

Abstract
The second member of the human ErbB family of receptor tyrosine kinases, HER2/hErbB2, is regarded as an exceptional case: The four extracellular subdomains could so far only be found in one fixed overall conformation, designated “open” and resembling the ligand‐bound form of the other ErbB receptors. It thus appears to be different from the extracellular domains of the other family members that show inter‐subdomain flexibility and exist in a “tethered” form in the absence of ligand. For HER2, there was so far no direct evidence for such a tethered conformation on the cell surface. Nonetheless, alternative conformations of HER2 in vivo could so far not be excluded. We now demonstrate the rigidity of HER2 on the surface of tumor cells by employing two orthogonal approaches of protein engineering: To directly test the potential of the extracellular domain of HER2 to adopt a pseudo‐tethered conformation on the cell surface, we first designed HER2 variants with a destabilized interface between extracellular subdomains I and III that would favor deviation from the “open” conformation. Secondly, we used differently shaped versions of a Designed Ankyrin Repeat Protein (DARPin) fusion, recognizing subdomain I of HER2, devised to work as probes for a putative pseudo‐tethered extracellular domain of HER2. Combining our approaches, we exclude, on live cells and in vitro, that significant proportions of HER2 deviate from the “open” conformation.
Keywords: HER2, ErbB receptors, protein engineering, conformational probe, DARPins
Introduction
Most multi‐domain proteins, especially transmembrane receptors anchored in the cell membrane, are not fixed in a single arrangement, but sample different conformations, usually directly related to their function. The four members of the human ErbB receptor tyrosine kinase family (EGFR/HER1/ErbB1, HER2/ErbB2/neu, HER3/ErbB3, and HER4/ErbB4) are important and well‐studied examples of such conformationally variable proteins. In a generalized model of ErbB signaling, supported by x‐ray structures of all four receptors, the three ligand‐binding receptors (i.e., all except ErbB2/HER2) undergo conformational changes within their extracellular domains (ECDs) upon ligand association, enabling them to form homo‐ or hetero‐dimers in the ligand‐bound state [Fig. 1(A–C)].1 Upon dimerization, the cytoplasmic kinase domains (KDs) of the receptors are then able to transphosphorylate intracellular tyrosine residues of the associated dimerization partner at its cytoplasmic domain. In turn, these phosphotyrosines become binding sites for downstream signaling molecules, and thereby feed into major signaling networks.
Figure 1.
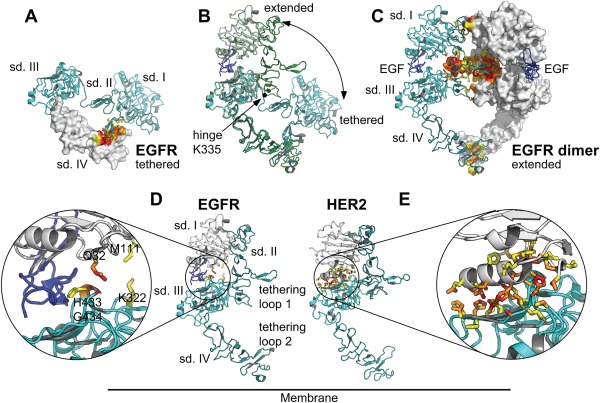
Conformational changes and domain interactions in EGFR (PDB ID 1YY9 and 3NJP) and HER2 (PDB ID 1N8Z and 3N85). (A) Tethered conformation of the EGFR ECD (PDB ID 1YY9). Contacts between the tethering loops in subdomain II and IV, stabilizing the tethered conformation, are highlighted in red (van‐der‐Waals contacts ≤3.6 Å) and orange (solvent‐excluding contacts ≤5 Å), non‐contact atoms in contact residues are shown in yellow. (B) Comparison of the tethered and extended conformations of EGFR. The hinge motion relating the two conformations is predominantly due to a change of the main‐chain torsion angles of Lys 335, located at the boundary between subdomains II and III and corresponding to Arg 340 in HER2. (C) Extended conformation of EGFR (PDB ID 3NJP) in the EGFR homodimer. The tethering loops in subdomains II and IV interact with the corresponding loops of the second molecule in the dimer. (D) The open conformation of EGFR is stabilized by the ligand EGF (dark blue) binding to both subdomain I (white) and III (cyan). The two subdomains barely touch. (E) The extended conformation of HER2 is stabilized by direct interactions of subdomains I and III.
The whole ECD of ErbB receptors is connected via a short flexible peptide to the transmembrane (TM) helix and via another flexible link at the cytoplasmic end of the TM helix to the intracellular kinase domain. Thus, the whole ECD and KD are free to rotate or tilt with respect to each other. There is direct evidence for this mobility from cryo‐EM studies of EGFR, which confirm the crystallographically determined shapes of the ECD and KD, but describe the relative orientation of the ECD to the KD as flexible.2
Regarding the arrangement of the four extracellular subdomains with respect to each other, the ECD of HER2 has, in contrast to the other family members, only been seen crystallographically in an extended conformation,3 resembling the ligand‐bound state of the other family members [Fig. 1(D,E)].4, 5, 6, 7 This conformation, frequently designated “open,” has hence been assumed to be constitutive and may explain why HER2 appears to be the preferred heterodimerization partner for the ligand‐activated ErbB receptors. Upon overexpression, HER2 can feed into pro‐proliferative signaling through spontaneous formation of signaling‐competent HER2 homodimers that assemble at high HER2 concentrations, as found in breast tumors.3, 8 Under these conditions of overexpression, HER2 thus becomes a key regulatory element driving cell proliferation, survival, migration and invasiveness of cancer cells.
