

Summary
Protein phosphorylation is one of the widely used post-translational modifications that alter protein function in vivo. We recently showed phosphorylation of Drosophila Plexin A by cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA) and subsequent inhibition of Plexin-mediated repulsive guidance. This phosphorylation occurs in the active site of the Plexin GTPase activating protein (GAP) domain, which in turn inhibits endogenous GAP activity toward Ras family small GTP binding proteins by recruiting the phospho-serine/threonine binding protein 14-3-3ε. Here we describe how phosphorylation of Plexin A can be detected and quantitated using an in vitro kinase assay and radioactive [γ-P32] adenosine 5’-triphosphate (ATP).
Keywords: Phosphorylation, Plexin, PKA, kinase assay
1. Introduction
Growing axons are attracted and repelled by guidance cues to precisely innervate their appropriate targets (1). To accomplish this process, the activity of guidance receptors is strictly regulated by multiple molecular mechanisms (2). For instance, members of one of the largest families of guidance receptors, Plexins, are activated by interacting with their ligands, semaphorins, exerting repulsive guidance effects and preventing growing axons from extending into inappropriate areas (3, 4). Semaphorins not only induce actin depolymerization through the activation of MICALs (5), which results in growth cone collapse, but they also inhibit Integrin-mediated cell adhesion by activating Plexin’s GAP activity toward Ras/Rap-family small GTPases (6, 7, 8, 9, 10, 11). Although repulsive guidance signaling enables axons to defasciculate or turn at a choice point, prolonged repulsion hinders axon growth/elongation. We recently showed that Plexin GAP activity from Drosophila Plexin A is subject to PKA-mediated post-translational modification and a subsequent protein-protein interaction, which results in inactivation of the Plexin GAP (7). Using in vitro kinase assays and mutagenesis studies we demonstrated that PKA phosphorylates a specific serine residue located in the active site of the Plexin GAP domain. This phosphorylation recruits the phosphorylated serine binding protein 14-3-3ε and blocks the association of the GAP with its substrate. This PKA-mediated interaction with 14-3-3ε inactivates the Plexin GAP and restores Ras/Integrin-mediated cell adhesion.
Phosphorylation is one of the most widely employed post-translational modifications and modulates protein function by multiple mechanisms in a rapid and reversible manner (12). In general, protein kinases specifically identify their recognition motif in a target protein and conjugate a phosphate from the gamma position in ATP as a phosphate donor to a hydroxyl group in a serine, threonine, or tyrosine residue in a target protein. Protein phosphorylation can be detected largely by two independent methods. One type of assay is an in vitro kinase assay, which involves a purified kinase and target substrates and measures the increased level of the phosphate group in a substrate (13, 14). In vitro kinase assays are useful for testing if a given kinase directly phosphorylates a target protein/peptide. Moreover, in conjunction with site-directed mutagenesis and protein purification, one can examine direct phosphorylation sites in a target protein. The other type of assay involves antibodies that specifically recognize the phosphorylated form of target protein (15). With these phospho-specific antibodies and immunolabeling approaches, the subcellular localization and/or temporal regulation of phosphorylation events can be monitored in tissue culture cells or living organisms.
To perform an in vitro kinase assay, candidate kinases and a partial or full-length target protein are required (13). As a source of phosphate, a mixture of ATP and a trace amount of radioactive ATP is used. Using precautionary measures, and applying personal protective equipment and aseptic technique to prevent unnecessary exposure to radioactive material, this assay provides a sensitive and quantitative means to measure phosphorylation events in vitro. Here we describe how the phosphorylation of Plexin can be examined by an in vitro kinase assay using purified proteins and radioactive ATP.
2. Materials
2.1 Purification of Plexin Protein
LB broth: add 10 g Bacto-tryptone, 5 g yeast extract, and 10 g NaCl in 800 ml of distilled water and adjust pH to 7.5 with NaOH. Adjust volume to 1L with distilled water. Autoclave to sterilize.
Ampicillin sodium salt: make a 50 mg/ml of stock solution with distilled water and store aliquots at -20°C.
LB agar plate: add 10 g Bacto-tryptone, 5 g yeast extract, and 10 g NaCl in 800 ml of distilled water and adjust pH to 7.5 with NaOH. Add 15 g of agar. Adjust volume to 1 L with distilled water. Autoclave to sterilize.
