

Abstract
The coronavirus nonstructural protein 3 (nsp3) is a multifunctional protein that comprises multiple structural domains. This protein assists viral polyprotein cleavage, host immune interference, and may play other roles in genome replication or transcription. Here, we report the solution NMR structure of a protein from the “SARS‐unique region” of the bat coronavirus HKU9. The protein contains a frataxin fold or double‐wing motif, which is an α + β fold that is associated with protein/protein interactions, DNA binding, and metal ion binding. High structural similarity to the human severe acute respiratory syndrome (SARS) coronavirus nsp3 is present. A possible functional site that is conserved among some betacoronaviruses has been identified using bioinformatics and biochemical analyses. This structure provides strong experimental support for the recent proposal advanced by us and others that the “SARS‐unique” region is not unique to the human SARS virus, but is conserved among several different phylogenetic groups of coronaviruses and provides essential functions.
Keywords: SARS‐unique domain, frataxin, double‐wing motif, NMR, coronavirus, protein functional annotation, viral protein, nonstructural protein
Short abstract
PDB Code(s): 5UTV
Introduction
Coronaviruses are single‐stranded, positive‐sense, enveloped RNA viruses that infect both humans and animals. Coronavirus infections have a range of severity and include upper and lower respiratory symptoms, with a low frequency of acute lung injury and acute respiratory distress syndrome.1 Acute gastrointestinal, hepatic, and neurological symptoms have also been observed.2 Since 2002, the human coronaviruses (CoVs) have emerged as significant public health threats. The severe acute respiratory syndrome (SARS) virus is the etiological agent of the 2003–2005 pandemic that affected more than 30 countries.3 In 2012, the Middle East respiratory syndrome (MERS) virus emerged in the Middle East, followed by the spread of the virus to other countries (e.g., the UK, South Korea). As of 2016, there had been 1728 confirmed cases of MERS affecting persons in 27 countries.4 Prior to these outbreaks, CoVs were known to be responsible for mild upper and lower respiratory infections. For example, human CoV 229E and OC43 cause a minority of respiratory tract infections.2 Based on phylogenetic and serological analyses, the International Committee for Taxonomy of Viruses has placed the CoVs in four genera, namely the Alphacoronaviruses, Betacoronaviruses, Gammacoronaviruses and Deltacoronaviruses.5 Under this classification, the betacoronavirus genus has been divided into groups a to d, whereby the SARS‐like CoVs are found in group B and MERS‐like CoVs in group C. The group D so far has been detected only in bats.6
Bats are reservoir hosts of multiple zoonotic viruses, including CoVs. Surveillance studies and phylogenetic analyses have shown that high genetic diversity exists among the SARS‐like viruses present in bats, allowing for the possibility of recombination and the evolution of new variants.7 A bat virus with 96% nucleotide sequence identity to the human SARS‐CoV was shown to be capable of using the human ACE2 enzyme as a receptor. This demonstrates the same mode of cell entry as the human SARS‐CoV.8 The bat SL‐CoV‐WIV1 could grow on human epithelial cells and Vero E6 cells, and was neutralized by human SARS convalescent sera. This virus is a possible direct progenitor of the human SARS‐CoV.8, 9
Several group c betacoronaviruses, such as the HKU4, HKU5, and PREDICT/PDF‐2180, have been identified in bats from distinct locations around the world. Some genome regions in these bat viruses are highly conserved with respect to the human MERS virus; for example, PREDICT/PDF‐2180 shares 97% sequence identity with the MERS virus in ORF1B.10 It is hypothesized that RNA recombination either in the bat or in an intermediate animal host gave rise to the MERS‐CoV.10 The HKU4 virus, which is derived from the lesser bamboo bat (Tylonycteris pachypus), shares 92.4% RNA polymerase, 67.4% spike protein, and 72.3% nucleocapsid amino acid identity with the MERS CoV and is able to use the same receptor for attachment and entry (the cell surface protein DPP4).11, 12 The group D betacoronavirus Hong Kong University 9 (HKU9) is also widely distributed, and has been detected in diverse species including Rousettus leschenaulti, Hipposidereos commersoni, Eidolon helvum, and Rousettus aegyptiacus from Asia to Africa.13, 14, 15, 16
Whether bat CoVs undergo adaptation to intermediate hosts, or are transmitted directly to humans, it is clear that they pose a threat to human health. Hence, it is imperative to understand bat CoV biochemical and biological functions. At present, only one high‐resolution structure of a BatCoV HKU9 protein domain is known, the spike protein external receptor‐binding domain (RBD).17 This structure revealed critical new information such as the external subdomain adopting a helical fold versus the beta‐sheet topology observed in other betaCoV receptor domains. As a result, the HKU9 RBD does not bind to the other betaCoV receptors, ACE2 and CD26, underlining the importance of carrying out structural studies on bat proteins. Hence, we have initiated a program to explore bat protein structure‐function relationships, with the goal of determining conserved versus divergent functions.