X‐ray crystal structures have suggested that the ligand‐independent signaling properties of HER2 result from the absence of a “closed” conformation of the extracellular region that is adopted by the other human ErbB receptors as long as they are not bound by a ligand.3, 5, 9 In this “tethered” form of EGFR,10 HER3,11 and HER4,12 intramolecular contacts between the “dimerization arm” in subdomain II and the “tethering arm” in subdomain IV, a loop similar but smaller than the loop forming the dimerization arm, constrain the relative orientation of the two subdomains I and II.10 For EGFR, HER3, and HER4, ligand binding to both subdomains I and III induces a conformational change from the closed and tethered to an open, extended conformation.9 The two conformations of the extracellular domain are related by a large hinge motion around a pivot point between subdomains II and III (Lys 335 in EGFR, Lys 329 in HER3 and Lys 332 in HER4).
For the case of EGFR, there is direct evidence suggesting that the adoption of a tethered form on the cell surface affects ligand binding. However, even when the tethering interactions are removed by introducing mutations or deleting extracellular subdomain IV, this does not lead to spontaneous kinase activation, and the tether is accordingly only one amongst several structural features governing EGFR function.13, 14, 15, 16 HER4 ECD is able to adopt a closed conformation even in the absence of the tethering extracellular subdomain IV.17
A closed conformation, let alone a tether, was not found in X‐ray structures of unliganded HER2 ECD,3, 4, 6, 7, 18 however. Rather, HER2 adopts an extended (open) conformation in the absence of a ligand, which strongly resembles the unliganded structure of the EGF receptor in Drosophila melanogaster (dEGFR).19 In dEGFR as well as HER2, extracellular subdomains I and III interact directly, forming a large, partially hydrophobic interface that stabilizes the extended conformation in the absence of a ligand. For dEGFR, ligand binding is sufficient to separate extracellular subdomains I and III, and induce a rather subtle conformational change (compared to HER1, HER3, and HER4) from an autoinhibited to the active state. It has therefore been hypothesized that the crystal structure of ligand‐free HER2 ECD, which does not homodimerize in solution, represents an autoinhibited state as well, which would be distinctly different from a (potentially ligand‐bound) active state.19 However, no activating ligand has been identified for HER2 in decades of intense research,20 the interactions between extracellular subdomain I and III are even more extensive than in dEGFR, and, in addition, molecular dynamics simulations evaluated by principal component analysis did not reveal large subdomain motions in HER2 ECD21 – therefore, the whole HER2 ECD may be considered very rigid. Nonetheless, the rigidity of the four extracellular HER2 subdomains in the arrangement found crystallographically has so far neither been addressed experimentally, nor has it been tested on cells.
Designed Ankyrin Repeat Proteins (DARPins) are artificial binding proteins that have been developed over the last decade22, 23 to expand the potential of classical binding proteins, namely antibodies and their fragments. DARPins can be generated to recognize their respective target with at least the same specificity and affinity as antibodies, but because of their robustness and extreme stability, they allow a multitude of more advanced formats and applications.22, 23 We have recently described bispecific DARPin constructs, consisting of two flexibly linked DARPins per molecule, binding to HER2 subdomains I and IV, which cause apoptosis in HER2‐dependent tumor cell lines.24, 25 One model for the biparatopic binding of one DARPin molecule to two receptor monomers based on X‐ray complex structures suggests that the intermolecular binding of these DARPin constructs bends HER2 ECD toward the plane of the membrane as an entire rigid body, using the short membrane‐proximal flexible peptide as a pivot. The only conceivable alternative model, however, presumes that the HER2 ECD can deviate from the extended conformation toward a conformation resembling the tethered conformation of EGFR, HER3, or HER4. This prompted us to explore the possible existence of alternative conformations of HER2 on the cell surface by two orthogonal approaches.
To our knowledge, no means to induce a conformational rearrangement of HER2 ECD – be it by mutagenesis, addition of a conformation‐specific affinity reagent, or any other conceivable way – has been unequivocally proven to actually achieve this. A recent report by Menendez et al.,26 however, might be regarded as some evidence challenging the exclusive rigidity of the HER2 ECD. Here, several deletion mutants in extracellular subdomain III of HER2 have been described, which were initially generated to test the hypothesis that HER2 activation requires a protein sequence in the HER2 ECD that mediates HER2 homo‐ and heterodimerization. According to this work, deletion of residues 451–466 in extracellular subdomain III surprisingly disrupted the oncogenic potential of HER2 completely, and generated a “dominant‐negative form” of HER2. The authors hypothesized that, on the molecular level, the dominant‐negative form might correspond to a tethered‐like conformation. Given, however, that the effect of deleting whole amino acid stretches within a folded subdomain of HER2 might be dramatic regarding its overall structural integrity, and HER2 was not detected on the cell surface in microscopic images,26 the results obtained by this approach might be difficult to interpret. We therefore chose a milder approach to explore the paradigm of HER2 ECD as being a largely inflexible subdomain assembly.