30 °C shaking incubator.
2 L flask.
IPTG (isopropyl-β-D-thiogalactopyranoside): make a 0.1 M stock in distilled water and store aliquots at -20 °C.
Rosetta™2(DE3)pLysS competent cells (Novagen).
PMSF (phenylmethylsulfonyl fluoride): prepare a 100 mM stock solution in isopropanol and store at -20°C.
Dithiothreitol (DTT): prepare a 100 mM stock solution in distilled water and store at -20 °C
Protease inhibitors: cOmplete Protease Inhibitor Cocktail Tablet, EDTA-free (Roche). Dissolve 1 tablet in 500 µl of distilled water and use as a 100x concentrated stock.
Lysis buffer: 50 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1x Protease inhibitors (16).
Elution buffer: 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 5 mM reduced glutathione (Sigma, G4251) (see Note 1).
0.45 µm nitrocellulose syringe filter (Nalgene).
GSTrap FF affinity column (GE Healthcare).
Dialysis buffer: 10 mM Tris-HCl pH7.5, 2 mM MgCl2, 0.1 mM DTT.
Dialysis Cassette: Slide-A-Lyzer Dialysis Cassette (Pierce) with 10K molecular weight cut-off.
Amicon Ultra Centrifugal filters (Millipore).
Bovine serum albumin (BSA) standards: 2 mg/ml solution (Thermo Scientific, PI-23209).
Liquid nitrogen.
2.2 In vitro kinase reaction
DNA encoding the C-terminal half domain of Drosophila Plexin A (Plexin-C2; amino acid 1702-1945) was ligated in frame into the pGEX4T vector to allow production of a GST fusion protein (7).
Purified bovine protein kinase A (Promega: cAMP-dependent protein kinase, catalytic subunit; V516A). Make 2 µl aliquots for single use and store at -80°C. Avoid multiple freeze-thaw cycles (see Note 2).
5mM ATP: dissolve 13.78 mg of ATP (GE Healthcare, 27-1006-03) in 5 ml of distilled water. Make and store 200µl aliquots at -20°C.
1M Tris-HCl pH7.5: dissolve 121.14 g of Tris base in 1L of distilled water. Once completely dissolved, adjust pH to 7.5 by adding drops of 12N HCl.
1M MgCl2: dissolve 10.17 g of MgCl2 in 50 ml of distilled water.
10 mg/ml BSA: dissolve 0.1 g of BSA (BSA fraction V) in 10 ml of distilled water.
PKA reaction buffer: prepare 10x concentrated PKA reaction buffer by adding 2 ml of 1 M Tris-HCl pH 7.5, 1ml of 1M MgCl2, and 0.5 ml of 10 mg/ml BSA to distilled water to make 50 ml.
Diluted [γ-32P] ATP: dilute [γ-32P] ATP (PerkinElmer) 1 to 7 with water if [γ-32P] ATP is used within the first half life from purchase. The dilution factor of [γ-32P] ATP can be decreased depending on radioactivity left (see Note 3).
A water bath is set to 30 °C for the kinase reaction and either a heat block or boiling water bath is set above 98 °C for stopping the kinase reaction by protein denaturation.
3x Laemmli sample buffer: 188 mM Tris-HCl pH 6.8, 6% SDS, 30% glycerol, 0.024% Bromophenol blue, 15 % β-Mercaptoethanol.
2.3 Detection of phosphorylation
SDS-PAGE: standard SDS-poly acrylamide gel electrophoresis is applied to separate the phosphorylated protein product from the kinase (see Note 4).
Precision™ Plus Protein All Blue Standards (Bio-Rad) molecular weight marker (see Note 5). This marker includes 10 blue-stained proteins in a molecular weight range of 10 – 250 kDa.
Coomassie blue staining solution: 0.5 % Coomassie Brilliant Blue R-250, 50% Methanol, 10% acetic acid in distilled water.
Destaining solution: 50% Methanol and 10% acetic acid in distilled water.
Microwave-safe plastic containers to cover protein gels.
A liquid radioactive waste container for discarding staining solution after washes.
Plastic film (Saran wrap).
A heated gel dryer connected with vacuum generator (see Note 6).
Phosphorimager screen and phosphorimager scanner (Storm machine; Molecular Dynamics).
Whatman filter paper.
