cell lysates obtained from the control cells and from cells preincubated with ceramide (20 μM) for 6 h followed by menadione (5 μM) treatment. Cells were washed and treated with HE (10 μM) as described in the Fig. 3 legend. (B) The actual concentrations of 2-OH-E+ and E+ peaks calculated by using the calibration shown in Fig. 4.
Limitations of HE/Fluorescence Assay for Intracellular Detection of 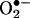 . The fluorescence maximum of 2-OH-E+ (shown in yellow in Fig. 8) corresponds to 567 nm, and that of E+ (shown in red in Fig. 8) corresponds to 590 nm. As shown in Fig. 8, because of the extensive overlap in fluorescence, suitable filters are essential for assessing the individual contribution of each species to the overall fluorescence. The overlapping fluorescence spectra at different excitation wavelengths are shown in Fig. 8. The relative fluorescence intensity of 2-OH-E+ and E+ in the presence and absence of DNA at different wavelengths are shown in Table 2. From the data shown in Table 2, the optimal excitation and emission wavelengths for detection of 2-OH-E+ by fluorescence should be 490 and 560–570 nm, respectively. Under this condition, the spectral interference from E+ is minimized. Detection of E+ will be favored when using the excitation wavelength of 510 nm and emission wavelength of 590 nm. In flow cytometry or fluorescence microscopy experiments, the use of different filters corresponding to regions 1, 2, and 3, as shown in Fig. 8, should be considered. For example, the filter corresponding to region 1 is optimal for detecting 2-OH-E+, and the filter corresponding to region 3 is optimal for detecting E+. However, the commonly employed filter (580–610 nm), using the excitation wavelength of 490 nm, will detect primarily E+ fluorescence (Fig. 8). Thus, under these conditions, it will be impossible to estimate the relative concentration of 2-OH-E+, the diagnostic intracellular marker product of
. The fluorescence maximum of 2-OH-E+ (shown in yellow in Fig. 8) corresponds to 567 nm, and that of E+ (shown in red in Fig. 8) corresponds to 590 nm. As shown in Fig. 8, because of the extensive overlap in fluorescence, suitable filters are essential for assessing the individual contribution of each species to the overall fluorescence. The overlapping fluorescence spectra at different excitation wavelengths are shown in Fig. 8. The relative fluorescence intensity of 2-OH-E+ and E+ in the presence and absence of DNA at different wavelengths are shown in Table 2. From the data shown in Table 2, the optimal excitation and emission wavelengths for detection of 2-OH-E+ by fluorescence should be 490 and 560–570 nm, respectively. Under this condition, the spectral interference from E+ is minimized. Detection of E+ will be favored when using the excitation wavelength of 510 nm and emission wavelength of 590 nm. In flow cytometry or fluorescence microscopy experiments, the use of different filters corresponding to regions 1, 2, and 3, as shown in Fig. 8, should be considered. For example, the filter corresponding to region 1 is optimal for detecting 2-OH-E+, and the filter corresponding to region 3 is optimal for detecting E+. However, the commonly employed filter (580–610 nm), using the excitation wavelength of 490 nm, will detect primarily E+ fluorescence (Fig. 8). Thus, under these conditions, it will be impossible to estimate the relative concentration of 2-OH-E+, the diagnostic intracellular marker product of 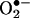 .
.
Fig. 8.

Optimal experimental conditions for detection of 2-OH-E+/DNA and E+/DNA complexes using fluorescence microscopy. Fluorescence spectra of 2-OH-E+ (shown in yellow) or E+ (shown in red) (10 μM) in the presence of DNA using the excitation wavelengths of 490 (A), 494 (B), and 510 (C) nm.
Table 2. Fluorescence intensities of 2-OH-E+ and E+ in the presence and absence of DNA at selected wavelengths.
| Samples | Ex, nm | Em, nm | Intensity, au |
|---|---|---|---|
| 2-OH-E+ | 510 | 590 | 3.09 |
| 490 | 590 | 5.55 | |
| E+ | 510 | 590 | 2.02 |
| 490 | 590 | 2.77 | |
| 2-OH-E+/DNA | 510 | 590 | 61.93 |
| 490 | 590 | 63.41 | |
| 494 | 567 | 99.99 | |
| E+/DNA | 510 | 590 | 30.95 |
| 510 | 610 | 24.25 | |
| 490 | 590 | 25.83 | |
| 494 | 567 | 18.16 |
2-OH-E+ (10 μM) and E+ (10 μM) were used in the measurement of intensities. Intensities of E+ and 2-OH-E+ were obtained from data shown in ref. 5, and those of E+/DNA and 2-OH-E+/DNA were obtained from this study. Ex, excitation wavelength; Em, emission wavelength; au, arbitrary units.