To this end, we designed HER2 variants with a destabilized subdomain interface between extracellular subdomain I and III using the Rosetta suite of programs (https://www.rosettacommons.org). This allowed us to introduce sets of only a few point mutations (three to eight), which were predicted to not affect the stability of the subdomains themselves, but only weaken their inter‐subdomain interactions. Furthermore, two residues located at the site corresponding to the “hinge” region in EGFR were replaced by the corresponding EGFR residues to allow flexibility. Together, destabilizing the subdomain interface and introducing a flexible hinge should allow the extracellular subdomains to rearrange more easily, and thereby reveal poorly sampled conformations alternative to the crystallographic, “open” conformation.
In a second and orthogonal approach, we exploited the rigid nature of DARPins to probe for alternative HER2 conformations on the cell surface. Based on the crystal structure of DARPin 929 in complex with isolated extracellular subdomain I (HER2_I),24 we have constructed a protein that, when binding to subdomain I on HER2 in extended conformation, would clash with subdomain III. However, even a small perturbation of the orientation of HER2 subdomains I and II relative to subdomains III and IV prevents this clash and restores binding. Binding experiments with this DARPin construct therefore allow direct detection of alternative HER2 conformations.
Results
Assuming that inherent rigidity of HER2 ECD prevents alternative conformations, e.g., a tethered‐like arrangement, we attempted to engineer HER2 variants with a destabilized subdomain I‐subdomain III interface together with the introduction of hinge flexibility.
To replace the hydrophobic, interacting residues at the subdomain I‐subdomain III interface with polar ones we used Rosetta fixed backbone design. In mutant mutQ (Table 1), we introduced residues which remove the hydrophobic domain interactions but are predicted to result in the strongest stabilization when considering only the isolated subdomains (Fig. 1). In an alternative approach, selected hydrophobic interface residues were replaced by charged residues to introduce electrostatic repulsion across the interface. In this case, the Rosetta interface analysis tool was used to identify combinations of mutations that were predicted to destabilize the interface, but only marginally affect the stability of subdomain I and III (mutants mut2R and mut3R, Table 1).
Table 1.
Overview of the Hinge and HER2 Extracellular Subdomain I and III Interface Mutationsa
| Abbreviation | EGFR hinge | Subdomain I | Subdomain III |
|---|---|---|---|
| flHER2_wt | – | – | – |
| flHER2_mutQ | A339R, R340K | M31K, A37S, M45E, L49R | A440Q, Y441H, L465K, L494E |
| flHER2_mut2R | A339R, R340K | L49R | L465R, A492R |
| flHER2_mut3R | A339R, R340K | T41R | L467R, H469R, L494R |
Residue numbering refers to the full‐length receptor including the signal sequence (UniProt ID P04626, ERBB2_HUM).
In EGFR, HER3, and HER4, the hinge regions, three‐residue segments between the last Cys of subdomain II and the first Cys of subdomain III, have the sequences RKV, PKA and PKA, respectively. In HER2, the corresponding sequence is ARV. Since the hinge motion mainly affects the main‐chain torsion angles of the central Lys, we changed the hinge sequence of HER2 to that of EGFR, thus changing residues 339 and 340 from AR to RK, for all mutant constructs.
Wild‐type and mutated HER2 were transiently overexpressed in HEK293T/17 cells with a C‐terminal fusion of GFP to monitor total protein levels, and these were very similar for wt and mutants. However, using DARPin 929, which recognizes a conformational epitope in subdomain I,24, 27 as probe for intact protein, very little of any of the mutant proteins could be detected at the cell surface (Fig. 2), in comparison to the wt. We therefore repeated the experiment with prior permeabilization in order to detect also receptor which did not reach the surface. Again, we obtained low signals for all the mutants (Supporting Information Fig. S1), indicating that, despite the mutants being well expressed as evidenced by the GFP fluorescence being similar to wt, only a very small fraction represents correctly folded protein.
Figure 2.
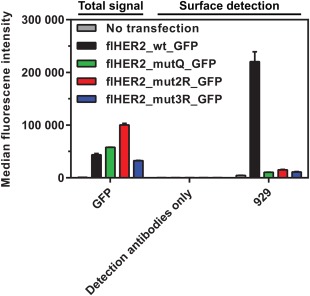
Mutations specifically destabilizing the open conformation of full‐length HER2 (flHER2) result in little functional protein at the cell surface. Binding of DARPin 929 to HEK293T/17 cells transiently transfected with wild‐type HER2 or mutants (see Table I for list of mutations) was detected in flow cytometry using anti‐DARPin serum. Note that despite at least equal overall protein production (monitored by the signal from the C‐terminally fused GFP), the surface binding signal of DARPin 929 for the HER2 mutants (green, red, blue bars) is maximally about 3‐fold over background (corresponding to endogenous HER2 of HEK293 cells, as seen in non‐transfected cells, gray bars). Error bars represent 1 SD of technical triplicates. The GFP signal was recorded in a separate channel and scaled for display.
The poor tolerance of HER2 ECD for engineering approaches motivated us to develop an orthogonal probe based on DARPins, which can sense alternative conformations of wild‐type receptor in its native membrane environment, on live cells. Two DARPins can be rigidly fused by continuing the C‐terminal helix of one DARPin into the N‐terminal helix of the second DARPin in such a way that the helix is embedded in at least one of the DARPins along its entire length (Y. Wu et al., submitted). When exploring different lengths of the shared helix, different relative orientations of the two DARPins are obtained [Fig. 3(A)].
Figure 3.

