

Abstract
Plasmodium falciparum, the causative agent of malaria, employs a diverse array of surface displayed proteins to promote dissemination and establish infection in the human host. Of these, Pf3D7_0606800 is highly immunogenic and has been designated a potential top 10 candidate for inclusion in a multicomponent malarial vaccine. The role of Pf3D7_0606800 in parasite biology, however, is unknown and its characterization has been complicated by a lack of sequence identity with proteins of known structure or function. Towards elucidating Pf3D7_0606800 function, we determined its structure to a resolution of 2.35 Å using selenium single wavelength anomalous dispersion. A bi‐lobed architecture displays the core structural hallmarks of Venus Flytrap (VFT) proteins prompting us to re‐annotate Pf3D7_0606800 as PfVFT1. Structural analysis further revealed an extended inter‐lobe groove that, when interrogated by molecular docking, appears well suited to bind peptide‐based ligands. Collectively, our structural characterization of the highly antigenic P. falciparum surface protein PfVFT1 provides intriguing functional insight and establishes a structural template that could prove valuable for malaria vaccine engineering studies.
Keywords: Plasmodium falciparum, X‐ray crystallography, bi‐lobed architecture, venus flytrap domain, conformational flexibility
Short abstract
Interactive Figure 1, Interactive Figure 2 | PDB Code(s): 5JKQ
Introduction
Plasmodium falciparum is a member of the phylum Apicomplexa and the etiological agent of severe human malaria.1 The widespread success of P. falciparum is largely due to its complex lifecycle supported by a diverse arsenal of parasite‐specific factors. Of particular interest are surface‐exposed parasitic antigens that may be linked to human virulence.2 Recent genomic and proteomic studies have identified several parasite‐specific antigens whose functions are unknown. Since antibodies to these proteins are found in naturally infected patients and are correlated with survival, they likely play a crucial role in P. falciparum biology.2, 3, 4 Thus, structural and functional characterization of these antigens has the potential to offer valuable insight into the molecular basis of P. falciparum pathogenesis and support the design of next generation malarial vaccines.
Pf3D7_0606800 is a parasite‐specific antigen expressed by the P. falciparum 3D7 strain in both sexual and asexual stages5 and was identified as a major candidate for inclusion in a multicomponent blood‐stage malaria vaccine along with well‐known targets such as AMA1.2, 6, 7, 8 Pf3D7_0606800 is readily accessible to the immune system, may contain heat‐labile conformational epitope(s),9 and is recognized by sera of malaria‐immune adults.6, 10 The function of Pf3D7_0606800, however is unknown. Bioinformatic analysis predicts a signal peptide with no identifiable membrane anchors suggesting that it coordinates a membrane‐associated partner to enable surface localization. Consistent with this hypothesis, fractionation of detergent‐resistance membranes showed that Pf3D7_0606800 clusters with GPI‐anchored parasite proteins including Pf38, Pf92, Pf113, PfMSP‐1, PfMSP‐4, PfASP, and PfRAMA.11
Defining the structural characteristics of Pf3D7_0606800 is a key step towards revealing its role in P. falciparum biology and understanding the molecular basis for its immunogenicity. To this end, we have recombinantly produced Pf3D7_0606800 in insect cells and determined the selenium‐phased 2.35 Å resolution crystal structure. Our study represents the first detailed characterization of this highly immunogenic, yet previously understudied malaria surface antigen and offers intriguing clues as to its biological function.
Results and Discussion
Pf3D7_0606800 adopts a compact bi‐lobed architecture
Toward defining the function of Pf3D7_0606800, we first sought to establish its overall architecture. Mature forms of native and selenomethionine derivatized Pf3D7_0606800 [Fig. 1(A)] were expressed in insect cells and purified by nickel affinity and size exclusion chromatography (SEC), which showed a monomeric distribution [Fig. 1(A)]. Pf3D7_0606800 crystallized in a primitive orthorhombic space group with four molecules in the asymmetric unit (AU). Selenium single wavelength anomalous dispersion (SAD) resulted in high quality phases that enabled modeling of the complete protein sequence from the signal peptide cleavage site through the C terminus for Chain A.
Figure 1.