The CoV virion is composed of four structural proteins, which are believed to assist genome packaging, cell entry and virus spread.2 In contrast, the replicase gene directs the expression of two large nonstructural polyproteins, pp1a and pp1ab, that become mature nonstructural proteins (nsps) after cleavage by viral proteases. These proteins assemble into a replicase‐transcriptase complex (RTC) that is responsible for RNA genome replication, processing and transcription of sub‐genomic RNAs. Interference with the innate immune system, and other interactions with functions of the host cell also localize to the nsps. Several of these functions are essential for viral replication, growth and virulence.18, 19, 20, 21, 22, 23, 24, 25
The nonstructural protein 3 (nsp3) is a multifunctional protein consisting of sixteen functional domains and 1,922 amino acid residues.18, 21, 26, 27, 28, 29, 30, 31, 32 This protein is the largest component of the RTC. Nsp3 is one of the most divergent regions of the CoV genome.33 The domain structure of nsp3 is variable among CoVs,32 with one or two papain‐like cysteine proteases, transmembrane regions, RNA‐binding proteins, and one or more macrodomains.27, 34, 35 Key functions of the nsp3 include protein/protein interactions involved in replicase assembly and function;36 polyprotein processing by the papain‐like cysteine protease domain;37 and deubiquitinase activity involved in innate immune system interference.38 There are one or more macrodomains in the protein, for which roles in countering the host cell innate immunity have been demonstrated21, 39 and roles in viral RNA synthesis have been proposed.40 A “SARS‐unique region” with a three‐domain structure was identified in the nsp3 of SARS.35 The macrodomains in the SARS‐unique region were shown to be G‐quadruplex binding proteins, and to interact with the RCHY ubiquitin ligase to target p53 for degradation.35, 41, 42 The smaller C‐terminal domain in this region adopts a frataxin‐like fold and has been shown to bind purine‐rich RNA sequences.35 In the human SARS‐CoV, the functions of this region were essential for viral replication.43 However, based on discoveries since 2002 and the emergence of other viruses, it has been hypothesized that the “SARS‐unique region” is in fact conserved in other viruses, in particular in the group B, C, and D betacoronaviruses.
We are investigating the “SARS‐unique region” of bat CoVs. Here, we report the solution structure of the small C‐terminal domain of this region, which we term HKU9 C. We describe for the first time the structural and functional analysis of a nonstructural protein domain from the betacoronavirus lineage D. We also discuss the conserved elements of the nsp3 C domain compared to other proteins in the frataxin fold family; including a possible functional site that is conserved relative to the human SARS‐CoV.
Results
NMR structure determination
NMR experiments were performed with uniformly 15N,13C‐labeled HKU9 C expressed and purified from E. coli. The construct used contains the entire predicted C domain spanning the residues 573–646 of the nonstructural protein 3 (nsp3), with an additional N‐terminal segment Ser‐His‐Met derived from fusion tag cleavage. These residues correspond to the residues 1345–1418 of the replicase polyprotein 1ab of BatCoV HKU9 (Uniprot ID: P0C6W5). The numbering differs because the viral polyprotein is cleaved by the viral protease PLpro to yield the mature viral nsp3.36, 44, 45, 46 We use the numbering of the mature nsp3 herein. Multidimensional NMR experiments were performed to assign 96% of the observable resonances of the peptide backbone and amino acid sidechains. All backbone 15N and 1HN resonances were assigned (Fig. 1). The structure determination was carried out based on 3D 15N‐ and 13C‐resolved [1H,1H]‐NOESY experiments that were analyzed with the J‐UNIO suite of programs.47
Figure 1.
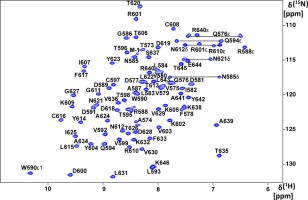
2D [15N,1H]‐HSQC spectrum of the HKU9 C domain. 1.2 mM 15N‐labeled HKU9 C in a 20 mM sodium phosphate (pH = 6.0), 150 mM NaCl, 3% d10‐DTT, 0.02% (w/v) NaN3 solution was measured on a Bruker Avance III 600MHz spectrometer. Backbone 15N–1H correlation peaks are indicated by single letter amino acid nomenclature. Arg and Trp assigned 15N‐1Hε correlation peaks are labeled. The amide side chain 15N–1H2 signals from Asn and Gln are shown with horizontal lines
Table 1 displays the statistics of the structure calculation, indicating a high‐quality structure determination. A dense network of long‐range NOEs was observed and the sequential and medium‐range NOE pattern was consistent with the secondary structures in the protein (Supporting Information Fig. S1). The ensemble of 20 conformers representing the solution structure of the HKU9 C domain (RMSD 0.34 Å) is well‐defined with the exception of the N‐terminal expression tag residues Ser −3 and His −2, and the C‐terminal residue Lys 646.