Discussion
Here we demonstrate that 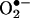 reacts with HE to form 2-OH-E+ as a specific product. To our knowledge, the product of the
reacts with HE to form 2-OH-E+ as a specific product. To our knowledge, the product of the 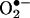 reaction has not been previously characterized. We have developed an HPLC method for detecting and quantifying intracellular
reaction has not been previously characterized. We have developed an HPLC method for detecting and quantifying intracellular 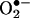 .
.
Intracellular Oxidation of HE: The Mechanism of Reaction Between 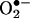 and HE. The assay based on HE oxidation has often been used to measure intracellular
and HE. The assay based on HE oxidation has often been used to measure intracellular 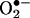 (1–4). HE can be oxidized by oxidants other than
(1–4). HE can be oxidized by oxidants other than 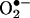 , such as the H2O2/redox-active metal ions peroxynitrite and cytochrome c (5, 14). However, these oxidants did not generate 2-OH-E+ as the product. Although the
, such as the H2O2/redox-active metal ions peroxynitrite and cytochrome c (5, 14). However, these oxidants did not generate 2-OH-E+ as the product. Although the 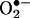 anion reacts with HE to form 2-OH-E+ as the major product in enzymatic systems (5), we found that E+ also was formed from HE in cells in a
anion reacts with HE to form 2-OH-E+ as the major product in enzymatic systems (5), we found that E+ also was formed from HE in cells in a 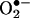 -dependent manner. At present, we do not know the mechanisms or cellular metabolism by which E+ could be formed from HE in cells. Commercial HE contains E+ as a contaminant, and attempts to eliminate the E+ impurity in commercial HE by using column chromatography were not successful. HE is light-sensitive, and the mechanism of photochemical oxidation of HE to E+ remains unclear. Because E+ has a positive charge, it does not readily diffuse into cells. It is likely that cellular formation of E+ from HE is associated with oxidative stress. The present data, however, clearly imply that intracellular formation of 2-OH-E+ is a diagnostic marker product of
-dependent manner. At present, we do not know the mechanisms or cellular metabolism by which E+ could be formed from HE in cells. Commercial HE contains E+ as a contaminant, and attempts to eliminate the E+ impurity in commercial HE by using column chromatography were not successful. HE is light-sensitive, and the mechanism of photochemical oxidation of HE to E+ remains unclear. Because E+ has a positive charge, it does not readily diffuse into cells. It is likely that cellular formation of E+ from HE is associated with oxidative stress. The present data, however, clearly imply that intracellular formation of 2-OH-E+ is a diagnostic marker product of 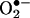 .
.
The exact mechanism of HE oxidation by 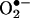 to 2-OH-E+ not clear. Although the is initial reaction between HE and
to 2-OH-E+ not clear. Although the is initial reaction between HE and 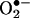 presumably involves the formation of a free radical (5, 14), the structure of this radical is not known. Previously, we suggested that the HE-derived radical could be a triaryl-type radical resulting from the loss of a tertiary hydrogen atom (5). However, on the basis of the present data, it is likely that the primary radical is derived from the loss of an aromatic amino hydrogen atom that, upon rearrangement, further reacts with another
presumably involves the formation of a free radical (5, 14), the structure of this radical is not known. Previously, we suggested that the HE-derived radical could be a triaryl-type radical resulting from the loss of a tertiary hydrogen atom (5). However, on the basis of the present data, it is likely that the primary radical is derived from the loss of an aromatic amino hydrogen atom that, upon rearrangement, further reacts with another 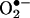 anion to form 2-OH-E+. Acetylation of the aromatic amino groups in HE totally inhibited its reaction with
anion to form 2-OH-E+. Acetylation of the aromatic amino groups in HE totally inhibited its reaction with 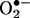 , lending additional support to the proposed mechanism (data not shown). Clearly, further studies are needed to fully understand this reaction mechanism.
, lending additional support to the proposed mechanism (data not shown). Clearly, further studies are needed to fully understand this reaction mechanism.