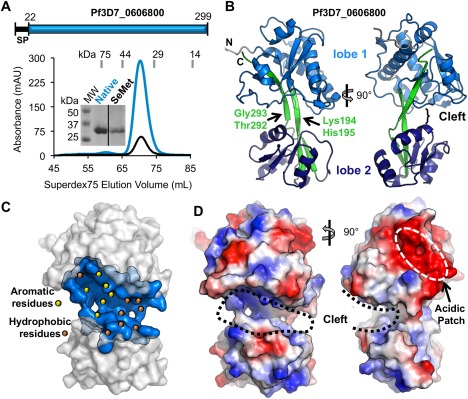
Structural characterization of Pf3D7_0606800 reveals a bi‐lobed domain architecture. A: Top: Schematic of the Pf3D7_0606800 protein sequence. Bottom: SEC profile of native (blue line) and SeMet (black line) Pf3D7_0606800 produced in insect cells. Inset, Coomassie‐stained SDS‐PAGE gel of Native and SeMet Pf3D7_0606800 (expected molecular weight: 32 kDa). B: Cartoon representation of orthogonal views of Pf3D7_0606800 structure. C: Surface representation of Pf3D7_0606800 highlighting the central cleft (blue) with the hydrophobic (orange dots) and aromatic (yellow dots) residues. D: Orthogonal views of Pf3D7_0606800 electrostatic surface representation. An interactive view is available in the electronic version of the article.
Structural analysis revealed a bi‐lobed architecture with the lobes linked by a pair of extended beta strands [Fig. 1(B)]. Both lobes exhibit similar packing of the secondary structure elements consisting of an α/β globular domain with a central β sheet of 4–5 strands flanked by clusters of α helices. Lobe 1 is formed predominantly by the N‐terminal portion of the protein and incorporates residues Lys30 to Pro191 and His294 to Lys299 making it nearly twice the size of Lobe 2, which extends from Lys198 to Asp287 [Fig. 1(B)]. The antiparallel beta strands that connect the two lobes form a scissor‐like overlap with Lys194–His195 and Thr292–Gly293, computationally predicted12 to be high probability articulation points [Fig. 1(B)]. Analysis of the four molecules in the AU also supports the potential for inter‐lobe flexibility; the overall root mean square deviation (RMSD) range of 1.04–1.27 Å over 223–261 Cαs is greater than for Lobe 1 (0.31 Å over 136 Cαs) or Lobe 2 (0.49 Å over 85 Cαs). Moreover, a structural superimposition of the four chains also revealed that Lobe 2 rotates ∼3°–5° relative to Lobe 1 for a total displacement of ∼4 Å.
The most striking feature of the Pf3D7_0606800 molecular surface is a prominent cleft located between Lobes 1 and 2 [Fig. 1(C)]. Several aromatic residues line the cleft [Fig. 1(C)] indicating the potential for π‐stacking interactions to coordinate a molecular partner. The most solvent‐exposed aromatic residues lining the cleft are Trp (85, 100), Tyr (41, 145), and Phe (132, 146) residues from Lobe 1 [Fig. 1(C), yellow dots]. The observed inter‐lobe flexibility [Fig. 1(B)] may also promote ligand binding by stabilizing the bound form of the complex through induced fit. A second notable surface feature in Lobe 1, located outside the central cleft, is a large acidic patch comprised of ten Asp and Glu residues (Glu44, Glu49, Glu50, Asp53, Asp56, Asp123, Glu130, Asp139, Glu140, Asp141) [Fig. 1(D), right] that might also support molecular interactions to enable surface localization or function.
Structural analysis of Pf3D7_0606800 reveals a Venus Flytrap domain
To overcome the lack of sequence identity with proteins of known structure or function, we next performed a structural homology search using the DALI server.13 The DALI search resulted in a significant hit with the Venus Flytrap (VFT)‐like ATP Binding Cassette (ABC) transporter from Streptococcus thermophilus (PDB ID 3HV1). A similar result was obtained when the search was performed with individual Pf3D7_0606800 lobes consistent with an overall conserved structural fold. Despite a sequence identity of only 14%, the two proteins align with a Z‐score of 11.3 (with scores less than 2 being spurious) and an RMSD of 5.0 Å over 254 Cα positions. A list of top six DALI hits is shown in Table 2. In addition to StABC, Pf3D7_0606800 exhibits strong structural similarity to more than 50 VFT domains. Based on this similarity, we propose re‐annotating Pf3D7_0606800 as PfVFT1, which reflects both its identification as a VFT protein and, to the best of our knowledge, as the first structurally validated VFT protein from Plasmodium.
Table 2.