Table 1.
Input for the Structure Calculation of HKU9 C and the Statistics of the 20 Energy‐Minimized Conformers Used to Represent the Solution Structure
| Quantity | Value |
|---|---|
| NOE upper distance limits | 1828 |
| Intraresidue (|i – j| = 0) | 311 |
| Sequential (|i – j| = 1) | 528 |
| Medium‐range (1<|i – j| < 5) | 329 |
| Long‐range (|i – j| ≥ 5) | 660 |
| Dihedral angle constraints | |
| Talos + | 132 |
| HABAS (CYANA) | 347 |
| NOEs per residue | 23.74 |
| Long‐Range NOEs per residue | 8.57 |
| CYANA minimized target function | 1.70 ± 0.40 |
| Residual NOE Violations | |
| Number ≥ 0.2 Å | 6 |
| RMS violation | 0.0214 |
| Residual dihedral angle violations | |
| Number ≥ 5.0° | 1 |
| RMS violation | 0.3553 |
| RMSD from Ideal Geometrya | |
| Bond Lengths, Å | 0.016 |
| Bond Angles, ° | 2.8 |
| RMSD to the mean coordinates, Åa | |
| Backbone (574–645)b | 0.34 ± 0.11 |
| Heavy Atom (574–645)b | 0.72 ± 0.08 |
| Ramachandran plot statisticsa | |
| Most favored regions (%) | 90.2 |
| Allowed regions (%) | 6.1 |
| Disallowed regions (%) | 3.7 |
As determined by MOLPROBITY [81]. Calculated using PSVS version 1.5 [78].
Residue range used to calculate the backbone and heavy‐atom RMSDs.
Solution structure of the HKU9 C domain
A fold consisting of six β‐strands arranged in an antiparallel β‐sheet, together with two α‐helices at the N‐ and C‐termini that pack on one side of the sheet is observed (Fig. 2). The fold is described as a double‐wing motif or frataxin‐like fold48 and is classified as similar to the N‐terminal domain of CyaY, a bacterial regulatory protein.49 The helices rest in the same plane antiparallel to each other and contribute to one side of the hydrophobic core [Fig. 2(A)]. The two helices, α1 and α2, are comprised of residues 574–585 and 636 − 644, respectively. The first beta strands β1 (591 − 592) and β2 (596 − 599) follow an extended loop after α1 and lead to the first β hairpin. The remaining beta strands β3–β6 span the residues 602 − 609, 613 − 616, 622 − 626, and 629 − 632 forming a curved β‐sheet. The topology of the frataxin fold is shown in Figure 2(C).
Figure 2.
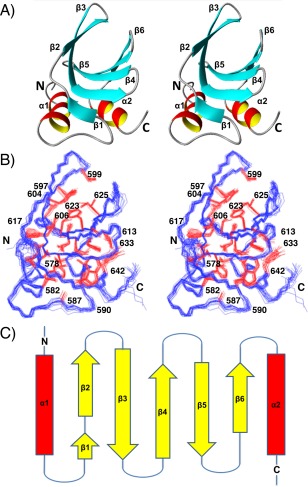
NMR solution structure of the HKU9 C domain. Wall‐eye stereo views are shown. (A) Ribbon representation of the representative conformer (nearest to the mean coordinates of the ensemble). Secondary structures are labeled. (B) Line representation of the 20‐conformer ensemble. The polypeptide backbone (blue) and selected side chains with solvent accessibility below 15% (red) are shown. (C) Topology diagram of HKU9‐C. The α‐helices (red) are indicated by rectangles and the β‐sheets (yellow) are indicated by arrows
The hydrophobic core is primarily defined by residues from the α‐helices and β‐strands [Fig. 2(B)]. The side chains from Val 575, Phe 578, Val 579, and Ile 582 in α1 and Val 636, Ala 639, Tyr 642, and Leu 643 in α2 encompass the α‐helix contribution to the hydrophobic core. The side chains from Cys 597 and Val 599 in β2; Tyr 604, Thr 606, Ile 607, and Cys 608 in β3; Thr 613, Leu 615, Cys 616, and Phe 617 in β4; and Leu 622, Tyr 623, Ala 624, and Ile 625 in β5 additionally contribute to the hydrophobic core together with Gly 586, Ala 587, Trp 590, Asp 618, Asn 621, and Phe 633 located in loop regions.