Potential Applications of HPLC/Fluorescence Assay for Intracellular Detection of 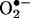 . Despite the unequivocal identification of oxy radicals by using the spin-trapping method, a major limitation of the use of this technique in cells is that it detects oxy radical adducts only in the extracellular milieu. Although it is possible that the intracellularly formed oxy radical adduct diffuses out of cells, it is very difficult to distinguish between the intracellular and extracellular formation of oxy radical adducts. The HPLC fluorescence method described here can distinguish intracellular and between extracellular
. Despite the unequivocal identification of oxy radicals by using the spin-trapping method, a major limitation of the use of this technique in cells is that it detects oxy radical adducts only in the extracellular milieu. Although it is possible that the intracellularly formed oxy radical adduct diffuses out of cells, it is very difficult to distinguish between the intracellular and extracellular formation of oxy radical adducts. The HPLC fluorescence method described here can distinguish intracellular and between extracellular 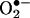 formation. The HPLC/fluorescence assay may be used as a tool for diagnosing and monitoring disease progression and clinical conditions where there exists increased likelihood for abnormal
formation. The HPLC/fluorescence assay may be used as a tool for diagnosing and monitoring disease progression and clinical conditions where there exists increased likelihood for abnormal 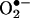 generation (15–25). For example, the formation of elevated levels of
generation (15–25). For example, the formation of elevated levels of 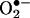 has been associated with hypertension (15, 21, 25). Hypercholesterolemia is another clinical condition that is associated with increased
has been associated with hypertension (15, 21, 25). Hypercholesterolemia is another clinical condition that is associated with increased 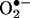 and decreased ·NO formation (16, 17).
and decreased ·NO formation (16, 17).
Summary
The ability to detect and quantify 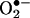 is essential for testing and screening drugs that mitigate intracellular
is essential for testing and screening drugs that mitigate intracellular 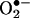 generation. Because of the overlapping fluorescence spectra from 2-OH-E+, the red fluorescence formed from HE cannot be used to quantitate intracellular
generation. Because of the overlapping fluorescence spectra from 2-OH-E+, the red fluorescence formed from HE cannot be used to quantitate intracellular 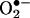 formation. Previous fluorescence microscopy studies with HE designed to detect and quantify
formation. Previous fluorescence microscopy studies with HE designed to detect and quantify 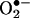 should be reexamined in light of the results described here. Our HPLC/fluorescence assay using HE as a probe is more suitable for quantifying intracellular
should be reexamined in light of the results described here. Our HPLC/fluorescence assay using HE as a probe is more suitable for quantifying intracellular 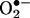 .
.
Acknowledgments
We thank Fuquan Yang and Wesley White (National Institute of Diabetes and Digestive and Kidney Diseases) for their assistance with the NMR and mass spectral data. This work was supported by National Institutes of Health Grants 5P01HL68769-01, 5R01HL067244, 2R01NS39958, and 5R01CA77822.
Author contributions: B.K., H.Z., and J.V.-V. designed research; H.Z. and J.V.-V. performed research; H.Z., J.J., H.M.F., E.A.S., R.L.L., and B.K. contributed new reagents/analytic tools; and H.Z., H.M.F., J.V.-V., and B.K. analyzed data; and B.K. wrote the paper.