Top 6 DALI Hits of PfVFT1
| PDB ID | Z‐score | Function |
|---|---|---|
| 3HV1 | 11.3 | Polar amino acid ABC uptake transporter substrate |
| 2V25 | 10.9 | Major cell binding factor |
| 5HMT | 10.4 | Cyclohexadienyl dehydratase |
| 3KBR | 10.3 | Cyclohexadienyl dehydratase |
| 2VHA | 10.2 | Periplasmic binding transport protein |
| 3K4U | 10.1 | Binding component of ABC transporter |
While the overall VFT architecture if conserved, PfVFT1 harbors several unique secondary structure elements, particularly in the N‐ and C‐termini [Fig. 2(A)]. Specifically, two helices (α1 and α2) in Lobe 1 extend from the N‐terminus, whereas their equivalents (α6 and α7) in all other VFTs are located at the C‐terminus [Fig. 2(A)]. This rearrangement in PfVFT1 appears to contribute to a more condensed packing of Lobe 1 as a 35‐residue loop connecting the C‐terminus of α2 to the N‐terminus of α3 wraps tightly around α1 and α2. Additionally, a helix located between β6 and β7 in Lobe 2 of most VFTs is absent in PfVFT1 [Fig. 2(A), gray rectangle with yellow border]. This helix, which plays a role in substrate recognition in select VFTs,17, 18 is replaced by a hydrophobic loop in PfVFT1 that forms a part of the hydrophobic cleft and may play a similar role in ligand recognition. The cleft of PfVFT1 is also larger (surface area: 928 Å2; volume: 2004 Å3) than that of StABC (surface area: 434 Å2; volume: 404 Å3) as shown by surface pocket analysis with CastP19 consistent with the ability to coordinate a larger ligand.
Figure 2.
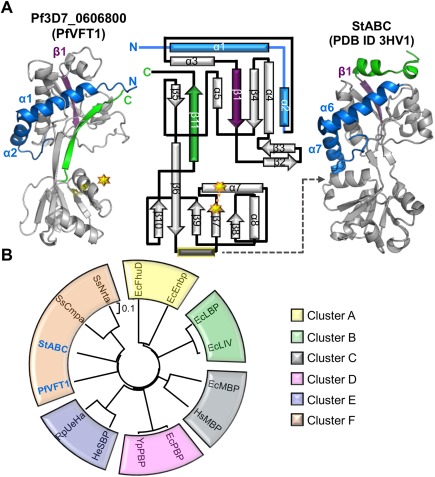
Pf3D7_0606800 adopts a Venus Flytrap (VFT) domain fold and is re‐annotated as PfVFT1. A: Secondary structure elements that are conserved (gray) and that diverge (purple, green, and blue) between PfVFT1 and StABC (PDB ID 3HV1). Topology diagram of PfVFT1 highlighting unique N‐terminus (blue components prior to β1, deep purple) and C‐terminus (green). Starburst and dotted connector indicate a disulfide bond. Gray dotted indicates lobe 2 helix (yellow outline) that is common to most VFTs, but absent in PfVFT1. B: A phylogenetic tree assembled using structure‐based (RMSD) alignments of PfVFT1 with two representative members for every cluster, obtained from previous evolutionary analysis on VFTs14 using PDBefold.15, 16 For simplicity, the recently identified Cluster G14 was not included. Escherichia coli (Ec); Homo sapiens (Hs); Yersinia pesits (Ys); Halomonas elongate (He); Ruegeria pomeroyi (Ru); Streptococcus thermophiles (St); Synechocystis spp. (Ss). EcFhuD‐1PSZ; EcFnbp‐2CHU; EcLBP‐1USK; EcLIV‐1Z16; EcMBP‐4MBP; HsMBP‐2ZVK; EcPBP‐1IXH, YsPBP‐2Z22; HeSBP‐2VPO; RuSBP‐3FXB; StABC‐3HV1; SsCmpa‐2I48; SsNrta‐2G29. An interactive view is available in the electronic version of the article.