ImageJ software (NIH).
3. Methods
3.1 Purification of the Plexin Protein
Transform Rosetta2LysS E. coli with pGEX4T-PlexinC2 plasmid DNA and plate on an LB agar culture plate containing 50µg/ml ampicillin (see Note 7).
Inoculate a single bacterial colony in 100 ml of LB broth containing 50 µg/ml of ampicillin and grow at 37 °C overnight with rapid shaking.
Dilute the overnight culture into 900 ml of pre-warmed LB media with 50 µg/ml ampicillin, divide the culture into two 2L flasks and incubate at 37 °C for 1 hour with shaking (see Note 8).
To induce Plexin protein expression, add 0.5 ml of 0.1 M IPTG to a final concentration of 0.1 mM, and incubate for 3 hours.
Collect bacterial cultures into 1 L centrifuge bottles and spin down at 4000×g for 20 min at 4°C (see Note 9).
Resuspend bacterial pellets by pipetting or vortexing gently in 20 ml of cold lysis buffer.
Lyse bacteria by sonication on ice with 3 sec/5 sec of on/off cycles at 10% of maximum power until the lysate turns to a non-viscous brown liquid.
Centrifuge the lysate at 31400×g for 30 min at 4 °C.
Transfer the supernatant into another centrifuge tube and repeat Step 8.
Filter the cleared lysates through a 0.45 µm nitrocellulose syringe filter.
Load the lysates onto a 1 ml GSTrap FF affinity column using an FPLC system (see Note 10).
Wash the column with 30 ml of cold lysis buffer (without PMSF and protease inhibitors) to removed non-specifically bound proteins.
Elute GST-PlexinC2 protein applying a gradient of lysis buffer (without PMSF and protease inhibitors) and elution buffer by setting up a gradient which reaches 100% of elution buffer at 20 ml. Collect 1 ml samples in separate tubes. Once the gradient reaches 100% of elution buffer, continue to elute with another 10 ml of elution buffer (see Note 11).
Run a small aliquot of each sample on 10% SDS-PAGE gels with a standard protein size marker for 50 min at 200 V.
Remove the gel and stain with Coomassie blue staining solution (see Note 12).
Destain the gel several times with destaining solution until clear bands appear from a dark blue background.
Identify fractions containing the GST-PlexinC2 protein band and pool those fractions together.
Dialyze pooled fractions against 2 L of dialysis buffer at 4 °C overnight with one buffer change.
Concentrate purified proteins using an Amicon Ultra Centricon centrifugal filter device.
Quantify the concentration of purified GST-PlexinC2 by comparing a series of diluted samples with different amounts of BSA standard after separation by SDS-PAGE, and staining with Coomassie Blue (see Note 13).
Adjust protein concentration to 1 to 5 mg/ml using dialysis buffer. Make 10 to 20 µl of aliquots and snap freeze in liquid nitrogen. Store aliquots at -80 °C.
3.2 In vitro kinase reaction
Thaw frozen GST-PlexinC2 protein and dilute with 1x PKA reaction buffer to a 20 µM concentration.
Prepare a master reaction tube with a final concentration of 10 µM substrate, 500 µM ATP, and 1x PKA reaction buffer, and keep the tube on ice. For instance, we prepared a mixture of 40µl of 20µM GST-PlexinC2 protein, 8µl of 5mM ATP, 8µl of 10x PKA reaction buffer, and 12µl of water (and the rest of the volume will be added in Step 4, below).
Prepare master reaction tubes with GST as a negative control and another protein (e.g., a known substrate of PKA such as (17)) as a positive control.
Initiate the kinase reaction by adding 10 µl of diluted [γ-32P] ATP and 2 µl of PKA into the master reaction tubes from Steps 2 and 3 (Fig. 1A) and incubate them in a 30 °C heat block or water bath (see Note 14).
Perform a time course experiment by removing 10 µl of reaction mixture from each master reaction tube separately to a new tube containing 3x Laemmli sample buffer to stop the PKA reaction, and keep the tube on ice until all samples from each time point are collected. Collect samples at 0, 5, 15, 30, 60, 120, and 180 minutes after the initiation of the kinase reaction (Fig. 1A).
When all the samples are collected, denature samples using a >98°C hot plate or boiling water bath (see Note 15).