Abbreviations: HE, hydroethidine; E+, ethidium; SOD, 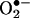 dismutase; BMPO, 5-tert-butoxycarbonyl-5-methyl 1-pyrroline N-oxide; BAEC, bovine aortic endothelial cell; DPBS, Dulbecco's PBS; MnTBAP, manganese (III) tetrakis (5,10,15,20-benzoic acid) porphyrin
dismutase; BMPO, 5-tert-butoxycarbonyl-5-methyl 1-pyrroline N-oxide; BAEC, bovine aortic endothelial cell; DPBS, Dulbecco's PBS; MnTBAP, manganese (III) tetrakis (5,10,15,20-benzoic acid) porphyrin
References
- 1.Rothe, G. & Valet, G. (1990) J. Leukocyte Biol. 47, 440–448. [PubMed] [Google Scholar]
- 2.Carter, W., Narayanan, P. K. & Robinson, P. (1994) J. Leukocyte Biol. 55, 253–258. [DOI] [PubMed] [Google Scholar]
- 3.Bindokas, V., Jordán, J., Lee, C. & Miller, R. (1996) J. Neurosci. 16, 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Budd, S., Castillo, R. & Nicholls, D. (1997) FEBS Lett. 415, 21–24. [DOI] [PubMed] [Google Scholar]
- 5.Zhao, H., Kalivendi, S., Zhang, H., Joseph, J., Nithipatikom, K., VásquezVivar, J. & Kalyanaraman, B. (2003) Free Radical Biol. Med. 34, 1359–1368. [DOI] [PubMed] [Google Scholar]
- 6.Fink, B., Laude, K., McCann, L., Dougham, A., Harrison, D. & Dikalov, S. (2004) Am. J. Physiol. 287, C895–C902. [DOI] [PubMed] [Google Scholar]
- 7.Zhao, H. T., Joseph, J., Zhang, H., Karoui, H. & Kalyanaraman, B. (2001) Free Radical Biol. Med. 31, 599–606. [DOI] [PubMed] [Google Scholar]
- 8.Tampo, Y., Kotamraju, S., Chitambar, C., Kalivendi, S., Keszler, A., Joseph, J. & Kalyanaraman, B. (2003) Circ. Res. 92, 56–63. [DOI] [PubMed] [Google Scholar]
- 9.Thomas, G. & Roques, B. (1972) FEBS Lett. 26, 169–175. [Google Scholar]
- 10.Day, B. T., Shawen, S., Liochev, S. L. & Crapo, J. (1995) J. Pharmacol. Exp. Ther. 275, 1227–1232. [PubMed] [Google Scholar]
- 11.Khan, N., Wilmot, C. M., Rosen, G. M., Demidenko, E., Suan, J., Joseph, J., O'Hara, J., Kalyanaraman, B. & Swartz, H. M. (2003) Free Radical Biol. Med. 34, 1473–1481. [DOI] [PubMed] [Google Scholar]
- 12.Matsunaga, T., Kotamraju, S., Kaliwendi, S. V., Dhanasekaran, A., Joseph, J. & Kalyanaraman, B. (2004) J. Biol. Chem. 279, 28614–28624. [DOI] [PubMed] [Google Scholar]
- 13.Cutler, R. G., Pedersen, W. A., Camandola, S., Rothstein, J. D. & Mattson, M. P. (2002) Ann. Neurol. 52, 448–457. [DOI] [PubMed] [Google Scholar]
- 14.Benov, L., Sztejnberg, L. & Fridovich, I. (1998) Free Radical Biol. Med. 25, 826–831. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki, H., Swei, A., Zweifach, B. & Schmid-Schönbein, G. (1995) Hypertension 25, 1083–1089. [DOI] [PubMed] [Google Scholar]
- 16.Ohara, Y., Peterson, T. E. & Harrison, D. G. (1993) J. Clin. Invest. 91, 2546–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller, F., Gutterman, D., Rios, D., Heistad, D. & Davidson, B. (1998) Circ. Res. 82, 1298–1305. [DOI] [PubMed] [Google Scholar]
- 18.Kawase, M., Murakami, K., Fujimura, M., Morita-Fujimura, Y., Gasche, Y., Kondo, T., Scott, R. & Chan, P. (1999) Stroke 30, 1962–1968. [DOI] [PubMed] [Google Scholar]
- 19.Kim, G., Kondo, T., Noshita, N. & Chan, P. (2002) Stroke 33, 809–815. [DOI] [PubMed] [Google Scholar]
- 20.Sorescu, D., Weiss, D., Lassègue, B., Clempus, R., Szöcs, K., Sorescu, G., Valppu, L., Quinn, M., Lambeth, D., Vega, D., et al. (2002) Circulation 105, 1429–1435. [DOI] [PubMed] [Google Scholar]
- 21.Dantas, A. P., Tostes, R., Fortes, Z., Costa, S., Nigro, D. & Carvalho, M. H. (2002) Hypertension 39, 405–411. [DOI] [PubMed] [Google Scholar]
- 22.Paravicini, T., Gulluyan, L., Dusting, G. & Drummond, G. (2002) Circ. Res. 91, 54–61. [DOI] [PubMed] [Google Scholar]
- 23.Luo, J., Li, N., Robinson, P. & Shi, R. (2002) J. Neurosci. Methods 120, 105–112. [DOI] [PubMed] [Google Scholar]
- 24.Schuchmann, S. & Heinemann, U. (2000) Free Radical Biol. Med. 28, 235–250. [DOI] [PubMed] [Google Scholar]
- 25.Zou, A.-P., Li, N. & Cowley, A. (2001) Hypertension 37, 547–553. [DOI] [PubMed] [Google Scholar]