Since VFT proteins are clustered into several different subgroups and often based on the nature of their ligands,14, 20 we next sought to determine in which subgroup PfVFT1 clusters. However, the sequence diversity of PfVFT1 relative to other VFTs significantly complicates a multiple sequence alignment‐based approach. We thus leveraged the PfVFT1 structure and generated a distance tree based on structural comparisons [Fig. 2(B)]. Though the tree cannot be statistically validated using bootstrap values, the topology of the tree is similar to that previously described for VFTs.14 Based on our analysis, PfVFT1 aligns with cluster F [Fig. 2(B)], which includes VFTs that bind amino acid ligands such as l‐glutamate, l‐histidine, l‐lysine, and l‐ arginine.14
The PfVFT1 cleft displays structural features consistent with peptide coordination
Homologs of PfVFT1 are not observed across all Plasmodium species, but instead predominate in Plasmodium parasites that cause malaria in apes (PgVFT1 and PrVFT1) and birds (PgalVFT1 and PrelVFT1). A sequence comparison of these VFT1 homologs reveals a clear bifurcated evolutionary profile [Fig. 3(A), left]. To better understand the implications of these sequence differences, we mapped them onto the structure of PfVFT1 using ConSurf23 [Fig. 3(A), right]. While the surface generally shows extensive divergence throughout the Plasmodium spp., the hydrophobic cleft is notably well conserved consistent with an important functional role. Additionally, a structure‐based sequence alignments with VFTs and mapping the conserved residues on PfVFT1 using consurf23 revealed no residues that appeared to be conserved in the hydrophobic cleft suggesting that the hydrophobic cleft may be unique to Plasmodium spp.
Figure 3.

Peptide coordination in the inter‐lobe cleft. A: Left: Plasmodium spp. VFT1 polypeptide sequences were aligned using Clustal Omega21 and a phylogenetic tree rooted to the divergent PfVFT1 homolog in T. gondii was generated using MEGA22 with 500 bootstrap replicates (See methods for accession numbers). Right: mapping of conserved (burgundy) and variable (cyan) residues of PfVFT1 homologs onto the PfVFT1 surface using ConSurf23; conservation of cleft region indicated by black border. B: Interactions between the N‐terminal tail of Chain A and the inter‐lobe cleft of Chain B. Inset: residues from Chains B and A are labeled green and yellow, respectively. C: Left: Weblogo of the consensus sequence of the 500 best‐scored tripeptides and respective docking result to PfVFT1. Right: Energy minimized docking pose of the consensus peptide.
Analysis of the conserved cleft amongst the four PfVFT1 monomers in the AU revealed that the N‐terminal tail of Chain A binds in the cleft of the neighboring Chain B. The Chain A N‐terminal tail (22GVEEVVNNK30) is stabilized through intermolecular hydrogen bonds between E24, E25 of the tail with Y62 and W105 of the cleft [Fig. 3(B)], and a salt bridge between E25 and K103 (cleft) [Fig. 3(B)]. To get an insight into the functional relevance of PfVFT1 cleft region, we compared the electron density of the cleft region with previously studied ligand bound VFTs by structurally overlaying PfVFT1 and amino acid bound VFTs belonging to the cluster F. While the N‐terminal peptide (from Chain B‐ PfVFT1) models into the electron density very well, structural overlays revealed no suitable amino acid modeling into the electron density clearly suggesting that the binding partner for PfVFT1 is not similar to classical VFTs.
With the electron density comparison failing to give any insight into the functional relevance of PfVFT1, we decided to investigate the relevance of the peptide–PfVFT1 bimolecular complex. Towards this, we synthesized the N‐terminal peptide and performed native gel shift binding assays and thermal stability assays with PfVFT1. No definitive interaction was measured, suggesting that the peptide may be too small or have an incorrect consensus sequence that is only able to bind in the artificially high protein concentration of a crystal lattice. To further probe the potential for a peptide ligand to bind in the hydrophobic PfVFT1 cleft, we screened in silico 8000 tripeptides without defining any constrained interactions. The energetically most favored 500 tripeptides bound to the hydrophobic patch of the binding pocket of Lobe 1. A consensus scoring for each of the positions was performed and displayed in Weblogo24 format [Fig. 3(B), left]. The consensus sequence was then docked against PfVFT1 to represent a potential binding mode of an enriched peptide sequence [Fig. 3(B), right]. Notably, the in silico generated consensus sequence is not anchored by specific interactions, indicating that shape complementarity may be a primary driving force for coordination within the PfVFT1 cleft.