Models of shared helix constructs in complex with HER2 ECD. (A) Shared helix constructs (DD) are rigid fusions of two DARPins, in which the C‐terminal helix of one DARPin continues into the N‐terminal helix of a second DARPin. The length of this shared helix determines the relative orientation of the two DARPins and the overall shape of the construct, and is indicated by Hnn. The N‐terminal DARPin (tan or yellow) is a non‐binding DARPin (N3C), the C‐terminal DARPin 929 (orange) recognizes HER2 subdomain I. (B) Five out of nine designed DD constructs display van‐der‐Waals clashes with HER2 subdomain I (shown as red spheres), as shown for the example of N3C_H06_929. (C) Three constructs are predicted to bind to both isolated HER2_I and the full HER2 ECD without any clashes. One of these, N3C_H12_929, was used as positive control in binding experiments. (D) One construct, N3C_H09_929, is predicted to bind to the isolated HER2_I, but is prevented from binding to the full HER2 ECD in extended conformation by clashes with subdomain III. (E) A hinge motion similar to the one relating the extended to the closed conformation in EGFR would allow the HER2 ECD to escape this clash and allow N3C_H09_929 to bind. The models of the extended and of a hypothetical pseudo‐tethered HER2 ECD were aligned by a least‐squares fit of the Cα atoms of subdomain I (residues 24–212).
We intended to exploit the fact that DARPin‐DARPin (DD) fusions of a second, non‐HER2‐binding DARPin (N3C) to the C‐terminal helix of DARPin 929 would differ in whether they clash with the rest of the ECD, depending on the geometry of the fusion. Some DD fusions were predicted to not interfere with their recognition of the HER2 ECDs. In contrast, most fusions to the N‐terminal helix of DARPin 929 lead to clashes: Five out of nine fusion constructs were predicted to no longer bind even to HER2_I, the isolated N‐terminal subdomain I (residues 24–219) of the HER2 ECD recognized by DARPin 929, due to heavy steric clashes with this subdomain [Fig. 3(B)]. Three fusion constructs were predicted to retain binding to both isolated HER2_I and to the HER2 ECD, as well as to full‐length HER2 on intact cells, since in these constructs the non‐binding DARPin points away from the receptor. Out of these we selected N3C_H12_929 as a positive binding control [Fig. 3(C)]. Here, H12 denotes the shared connector helix. The most interesting construct was N3C_H09_929: In this fusion, the non‐binding DARPin does not interfere with binding to HER2_I, but clashes into subdomain III of the HER2 ECD [Fig. 3(D)], as it bridges the interface between subdomain I and subdomain III. However, this clash is light enough that even a minor hinge motion in HER2 would relieve the clash, when the interface between subdomains I and III is no longer intact [Fig. 1(E)]. We thus explored whether N3C_H09_929 would act as a conformational sensor as it can only bind to the HER2 ECD or full length HER2 if there is hinge mobility.
Therefore, our designed HER2 ECD conformational sensor was first tested in in vitro binding assays [Fig. 4(A)]. To produce the well‐folded isolated extracellular subdomain I (HER2_I) utilized as control in this assay, the “classical” sequence‐based subdomain boundary definition in HER23, 5, 9 had to be amended: The first module of subdomain II contributes a Trp side chain to the hydrophobic core of subdomain I, consequently the first four cysteines of the “classical” definition of subdomain II are structurally a part of subdomain I and have to be included in the construct. Therefore, a structural definition of HER2_I has to at least include Cys212 (Supporting Information Fig. S2). We recently reported successful expression and purification of a HER2_I construct, comprising amino acids 24–219 (numbering of the precursor), from Sf9 insect cells.24 This protein elutes as a monomer in size exclusion chromatography (Supporting Information Fig. S3) and is sufficiently stable to have allowed determination of its crystal structure (PDB ID 4HRL, 4HRM, 3H3B), which was shown to agree well with the structure of the corresponding domain in the full HER2 ECD.24
Figure 4.
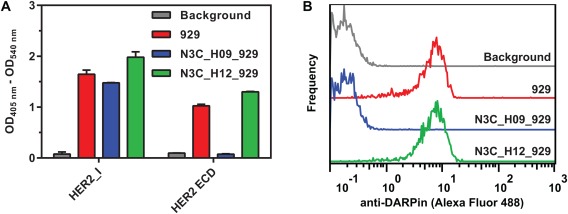
Binding experiments with the engineered conformational probe. (A) DD constructs and controls (100 nM; detection antibody background, gray; 929, red; N3C_H09_929, blue; and N3C_H12_929, green bars) binding to immobilized HER2_I or HER2 ECD in ELISA; error bars represent 1 SD of technical triplicates. (B) Binding of DD constructs and controls (100 nM; detection antibody background, gray; 929, red; N3C_H09_929, blue; and N3C_H12_929, green solid line) to live BT474 cells in flow cytometry. Bound DARPin constructs were detected using rabbit anti‐DARPin serum and fluorescently labeled secondary antibody.
As expected, the unfused (and thus unhindered) DARPin 929 and the conformation‐insensitive control construct N3C_H12_929 bound to the HER2 ECD, while the conformation‐sensitive construct N3C_H09_929 failed to bind, consistent with only an extended, fully rigid form of HER2 being present. All three constructs bound equally well to HER2_I used as positive control, in which the clash with the absent subdomain III is impossible.
Finally, flow cytometry was used to measure binding of the 929‐derived DD constructs to HER2 in its native environment on the cell surface of the HER2‐overexpressing cancer cell line BT474. It demonstrated clear binding of N3C_H12_929 and unfused DARPin 929, but no binding for N3C_H09_929 [Fig. 4(B)], supporting the absence of hinge flexibility also in full‐length, membrane‐embedded HER2 on cells.