Fig. 1. A time course kinase reaction.
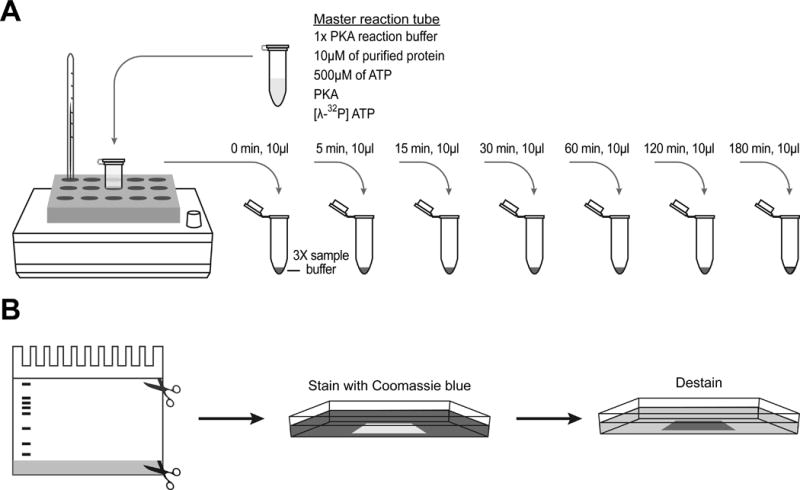
(A) A master kinase reaction mixture is prepared by adding kinase buffer, purified protein, and ATP. Before starting the reaction, add kinase and [γ-32P] ATP last. Incubate the master mixture in a heat block set at a desired temperature depending on the kinase. At each time point, remove 10 µl of the master reaction mixture, transfer to a new tube containing 3x Laemmli sample buffer to stop the reaction, and keep the tubes on ice until all the samples are collected.
(B) Left: when running a SDS-PAGE to separate phosphorylated proteins from unincorporated [γ-32P] ATP, stop running the gel before the dye front runs through the gel. Trim the gel to remove the stacking gel and the dye front. Middle and right: stain the trimmed gel with Coomassie blue solution and destain until desired contrast is reached.
3.3 Separation of phosphorylated protein
Prepare a 10% SDS-polyacrylamide gel with a 4% stacking gel (see Note 16).
Briefly spin down the samples and load the denatured protein and a protein size marker.
Run the gel at a constant voltage and monitor closely. Put a Plexiglas shield around the gel tank containing radioactive materials and frequently monitor radioactivity using Geiger-Müller counter.
Stop running the gel when the dye front is close to the bottom of the gel (see Note 17).
Trim the upper stacking gel and the bottom of the separation gel including the dye front (see Note 18) (Fig. 1B).
Put the trimmed gel in a plastic container and add Coomassie blue staining solution to cover the gel (Fig. 1B).
Cover the container with a lid or plastic film and heat up the container in a microwave for a minute.
Gently rock the container for 10 minutes. Beware not to spill the solution containing radioactive materials.
Discard used staining/destaining solution in a designated radioactive waste container.
Rinse the gel twice with destaining solution (Fig. 1B). Add more destaining solution and heat up the container in a microwave for a minute.
Gently rock the container until substrate protein is seen with good contrast (Fig. 1B). Repeat step 10 if necessary to more clearly visualize the substrate protein.
Once the gel turns to a pale blue color, while leaving the substrate protein as dark blue, rinse the gel with distilled water several times (see Note 19).
Cut Whatman filter paper to approximately 5cm larger than the gel on each side and wet with distilled water.
Place the stained gel in the center of the pre-wet Whatman filter paper and cover with plastic film (Fig. 2A) (see Note 20).
Dry the gel on the filter paper using a heated gel dryer connected to a vacuum generator. To do this, first set the temperature at 60°C (see Note 21). Then, place the gel in the center of the dryer and cover with a rubber sealing gasket. Next, turn on the dryer and make sure that the vacuum is applied evenly over the gel and that the seal is tight (see Note 22). Finally, dry the gel for 3 hours (or longer) to obtain a completely dried gel that looks like a flat piece of paper (Fig. 2B).
Label the border of the plastic film cover with the details of the experiment and date.
Fig. 2. Detection of phosphorylated protein using a phosphorimager screen.