Materials and Methods
Cloning, protein production, and purification
A construct encoding the mature form of Pf3D7_0606800 (Val23 to Lys299) with three predicted N‐linked glycosylation sites mutated to NxA was codon optimized for insect cells and synthesized by GenScript. The construct was sub‐cloned into a modified pAcGP67b vector with an N‐terminal His6 tag and produced in insect cells using previously established protocols.25 Recombinant PfVFT1 was purified by nickel‐affinity and SEC (Superdex 16/600 75) in 20 mM HEPES pH 7.5, 150 mM NaCl. Selenomethionine (SeMet) labeled protein was generated using established protocols.26
Bioinformatics
Phylogenetic analysis for Plasmodium VFTs based on the sequences in PlasmoDB,5 NCBI,27 and Genbank28 was performed in MEGA 6.0.22 A neighbor‐joining tree with 500 bootstrap replicates was constructed and rooted to the PfVFT1 homolog in Toxoplasma gondii (NCBI: XP_002365273.2). Accession numbers: P. falciparum VFT1 (PfVFT1), Pf3D7_0606800 (PlasmoDB); Plasmodium reichenowi VFT1 (PrVFT1), PRCDC_0605400 (PlasmoDB); Plasmodium gaboni VFT1 (PgVFT1), XP_018642701.1 (NCBI); Plasmodium gallinacuem VFT1 (PgalVFT1), CRG93648.1 (Genbank); Plasmodium relictum VFT1 (PrelVFT1), CRH01066.1 (Genbank). Sequences were aligned in Clustal Omega21 and surface mapped using ConSurf.23 Two representative structures of each cluster were pairwise structurally aligned to PfVFT1 with PDBefold15, 16 and the RMSD scores were input to the kitsch program in phylips package29 to generate the distance tree.
Crystallization and data collection
Crystals of selenomethionine‐labeled PfVFT1 were grown in 0.1M Bis–Tris:HCl pH 6.5, 2.0M ammonium sulfate using sitting drops. Crystals were cryoprotected in paratone or saturated lithium sulfate and flash cooled in liquid nitrogen. Diffraction data were collected on beamline 7–1 at the Stanford Synchrotron Radiation Lightsource (SSRL) using a wavelength of 1.127 Å, or an optimized wavelength of 0.9792 Å for the f” Se edge.
Data processing, structure solution, and refinement
Diffraction data were processed to 2.50 Å (PfVFT1‐SeMet SAD) or 2.35 Å (PfVFT1‐SeMet) resolution using Imosflm30 and Aimless.31 The structure of PfVFT1‐SeMet SAD was solved by SeMet SAD with 13 high confidence Se sites identified using ShelxC/D32 and refined using Phenix.AutoSol (Supporting Information Fig. S1).33 High quality phases enabled building and registering of ∼80% of the backbone using Phenix.autobuild.34 The higher resolution PfVFT1‐SeMet structure was solved by molecular replacement using the Se‐phased model in Phaser.35 COOT36 was used for model building and selection of solvent atoms and the model was refined in Phenix.refine.37 Structural validation was performed with Molprobity38 with greater than 98% of residues in the most favored conformations. Data collection and refinement statistics are presented in Table 1. Atomic coordinates and structure factors of PfVFT1 have been deposited in the PDB with accession code 5JKQ.
Table 1.
Data Collection and Refinement Statistics
| PfVFT1‐SeMet SAD | PfVFT1‐SeMet | |
|---|---|---|
| A. Data collection statistics | ||
| Space group | P21212 | P21212 |
| a, b, c (Å) | 84.62, 201.01, 77.53 | 84.77, 201.43, 77.56 |
| α, β, γ (deg.) | 90, 90, 90 | 90, 90, 90 |
| Wavelength | 0.9792 | 1.127 |
| Resolution range (Å) | 84.62–2.50 (2.59–2.50) | 84.77–2.35 (2.42–2.35) |
| Measured reflections | 667,898 (65,916) | 240,477 (19,464) |
| Unique reflections | 46,651 (4505) | 55,998 (4514) |
| Redundancy | 14.3 (14.6) | 4.3 (4.3) |
| Completeness (%) | 100.0 (100.0) | 99.8 (99.8) |
| I/σ(I) | 15.8 (5.1) | 8.6 (2.3) |
| R merge | 0.162 (0.603) | 0.125 (0.546) |
| B. Refinement statistics | ||
| Resolution (Å) | 78.18–2.35 | |
| R work/R free | 0.210/0.263 | |
| No. of atoms | ||
| Protein (A/B/C/D) | 2260/2265/2201/2126 | |
| Sulfate | 30 | |
| Solvent | 243 | |
| B‐values (Å2) | ||
| Protein (A/B/C/D) | 25.9/29.6/23.6/26.8 | |
| Sulfate | 38.6 | |
| Solvent | 22.8 | |
| r.m.s. deviation from ideality | ||
| Bond lengths (Å) | 0.004 | |
| Bond angles (deg.) | 0.721 | |
| Ramachandran statistics (%) | ||
| Most favored | 98.1 | |
| Allowed | 1.9 | |
| Disallowed | 0.0 |
Values in parentheses are for the highest resolution shell.