Discussion
Overall, a new approach was developed to investigate the conformation of the extracellular part of a receptor, exemplified by HER2. Our first direct engineering attempt to destabilize the subdomain interface between extracellular subdomains I and III, in order to facilitate adoption of conformations different from the canonical “open” arrangement, did impede surface expression of the receptor. We believe that this simply results from the strict intracellular quality control for this transmembrane receptor and is ultimately a result of the intrinsic rigidity of HER2, which makes it completely inaccessible even for mild engineering presumed to favor domain rearrangements. This also suggests that the dominant‐negative, putatively tethered‐like deletion mutants, previously described by Menendez and colleagues,26 might in fact have led to misfolded receptor as well, which accumulates intracellularly, rather than in truly pseudo‐tethered HER2 on the cell surface. We would like to point out the absence of surface stain for the deletion mutants in the microscopy images in this article,26 which is consistent with our observations on the interface mutants described here.
We subsequently tried to assess the receptor's potential to deviate from the open conformation with protein binders, designed to act as “conformational probes.” This procedure makes use of the rigid nature of DARPins, which can be inflexibly linked to each other using several different shared helices, thereby yielding different shapes. Depending on the resulting relative orientation, binding to particular target conformations is prevented through steric clashes. We also made use of the previously obtained structural knowledge on the precise mode of binding of one particular DARPin on subdomain I of HER2.24
The change from an extended to a tethered conformation in the ErbB receptors EGFR, HER3, and HER4 requires flexibility of the hinge region between subdomains II and III. In these receptors, the interaction between extracellular subdomains I and III is not sufficient to stabilize a defined extended structure in the absence of the ligand. The interaction of the tethering loop in subdomain IV with subdomain II in turn stabilizes a defined tethered conformation. Binding of the ligand to both subdomain I and III shifts the conformational equilibrium to an extended conformation.
In HER2, a direct interface between subdomain I and III stabilizes the extended conformation in the absence of ligand, and no defined alternative conformation has been observed. To escape the clash with the rigidly fused DARPins, hinge flexibility would be sufficient, even if the receptor were unable to lock into a defined tether. Our data confirm that in the HER2 ECD, the interface between subdomains I and III is too strong to allow hinge flexibility. A subdomain arrangement that would dislodge subdomains I and III from each other, as observed in the tethered conformations of the extracellular domains of EGFR, HER3, and HER4, can now experimentally be excluded. Accordingly, the paradigmatic constitutively extended and rigid conformation of HER2 ECD was confirmed experimentally, both in vitro and on the surface of tumor cells. Furthermore, these findings illustrate that a putative tethered form of HER2 cannot be induced (at least not by the binding energy of the DARPin 929 with K D=3.8 nM28). Also, we found that a non‐open form of HER2 cannot be engineered when staying close to the HER2 sequence, as no significant amounts of folded HER2 are detected even in intracellular stores, nor on the surface, even though the protein is well expressed, but presumably prevented to reach the plasma membrane by the secretory quality control.
It is tempting to speculate whether even more advanced engineering may eventually succeed in conveying conformational flexibility to HER2. We show that the isolated extracellular subdomain I can be expressed as a well‐folded monomer. It is therefore likely that the exposure of the surface of extracellular subdomain III through hinge motion is the cause of instability, as it would abolish the mutual stabilization of the two domains through the interface. We have applied two different strategies toward destabilizing the interface between subdomains I and III, both predicted to not compromise the intrinsic stability of the individual domains: in one, we aimed to optimize interactions with the solvent to optimally stabilize the solvated domain in the absence of the partner domain, in the second we looked for a minimal number of mutations causing destabilization of the interface.
Our results suggest that to compensate for the loss of interface stabilization, we would first have to improve the folding and intrinsic stability of the isolated domains, in particular of subdomain III, by in‐vitro evolution and/or by design, and then prove that both individual domains are robust in the absence of the partner domain. However, this would obviously introduce mutations throughout the domains, and the sequence of such a construct would clearly deviate significantly from that of the wild‐type receptor. Nonetheless, such a flexible HER2 variant may still represent an interesting further control for our conformational probe.
Conclusions
We developed an alternative strategy for sensing the conformation of proteins, as the secretory quality control can preclude testing by mutagenesis. We turned to shared helix‐linked DARPin‐DARPin fusions as sensors, which present a valuable blueprint for tackling various protein engineering challenges. Together, our experiments with engineered HER2 variants and the engineered binding probe strongly support the view that the extended conformation of HER2, in contrast to other epidermal growth factor receptor family members, always remains unperturbed. The strategy of an external conformational sensor may help in the investigation of other proteins in their natural environment.
Material and Methods
Design of less rigid HER2 mutants
The Rosetta suite of programs (https://www.rosettacommons.org) was used to identify potential mutations in the interface between HER2 subdomains I and III that were predicted to destabilize the interface between the two domains without significant destabilization of the isolated domains. Subdomains I and III (res. 23–212, res. 341–528) were taken from the structure of the HER2 extracellular domain (PDB ID 1N8Z, 2.52 Å resolution). The partially unresolved loops in 1N8Z (missing residues 124–132 in subdomain I and 383–386 in subdomain III) were replaced by the fully resolved loops in PDB ID 3H3B (2.45 Å resolution) and 2A91 (2.50 Å resolution) using the Homology module in Insight II (Accelrys). The resulting model of the subdomain I‐III complex, omitting cysteine‐rich subdomains II and IV, was submitted to Rosetta Relax with all‐heavy‐atom constraints to prepare for further analysis and computation (https://www.rosettacommons.org/docs/latest/rosetta_basics/preparation/preparing-structures#relax-with-all-heavy-atom-constraints-protocol).