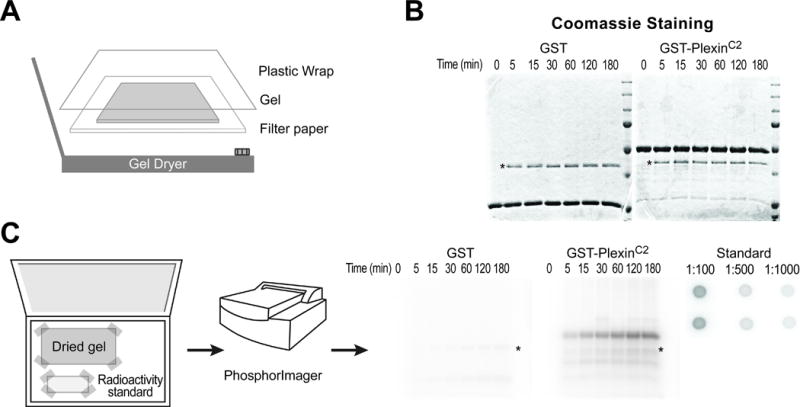
(A) To dry a destained gel, put the gel on a wet filter paper, wrap with a plastic film and place on the center of a heated gel dryer connected to a vacuum generator.
(B) A representative dried gel shows two major protein bands, a substrate with the strongest intensity and a protein kinase with a weaker intensity (asterisk, PKA in this example).
(C) Left: place a dried gel and a radioactivity standard spotted on filter paper in a phosphorimager cassette and tape the corners. Middle: detect radioactivity using a phosphorimager scanner. Right: representative images of an autoradiograph from the dried gel and a radioactivity standard. GST-PlexinC2 protein, but not GST alone, is phosphorylated. Note that the signal intensity increases over the reaction time. Autophosphorylated PKA is also detected (asterisk). The numbers in the radioactivity standard indicate dilution factors.
3.4 Detection and quantification of phosphorylation
Prepare a radioactivity standard of [γ-32P] ATP used in the kinase reaction by adding 2 µl of diluted [γ-32P] ATP to 198 µl of water (generating a 1:100 standard tube).
Transfer 20µl from the 1:100 standard tube in Step 1 to a new tube containing 80µl of water (generating a 1:500 standard tube).
Transfer 50µl from the 1:500 standard tube to a new tube containing 50µl of water (generating a 1:1000 standard tube).
Spot 5µl of each of these standards in duplicate on Whatman filter paper and wrap this filter paper with plastic film (Fig. 2C, far right).
Place both the dried gel from Method 3.3, Step 16 and the standards in a phosphorimager cassette and tape each of the corners to secure the gel and standards within the cassette (Fig. 2C, far left) (see Note 23).
Cover with an erased phosphorimager screen and lock the cassette. Place the cassette overnight in a dark place.
Scan the phosphorimager screen using a phosphorimager scanner (Fig. 2C, middle) (see Notes 24–25).
Quantitate the amount of incorporated [32P] into the substrate protein (Fig. 2C, right) using ImageJ software.
Using ImageJ, invert the scanned image of the dried gel and radioactivity standards so as to assign higher values to the stronger signal (i.e., white signals in a black background).
To measure the background signal, use the selection tool within ImageJ and draw a shape that includes a nonspecific area with a similar size to the protein band of interest. Measure both the size and integrated intensity of the selected area. A unitary background noise can be obtained by dividing the integrated intensity of the selected area by the size (background noise=IntensityBackground / AreaBackground).
To measure the signal of the protein band of interest, use the Image J selection tool (similar to the step above) to draw an area containing the phosphorylated substrate (with a similar size to the nonspecific area measured above), and measure both its size and integrated intensity. Calculate the noise corresponding to the area of each protein band (noise of a protein band=AreaProtein×IntensityBackground / AreaBackground). Subtract the noise from the signal for each protein band to get a normalized intensity (normalized intensity of a protein band=IntensityProtein – AreaProtein×IntensityBackground / AreaBackground).