In silico peptide docking
In silico peptide‐mimetic screens were performed using ROCS.39 All 8000 possible tripeptides were generated using OMEGA2 allowing 500 conformers per tripeptide to exhaustively sample rotational space. The library was then docked using FRED and visually inspected with VIDA. A consensus fingerprint was derived from the proximity of each docked amino acid to a defined point in space. Equidistant points along the binding pocket were assigned and residues within 1.5 Å were assigned to that particular position. The final consensus peptide was then docked to the PfVFT1 structure.
Conclusion
Severe human malaria, caused by P. falciparum, remains a major global human health concern. Improved therapeutic strategies are desperately needed as parasites continue to develop resistance to front‐line drug treatments and evade the development of a highly effective vaccine. In this study, we report the first structural characterization of PfVFT1 (previously Pf3D7_0606800), a P. falciparum protein identified as a top candidate for a multicomponent malaria vaccine and a putative factor in human virulence.2, 6 Intriguingly, while the VFT domain is generally well‐characterized, the PfVFT1 structure presents a novel topological organization, thereby expanding our understanding of this important family of ligand‐binding proteins. Structural analysis and in silico studies reveal a possible peptide binding function for PfVFT1 and provide a high‐resolution framework for future epitope mapping studies and functional characterization.
Supporting information
Supporting Information Figure 1.
Acknowledgment
Authors thank the staff at the Stanford Synchrotron Radiation Lightsource. The authors do not have a conflict of interest to declare.
Michelle L. Parker and Raghavendran Ramaswamy contributed equally to this work
References
- 1. World Health Organization (2014) World malaria report. Geneva, Switzerland: World Health Organization Press. [Google Scholar]
- 2. Frech C, Chen N (2011) Genome comparison of human and non‐human malaria parasites reveals species subset‐specific genes potentially linked to human disease. PLoS Comput Biol 7:e1002320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sanders PR, Gilson PR, Cantin GT, Greenbaum DC, Nebl T, Carucci DJ, McConville MJ, Schofield L, Hodder AN, Yates JR, III , Crabb BS (2005) Distinct protein classes including novel merozoite surface antigens in Raft‐like membranes of Plasmodium falciparum . J Biol Chem 280:40169–40176. [DOI] [PubMed] [Google Scholar]
- 4. Gilson PR, Nebl T, Vukcevic D, Moritz RL, Sargeant T, Speed TP, Schofield L, Crabb BS (2006) Identification and stoichiometry of glycosylphosphatidylinositol‐anchored membrane proteins of the human malaria parasite Plasmodium falciparum . Mol Cell Proteom 5:1286–1299. [DOI] [PubMed] [Google Scholar]
- 5. Aurrecoechea C, Brestelli J, Brunk BP, Dommer J, Fischer S, Gajria B, Gao X, Gingle A, Grant G, Harb OS, Heiges M, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Miller JA, Nayak V, Pennington C, Pinney DF, Roos DS, Ross C, Stoeckert CJ, Jr , Treatman C, Wang H (2009) PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res 37:D539–D543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Osier FH, Mackinnon MJ, Crosnier C, Fegan G, Kamuyu G, Wanaguru M, Ogada E, McDade B, Rayner JC, Wright GJ, Marsh K (2014) New antigens for a multicomponent blood‐stage malaria vaccine. Sci Transl Med 6:247ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Srinivasan P, Ekanem E, Diouf A, Tonkin ML, Miura K, Boulanger MJ, Long CA, Narum DL, Miller LH (2014) Immunization with a functional protein complex required for erythrocyte invasion protects against lethal malaria. Proc Natl Acad Sci USA 111:10311–10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vulliez‐Le Normand B, Tonkin ML, Lamarque MH, Langer S, Hoos S, Roques M, Saul FA, Faber BW, Bentley GA, Boulanger MJ, Lebrun M (2012) Structural and functional insights into the malaria parasite moving junction complex. PLoS Pathog 8:e1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Crosnier C, Wanaguru M, McDade B, Osier FH, Marsh K, Rayner JC, Wright GJ (2013) A library of functional recombinant cell‐surface and secreted P. falciparum merozoite proteins. Mol Cell Proteom 12:3976–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chia YS, Badaut C, Tuikue Ndam NG, Khattab A, Igonet S, Fievet N, Bentley GA, Deloron P, Klinkert MQ (2005) Functional and immunological characterization of a duffy binding‐like‐gamma domain from Plasmodium falciparum erythrocyte membrane protein‐1 expressed by a placental isolate. J Infect Dis 192:1284–1293. [DOI] [PubMed] [Google Scholar]