Two different strategies of destabilizing the hydrophobic interface were explored. In the first one, Rosetta fixed backbone design (fixbb) was used to replace the hydrophobic residues of the subdomain I‐III interface by the combination of polar residues predicted to yield the best stability for the isolated domains, resulting in flHER2_mutQ with four mutations each in subdomains I and III (Table 1). These were predicted to stabilize the domain in the absence of the partner domain, but significantly destabilize the interface between the domains.
In the second strategy, we explored which of the interface residues could be replaced by charged residues in such a way that the interface would be destabilized by charge repulsion, with only very mild destabilization of the free domains. To assess the influence of different combinations of mutations, the sum of the scores for the two domains was compared to the score of the complex of the two domains carrying the same mutations using the Rosetta interface analysis tool (flHER2_mut2R and flHER2_mut3R, Table 1).
The extended and the tethered conformation of EGFR are related by a change of the torsion angles of lysine 335. The corresponding residue in HER2 is arginine 340. To ensure similar flexibility of this hinge region between the last cysteine of subdomain II and the first cysteine of subdomain III, the Ala‐Arg‐Val sequence of HER2 was replaced by the Arg‐Lys‐Val of EGFR for all mutants.
Cloning, transfection of HER2 mutants
Wild‐type HER2 expression vector
Wild‐type HER2 (amplified from a Mammalian Gene Collection clone, GenBank accession number BC156755.1), preceded by a full‐consensus Kozak sequence, was assembled with a C‐terminal superfolder GFP29 fusion by PCR, connected by a G2SGSG2 linker. Subsequently, it was inserted using NheI and XbaI into a vector derived from pcDNA3.1/myc‐His(‐) A (ThermoFisher Scientific Inc., Carlsbad, USA). After this, the AhdI site within the β‐lactamase gene was removed, introducing silent mutations, employing sequence‐ and ligation‐independent cloning.30
DNA fragments for the generation of HER2 mutants
DNA fragments containing hinge and interface mutations in HER2 (see Table 1) were obtained from Integrated DNA Technologies (Leuven, Belgium) or GeneArt (Regensburg, Germany) and used as templates in PCR reactions as described below.
Introduction of extracellular subdomain III mutations and hinge residues
First, an N‐terminal fragment introducing the hinge residue mutations, (A339R and R340K) as in EGFR, was generated by a PCR on the HER2 gene (see above) using primers dIII_hinge_for (5′‐TGA ACA ATA CCA CCC CTG TC‐3′) and dIII_hinge_rev (5′‐CTT TCG ACA GGG CTT GCT GCA CTT C‐3′). Secondly, commercially obtained synthetic fragments containing several point mutations (Table 1) were amplified with the primers QE_for (5′‐CGG ATA ACA ATT TCA CAC AG‐3′) and QE_rev (5′–GTT CTG AGG TCA TTA CTG G–3′), for which binding sites had been added in the design of the fragments. Third, a C‐terminal fragment was generated by PCR on the HER2 gene using primers dIIIc_for (5′‐CAC ACT GCC AAC CGG CC‐3′) and dIIIc_rev (5′‐TCA CCT TCC TCA GCT CCG‐3′). Finally, all three combinations of the three sub‐fragments (Table 1) were assembled using primers dIII_hinge_for and dIIIc_rev and used to replace the respective fragments in wild‐type HER2, excised by AhdI and BsmBI (Esp3I).
Introduction of mutations in HER2 extracellular subdomain I
PCR fragments containing mutations within domain I were amplified using primers ECDI_for1.0 (5′‐TAC GAC TCA CTA TAG GGA GAC CCA AGC TGG‐3′) and ECDI_rev1.0 (5′‐AGC ACG TAG CCC TGC ACC TCC T‐3′) and then used to replace the respective fragments excised by NheI and SdaI. All fully assembled constructs were verified by Sanger sequencing (Microsynth, Balgach, Switzerland).
Transfection
HEK293T/17 cells (ATCC/LGC Standards, Wesel, Germany) were seeded into 25 cm2 flasks at a density of 80,000 cells cm−2 and one day later transfected using TransIT‐293 reagent (MirusBio, Madison, USA) according to the instructions by the supplier.
Flow cytometry
BT474 or HEK293T/17 cells (one day after transfection in this case) were detached with trypsin or Accutase (Sigma‐Aldrich, Buchs, Switzerland), resuspended in 4 mL PBSBA (Dulbecco's phosphate‐buffered saline supplemented with 1% w/v bovine serum albumin and 0.1% w/v sodium azide) and incubated at 37°C for 30 min. Subsequently, cells were resuspended, concentrations adjusted for equal final cell numbers (∼1–5 × 105 per sample) and dispensed in triplicates of 100 μL each for each condition in 96‐well plates or 1 mL for experiments in tubes. Cells were pelleted, resuspended in a 100 nM solution of the respective DARPin construct in PBSBA and incubated for 1 h at 37°C or 2 h on ice. Afterwards, cells were washed one to three times in ice‐cold PBSBA and strictly kept on ice from then on until measurement. Samples were then resuspended in 100 μL of anti‐DARPin serum obtained from rabbit immunization (Dreier et al., unpublished), diluted 1:1,000 in PBSBA or buffer controls and incubated for 45 min. Subsequently, cells were washed once to twice in ice‐cold PBSBA, resuspended in 100 μL goat anti‐rabbit APC‐conjugated antibody (Life Technologies, Zug, Switzerland) diluted 1:1,000 in PBS‐BA and incubated for a further 45 min. Finally, cells were washed one to three times in 200 μL ice‐cold PBSA (Dulbecco's phosphate‐buffered saline supplemented with 0.1% sodium azide), resuspended in PBSA and directly measured on a CyFlow Space (Partek, Görlitz, Germany) or a LSR Fortessa (BD Biosciences, Allschwil, Switzerland) flow cytometer with a high‐throughput sampler at the Flow Cytometry Facility, University of Zurich. Data were analyzed using FlowJo v10.0.8 software (FlowJo, Ashland, USA) and Prism v6.03 (GraphPad Software, La Jolla, USA).