Calculate the normalized intensity for the radioactivity standards by following a similar approach as in Step 11. Calculate the unitary intensity of each microliter of radioactive standard by multiplying by the dilution factor and dividing by 5 (which is the volume spotted on the filter paper). Obtain an average unitary intensity by calculating the unitary intensity from three different radioactive standards (and then calculating their average) and this value is the Standard radioactivity of [γ-32P] per microliter. Since we added only a miniscule amount of [γ-32P] ATP to the kinase reaction, this miniscule amount does not contribute appreciably to the final concentration of ATP that was used in the kinase reaction. Therefore, the radioactivity of each mole of ATP in the master reaction tube can be calculated by dividing the Standard radioactivity by the amount of ATP used (radioactivity of each mole of ATP = Standard radioactivity / (5 mM ATP×8 µl)).
To convert the radioactivity that is determined for each protein band into the number of incorporated phosphate molecules, divide the normalized intensity of each protein band by the radioactivity of each mole of ATP. This value also indicates how many molecules of protein substrate are conjugated with phosphate in the kinase reaction. The detected signal may vary depending on the radioactivity left in a stock of [γ-32P] ATP, but the unitary amount of incorporated phosphate into a given substrate would be consistent as long as the kinase activity remains the same.
Acknowledgments
This work was supported by NIH (MH085923) and Welch Foundation (I-1749) grants to Jonathan Terman.
Footnotes
Prepare elution buffer and add reduced glutathione just before use.
This purified catalytic subunit of PKA does not require addition of cAMP for kinase activation, which is necessary in vivo. Different isotypes of the PKA catalytic subunit (alpha, beta, and gamma) can be obtained from Cell Signaling Technology. If one cannot find a good commercial source of the kinase, the kinase of interest can be purified by performing standard recombinant protein purification procedures. Alternatively, immunoprecipitated kinase from homogenized lysates of tissue culture cells or animal tissues can be used if an antibody against the candidate kinase is available. If a specific antibody is not commercially available, one can overexpress the kinase with a fusion tag, such as hemagglutinin (HA) or FLAG, and use a commercially available HA or FLAG antibody. This immunoprecipitation method can be used to assess the activation states of the kinase in a physiological context.
To obtain a high signal to noise ratio, the assay should be performed within the first half life of [γ-32P] ATP, which is 14 days. Special care is required when handling radioactive materials.
The percentage of the gel is determined by the molecular weight of the protein to be separated. 10% PAGE is a good starting point to separate proteins with a size range from 20 kDa to 100 kDa. To prevent contamination by radioactive materials, a designated electrophoresis system including a gel tank and glass plates should be utilized for this assay.
A prestained protein marker is useful for monitoring protein migration and separation while running a gel.
A heated gel dryer connected with a vacuum generator will accelerate the fixation of the acrylamide gel on Whatman filter paper while minimizing gel cracking.
Similar purification approaches using different fusion tags and corresponding affinity columns can be applied to yield an equivalent quality of the protein substrate for the kinase reaction. If a protein of interest is insoluble after lysis, an optimization process is required, such as changing host cells, fusion tags, or domains to express.
At this point, optical density will reach between 0.6 and 0.8 where bacteria grow in log phase.
After centrifugation, keep samples on ice for the rest of the protein purification steps.
Batch-application of glutathione-conjugated beads can yield an similar quality of protein substrate for a kinase reaction. If an FPLC system is not available, a GSTrap FF column can be connected to a syringe using a proper adaptor. If an FPLC system is used, the flow rate should not exceed 1 ml/min throughout the purification process.
If generating a gradient is not applicable/possible with your purification set-up, several steps of increasing glutathione concentration, such as 1 mM, 3 mM, and 5 mM, will work as well.
Heating up the staining solution for 1 min with a microwave reduces incubation time.
One advantage of quantification by comparing with BSA standards is to check the purity of the purified “protein” and allow a close estimation of the quantity of the desired species if the purified “protein” is a mixture of multiple different proteins.
The reaction temperature may vary depending on the kinase.
Caps should be used to prevent heat-induced opening of the tubes and dispersal of the radioactive reaction mixture.
Large wells are preferred to prevent leaking of samples to adjacent wells when loading. Before loading samples, clean each well by pipetting or rinsing with a syringe to remove partially polymerized gel.
A thick blue dye front (band), which runs the quickest on the gel and leaves other smaller proteins behind, includes unincorporated [γ-32P] ATP and should not be allowed to run through/exit the gel.
Prestained protein size markers help locate the position of the substrate and prevent it from being cut out of the gel.
The more the gel is equilibrated in distilled water, the less the gel will shrink and crack when it is dried.
If plastic film is wrapped around the filter paper, the gel will not dry evenly.