- 11. Sanders PR, Cantin GT, Greenbaum DC, Gilson PR, Nebl T, Moritz RL, Yates JR, III , Hodder AN, Crabb BS (2007) Identification of protein complexes in detergent‐resistant membranes of Plasmodium falciparum schizonts. Mol Biochem Parasitol 154:148–157. [DOI] [PubMed] [Google Scholar]
- 12. Emekli U, Schneidman‐Duhovny D, Wolfson HJ, Nussinov R, Haliloglu T (2008) HingeProt: automated prediction of hinges in protein structures. Proteins 70:1219–1227. [DOI] [PubMed] [Google Scholar]
- 13. Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scheepers GH, Lycklama ANJA, Poolman B (2016) An updated structural classification of substrate‐binding proteins. FEBS Lett 590:4393–4401. [DOI] [PubMed] [Google Scholar]
- 15. Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. [DOI] [PubMed] [Google Scholar]
- 16. Krissinel E, Henrick K (2005) Multiple alignment of protein structures in three dimensions In: International Symposium on Computational Life Science. Springer, Berlin, Heidelberg, pp 67–78. [Google Scholar]
- 17. Oh BH, Pandit J, Kang CH, Nikaido K, Gokcen S, Ames GF, Kim SH (1993) Three‐dimensional structures of the periplasmic lysine/arginine/ornithine‐binding protein with and without a ligand. J Biol Chem 268:11348–11355. [PubMed] [Google Scholar]
- 18. Keegan R, Waterman DG, Hopper DJ, Coates L, Taylor G, Guo JX, Coker AR, Erskine PT, Wood SP, Cooper JB (2016) The 1.1 angstrom resolution structure of a periplasmic phosphate‐binding protein from Stenotrophomonas maltophilia: a crystallization contaminant identified by molecular replacement using the entire Protein Data Bank. Acta Crystallogr D Struct Biol 72:933–943. [DOI] [PubMed] [Google Scholar]
- 19. Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 34:W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berntsson RP, Smits SH, Schmitt L, Slotboom DJ, Poolman B (2010) A structural classification of substrate‐binding proteins. FEBS Lett 584:2606–2617. [DOI] [PubMed] [Google Scholar]
- 21. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ashkenazy H, Erez E, Martz E, Pupko T, Ben‐Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tonkin ML, Grujic O, Pearce M, Crawford J, Boulanger MJ (2010) Structure of the micronemal protein 2 (MIC2) A/I domain from Toxoplasma gondii . Protein Sci 19:1985–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parker ML, Penarete‐Vargas DM, Hamilton PT, Guerin A, Dubey JP, Perlman SJ, Spano F, Lebrun M, Boulanger MJ (2016) Dissecting the interface between apicomplexan parasite and host cell: insights from a divergent AMA‐RON2 pair. Proc Natl Acad Sci USA 113:398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geer LY, Marchler‐Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, Bryant SH (2010) The NCBI BioSystems database. Nucleic Acids Res 38:D492–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benson DA, Cavanaugh M, Clark K, Karsch‐Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2013) GenBank. Nucleic Acids Res 41:D36–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Felsenstein J (2005) PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Seattle: Dep Genome Sci Univ Washington. [Google Scholar]
- 30. Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction‐image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Evans PR, Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sheldrick GM (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr 66:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse‐Kunstleve RW, Afonine PV, Zwart PH, Hung LW (2009) Decision‐making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr 65:582–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McCoy AJ (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63:32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen VB, Arendall WB, III , Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hawkins PCD, Skillman AG, Nicholls A (2007) Comparison of shape‐matching and docking as virtual screening tools. J Med Chem 50:74–82. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.