Functional analysis and predictions
Structural alignment of HKU9 C to other proteins using the programs TM‐Align50 and Dali51 revealed structural similarity to betacoronavirus (β‐CoV) C domains, frataxins, and hypothetical proteins (Table 2A). The most structurally similar proteins originate from other β‐CoV C domains, namely those of the human SARS‐CoV and murine hepatitis virus (MHV) C.30 The HKU9 C fold is similar to these viral domains, with a similar topology and overall backbone RMSD values of 1.7 Å and 2.2 Å, respectively. These viral domains have conserved residues and a highly similar fold despite their low sequence identity. Similarity to the frataxins is also evident, with RMSD values of approximately 3 Å and 1–10% sequence identity. These proteins also show slightly different topologies, with longer loops and secondary structure insertions between several secondary structure elements [Fig. 3(C)].
Table 2.
Bioinformatics Results from HKU9 C Structure‐Based Alignment and Protein Function Prediction
| Identifier | Protein Name | RMSD | %ID | ||
|---|---|---|---|---|---|
| A | |||||
| TM‐Align a | PDB ID: 2KAF | SARS nsp3 C domain | 1.66 | 18 | |
| PDB ID: 4YPT | MHV nsp3 domain | 2.21 | 15 | ||
| PDB ID: 1KAF | Phage T4 MotA | 2.84 | 7 | ||
| PDB ID: 1YB3 | P. furiosus 178653–001 | 3.12 | 1 | ||
| DALI b | PDB ID: 2KAF | SARS nsp3 C domain | 1.8 | 18 | |
| PDB ID: 4YPT | MHV nsp3 domain | 2.3 | 15 | ||
| PDB ID: 1KAF | Phage T4 MotA | 2.7 | 9 | ||
| PDB ID: 4HS5 | P. ingrahamii FTXN | 2.8 | 9 | ||
| PDB ID: 3T3X | Friedreich's Ataxia FTXN | 2.9 | 6 | ||
| PDB ID: 1YB3 | P. furiosus 178653–001 | 3.3 | 1 | ||
| Server Prediction | Structurally Similar Proteins | Alignment Scoresd | ||||
|---|---|---|---|---|---|---|
| Ligand | Binding Sitec | C | BS | |||
| B | ||||||
| COFACTOR | DNA | β | Ring 1B/Bmi1/UbcH5c PRC1 (4R8P) | 0.01 | 0.86 | |
| Peptide | α/β | Glycyl‐tRNA Synthetase (1ATI) | 0.01 | 0.77 | ||
| Cyanocobalamin (Vitamin B12) | CF | Glycerol Dehydratase (1MMF) | 0.01 | 0.20 | ||
| ATP | β | Human Glycyl‐tRNA synthetase (2ZT7) | 0.01 | 0.15 | ||
| C | ||||||
| COACH |
Calcium ions Zinc ions |
α α |
E. coli ROM variant (1F4M) Acyl Carrier Protein (2QNW) |
0.12 0.11 |
||
| “GAANDENY” | CF | AAA+ delivery protein (1OU8) | 0.09 | |||
| “QRKWYPLRP” | CF | KnI1/NsI1 complex (4NF9) | 0.05 | |||
| Peptide | α/β | Glycyl‐tRNA Synthetase (1ATI) | 0.04 | |||
| DNA | β | Ring 1B/Bmi1/UbcH5c PRC1 (4R8P) | 0.02 | |||
| Serotonin | α | AM182 Serotonin Complex (3BRN) | 0.02 | |||
| TM‐Site | Zinc(II) ions | α | Ferric Enterobactin (2CHU) | 0.37 | ||
| Apcin | β | Cdc20 (4N14) | 0.18 | |||
| FINDSITE | “GAANDENY” | CF | AAA+ delivery protein (1OU8) | 0.27 | ||
| N‐acetyl‐mannosamine | α | L‐Ficolin protein (2J0G) | 0.13 | |||
| Serotonin | α | AM182 Serotonin Complex (3BRN) | 0.06 | |||
TM‐Align[50] scales structure similarity to protein templates from residue‐specific alignment.
DALI[51] server uses structure‐based templates to provide matches in structure and function.
Predicted binding regions of the HKU9 C protein are described: residues from the α‐helices (α), residues from the solvent‐accessible β‐sheet (β), and the conserved polar face containing the Arg‐Asp‐Trp and Lys‐Arg‐Gly motif (CF).