Higher temperature can reduce the drying time but may cause cracking.
Place a heavy book on the top of the dryer cover to facilitate drying.
An alternative to the Phosphorimager screen is standard X-ray film. However, a Phosphorimager has superior sensitivity, takes less exposure time, and detects signals in a linear dynamic range.
If the protein used in the reaction is a substrate of the kinase, the intensity of dark bands corresponding to the molecular weight of the protein will increase with an increasing duration of the reaction.
Most kinases are autophosphorylated by another molecule of the same kinase. Therefore, one additional protein band may appear corresponding to the molecular weight of the kinase used in the assay (Fig. 2B and 2C right, asterisks).
References
- 1.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 2.Kolodkin AL, Tessier-Lavigne M. Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb Perspect Biol. 2011:3. doi: 10.1101/cshperspect.a001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yazdani U, Terman JR. The semaphorins. Genome Biol. 2006;7:211. doi: 10.1186/gb-2006-7-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hota PK, Buck M. Plexin structures are coming: opportunities for multilevel investigations of semaphorin guidance receptors, their cell signaling mechanisms, and functions. Cell Mol Life Sci. 2012 doi: 10.1007/s00018-012-1019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hung RJ, Yazdani U, Yoon J, et al. Mical links semaphorins to F-actin disassembly. Nature. 2010;463:823–827. doi: 10.1038/nature08724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang T, Terman JR. Regulating small G protein signaling to coordinate axon adhesion and repulsion. Small GTPases. 2013;4:34–41. doi: 10.4161/sgtp.22765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang T, Terman JR. 14-3-3epsilon couples protein kinase A to semaphorin signaling and silences plexin RasGAP-mediated axonal repulsion. Neuron. 2012;74:108–121. doi: 10.1016/j.neuron.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He H, Yang T, Terman JR, et al. Crystal structure of the plexin A3 intracellular region reveals an autoinhibited conformation through active site sequestration. Proc Natl Acad Sci U S A. 2009;106:15610–15615. doi: 10.1073/pnas.0906923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, He H, Srivastava N, et al. Plexins Are GTPase-Activating Proteins for Rap and Are Activated by Induced Dimerization. Sci Signal. 2012;5:ra6. doi: 10.1126/scisignal.2002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oinuma I, Ishikawa Y, Katoh H, et al. The Semaphorin 4D receptor Plexin-B1 is a GTPase activating protein for R-Ras. Science. 2004;305:862–865. doi: 10.1126/science.1097545. [DOI] [PubMed] [Google Scholar]
- 11.Ito Y, Oinuma I, Katoh H, et al. Sema4D/plexin-B1 activates GSK-3beta through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep. 2006;7:704–709. doi: 10.1038/sj.embor.7400737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubin CS, Rosen OM. Protein phosphorylation. Annu Rev Biochem. 1975;44:831–887. doi: 10.1146/annurev.bi.44.070175.004151. [DOI] [PubMed] [Google Scholar]
- 13.Hawes BE, Van Biesen T. Protein tyrosine kinase activity assays. Curr Protoc Pharmacol. 2001 doi: 10.1002/0471141755.ph0305s05. Chapter 3, Unit3 5. [DOI] [PubMed] [Google Scholar]
- 14.Carter AN. Current Protocols in Molecular Biology. John Wiley and Sons; 2001. Assays of Protein Kinases Using Exogenous Substrates; pp. 18.17.11–18.17.22. [DOI] [PubMed] [Google Scholar]
- 15.Archuleta AJ, Stutzke CA, Nixon KM, et al. Optimized protocol to make phospho-specific antibodies that work. Methods Mol Biol. 2011;717:69–88. doi: 10.1007/978-1-61779-024-9_4. [DOI] [PubMed] [Google Scholar]
- 16.Self AJ, Hall A. Purification of recombinant Rho/Rac/G25K from Escherichia coli. Methods Enzymol. 1995;256:3–10. doi: 10.1016/0076-6879(95)56003-3. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen C, Nishi A, Kansy JW, et al. Regulation of protein phosphatase inhibitor-1 by cyclin-dependent kinase 5. J Biol Chem. 2007;282:16511–16520. doi: 10.1074/jbc.M701046200. [DOI] [PMC free article] [PubMed] [Google Scholar]