DARPin cloning, expression & purification
To clone the DD constructs, the DARPin ORFs were digested with BamHI and KasI (for the N‐terminal DARPin) or HpaI and HindIII (for the C‐terminal DARPin) and ligated into compatible pQIq vectors (a lacIq‐encoding variant of pQE30 (Qiagen, Hilden, Germany) with double stop codon), coding for the respective shared helix. All DD‐constructs were expressed at 30°C overnight and purified essentially as described previously.31 Briefly, proteins were overexpressed in E. coli XL1‐Blue and purified via their N‐terminal MRGSH6 tag on nickel‐nitrilotriacetic acid superflow resin (Qiagen, Hilden, Germany).
Expression of HER2‐I and HER2‐ECD as target for ELISA
Recombinant HER2‐ectodomains carrying an N‐terminal melittin signal sequence and a N‐terminal His6 tag were expressed in Spodoptera frugiperda (Sf9) cells. Baculoviruses for infection of Sf9 cells were generated using the Multibac system as described.32 Sf9 cells were grown to a density of 4 × 106 cells/mL and coinfected with the respective virus at a MOI of 1. 72 h post infection, cells were harvested by centrifugation (30 min, 5,000g, 4°C), and the cleared medium was subjected to immobilized metal ion affinity chromatography (IMAC) purification on nickel‐nitrilotriacetic acid superflow resin (Qiagen, Hilden, Germany).
ELISA
For ELISAs, HER2‐domains (200 nM in PBS, 100 μL/well) were immobilized on MaxiSorp plates (Thermo Scientific, Zug, Switzerland) by overnight incubation at 4°C. Wells were blocked with 300 μL of PBSTB (PBS, 0.1% Tween‐20, 0.2% BSA) for 1 h at room temperature. Dilutions of purified DARPin constructs were incubated with the target domains for 1 h at 4°C, followed by three washing steps with 300 μL of ice‐cold PBSTB. For detection of bound DARPin constructs, a polyclonal rabbit anti‐DARPin serum (Dreier et al., unpublished) was added (1:5,000 in PBSTB, 1 h at 4°C). After incubation with a secondary anti rabbit‐IgG antibody alkaline phosphatase conjugate (Sigma Aldrich, Buchs, Switzerland) (1:10,000 in PBSTB, 1 h at 4°C) and washing, pNPP substrate (Fluka, Buchs, Switzerland) was added to measure alkaline phosphatase activity.
Supporting information
Supporting Information.
Acknowledgments
Flow cytometry was performed with equipment of the Flow Cytometry Facility, University of Zurich.
A.P. is a co‐founder and shareholder of Molecular Partners AG, which commercializes the DARPin technology.
Statement of Importance HER2/hErbB2 is a particularly potent member of the ErbB family of surface receptors, which are directly involved in the genesis of various types of cancer. We explore here experimentally whether an alternative three‐dimensional arrangement of the extracellular part of the protein might exist. The absence of such a conformer, found for all other members of the ErbB family, might be an important factor contributing to the exceptional malignant potential of HER2, with implications for the design of therapeutics.
References
- 1. Baselga J, Swain SM (2009) Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer 9:463–475. [DOI] [PubMed] [Google Scholar]
- 2. Mi L‐Z, Lu C, Li Z, Nishida N, Walz T, Springer TA (2011) Simultaneous visualization of the extracellular and cytoplasmic domains of the epidermal growth factor receptor. Nat Struct Mol Biol 18:984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Jr , Leahy DJ (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421:756–760. [DOI] [PubMed] [Google Scholar]
- 4. Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX (2004) Insights into ErbB signaling from the structure of the ErbB2‐pertuzumab complex. Cancer Cell 5:317–328. [DOI] [PubMed] [Google Scholar]
- 5. Garrett TPJ, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Kofler M, Jorissen RN, Nice EC, Burgess AW, Ward CW (2003) The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell 11:495–505. [DOI] [PubMed] [Google Scholar]
- 6. Bostrom J, Yu S‐F, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, Fuh G (2009) Variants of the antibody Herceptin that interact with HER2 and VEGF at the antigen binding site. Science 323:1610–1614. [DOI] [PubMed] [Google Scholar]
- 7. Fisher RD, Ultsch M, Lingel A, Schaefer G, Shao L, Birtalan S, Sidhu SS, Eigenbrot C (2010) Structure of the complex between HER2 and an antibody paratope formed by side chains from tryptophan and serine. J Mol Biol 402:217–229. [DOI] [PubMed] [Google Scholar]
- 8. Graus‐Porta D, Beerli RR, Daly JM, Hynes NE (1997) ErbB‐2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burgess AW, Cho H‐S, Eigenbrot C, Ferguson KM, Garrett TPJ, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S (2003) An open‐and‐shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell 12:541–552. [DOI] [PubMed] [Google Scholar]
- 10. Ferguson KM, Berger MB, Mendrola JM, Cho H‐S, Leahy DJ, Lemmon MA (2003) EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell 11:507–517. [DOI] [PubMed] [Google Scholar]
- 11. Cho H‐S, Leahy DJ (2002) Structure of the extracellular region of HER3 reveals an interdomain tether. Science 297:1330–1333. [DOI] [PubMed] [Google Scholar]
- 12. Bouyain S, Longo PA, Li S, Ferguson KM, Leahy DJ (2005) The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc Natl Acad Sci USA 102:15024–15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walker F, Orchard SG, Jorissen RN, Hall NE, Zhang H‐H, Hoyne PA, Adams TE, Johns TG, Ward C, Garrett TPJ, Zhu HJ, Nerrie M, Scott AM, Nice EC, Burgess AW (2004) CR1/CR2 interactions modulate the functions of the cell surface epidermal growth factor receptor. J Biol Chem 279:22387–22398. [DOI] [PubMed] [Google Scholar]
- 14. Mattoon D, Klein P, Lemmon MA, Lax I, Schlessinger J (2004) The tethered configuration of the EGF receptor extracellular domain exerts only a limited control of receptor function. Proc Natl Acad Sci USA 101:923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kovacs E, Zorn JA, Huang Y, Barros T, Kuriyan J (2015) A structural perspective on the regulation of the EGF receptor. Annu Rev Biochem 84:739–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pines G, Huang PH, Zwang Y, White FM, Yarden Y (2010) EGFRvIV: a previously uncharacterized oncogenic mutant reveals a kinase autoinhibitory mechanism. Oncogene 29:5850–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu P, Bouyain S, Eigenbrot C, Leahy DJ (2012) The ErbB4 extracellular region retains a tethered‐like conformation in the absence of the tether. Protein Sci 21:152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eigenbrot C, Ultsch M, Dubnovitsky A, Abrahmsén L, Härd T (2010) Structural basis for high‐affinity HER2 receptor binding by an engineered protein. Proc Natl Acad Sci USA 107:15039–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alvarado D, Klein DE, Lemmon MA (2009) ErbB2/HER2/Neu resembles an autoinhibited invertebrate EGF receptor. Nature 461:287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Citri A, Skaria KB, Yarden Y (2003) The deaf and the dumb: the biology of ErbB‐2 and ErbB‐3. Exp Cell Res 284:54–65. [DOI] [PubMed] [Google Scholar]
- 21. Franco‐Gonzalez JF, Cruz VL, Ramos J, Martínez‐Salazar J (2013) Conformational flexibility of the ErbB2 ectodomain and trastuzumab antibody complex as revealed by molecular dynamics and principal component analysis. J Mol Model 19:1227–1236. [DOI] [PubMed] [Google Scholar]
- 22. Jost C, Plückthun A (2014) Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Curr Opin Struct Biol 27:102–112. [DOI] [PubMed] [Google Scholar]
- 23. Plückthun A (2015) Designed Ankyrin Repeat Proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol 55:489–511. [DOI] [PubMed] [Google Scholar]
- 24. Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Plückthun A (2013) Structural basis for eliciting a cytotoxic effect in HER2‐overexpressing cancer cells via binding to the extracellular domain of HER2. Structure 21:1979–1991. [DOI] [PubMed] [Google Scholar]
- 25. Tamaskovic R, Schwill M, Nagy‐Davidescu G, Jost C, Schaefer DC, Verdurmen WPR, Schaefer JV, Honegger A, Plückthun A (2016) Intermolecular biparatopic trapping of ErbB2 prevents compensatory activation of PI3K/AKT via RAS‐p110 crosstalk. Nat Commun 7:11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Menendez JA, Schroeder B, Peirce SK, Vellon L, Papadimitropoulou A, Espinoza I, Lupu R (2015) Blockade of a key region in the extracellular domain inhibits HER2 dimerization and signaling. J Natl Cancer Inst 107:djv090. [DOI] [PubMed] [Google Scholar]
- 27. Epa VC, Dolezal O, Doughty L, Xiao X, Jost C, Plückthun A, Adams TE (2013) Structural model for the interaction of a designed ankyrin repeat protein with the human epidermal growth factor receptor 2. PLoS One 8:e59163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steiner D, Forrer P, Plückthun A (2008) Efficient selection of DARPins with sub‐nanomolar affinities using SRP phage display. J Mol Biol 382:1211–1227. [DOI] [PubMed] [Google Scholar]
- 29. Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS (2006) Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24:79–88. [DOI] [PubMed] [Google Scholar]
- 30. Li MZ, Elledge SJ (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods 4:251–256. [DOI] [PubMed] [Google Scholar]
- 31. Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy‐Davidescu G, Dreier B, Schibli R, Binz HK, Waibel R, Plückthun A (2010) Efficient tumor targeting with high‐affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res 70:1595–1605. [DOI] [PubMed] [Google Scholar]
- 32. Fitzgerald DJ, Berger P, Schaffitzel C, Yamada K, Richmond TJ, Berger I (2006) Protein complex expression by using multigene baculoviral vectors. Nat Methods 3:1021–1032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.