The confidence score (C‐score) is used to evaluate the reliability of the prediction. The binding site score (BS‐score) evaluates how significant is the match between the predicted binding site and the template binding site. Alignment score values range from 0 to 1, with higher values having greater significance.
Figure 3.
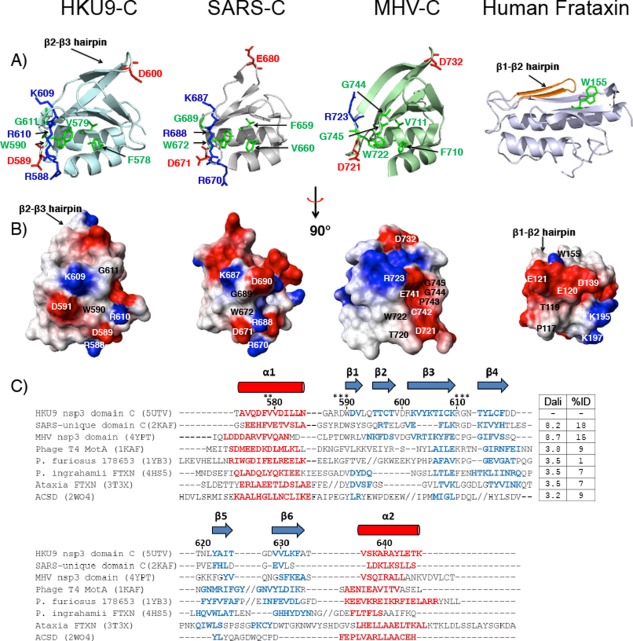
(A) Ribbon representations of nonstructural protein 3 C domains. Conserved residues or conservative substitutions relative to HKU9 are highlighted in green (aliphatic), blue (basic), and red (acidic). Residue numbers are indicated with respect to the first residue in each protein. Left to right: HKU9 C, SARS SUD‐C (PDB ID: 2KAF),35 MHV C domain (PDB ID: 4YPT),30 human frataxin (PDB ID: 3T3X).52 (B) C domain electrostatic potential surfaces. Red areas represent positively charged regions, blue areas represent negatively charged areas, and white areas represent neutral areas. (C) Structure‐based sequence alignment of the C domain in the Rousettus bat coronavirus HKU9, related viral proteins, and with other proteins in the frataxin fold family: phage T4 MotA (PDB ID: 1KAF),53 hypothetical protein (PDB ID: 1YB3), Psychromonas ingrahamii FTXN (PDB ID: 4HS5),52 Ataxia FTXN (PDB ID: 3T3X),54 AcsD (PDB ID: 2W04).55 The alignment is based on structural alignments obtained with TM‐Align.50 PDB codes are included after each protein name. The residue numbers for HKU9‐C are indicated. Alpha helix regions are displayed in red (cylinders) and beta strands are shown in blue (arrows). Gaps are shown as dashes (‐) and insertions where additional secondary structures are present are indicated by forward slash marks (//). Residues indicated by stars (*) discussed in the text are involved in potential functional sites. The corresponding Dali scores for the pairwise alignment of each protein with HKU9 C and the percent amino acid identity between each protein and HKU9 C domain are listed. Dali scores of 2.0 and higher indicate significant sequence identity51
Functional predictions of HKU9 C were based on an analysis of β‐CoV C domain structure‐function relationships, together with COACH meta‐server results.56 COACH creates a complementary profile and binding site prediction from TM‐SITE and S‐SITE and utilizes multiple structure‐based programs (COFACTOR, FINDSITE, and Concavity)56 to derive ligand binding predictions. We used this consensus server approach to predict functional characteristics of HKU9‐C (Table 2B).
Based on similarities to the human SARS‐CoV C domain, a possible function for HKU9 C is nucleic acid binding.35 To investigate this possibility, we conducted electrophoretic mobility shift assays (EMSA) with a panel of RNA and DNA oligonucleotides including purine‐rich, pyrimidine‐rich and G‐quadruplex sequences. However, no oligonucleotide binding was detected. A second possibility is that HKU9 C functions in concert with the neighboring macrodomains, which are binding proteins and enzymes acting on ADP‐ribose and related metabolites.57, 58, 59 Structural similarity and binding site similarity to adenylate‐binding proteins is also present. Chemical shift perturbation analysis was employed by titrating to 20 times the protein concentration of ADP and ADP‐ribose, which are known ligands for macrodomains.57 No chemical shift changes or line broadening in the [15N,1H]‐HSQC spectrum were observed, indicating no interactions or complexes formed.
Functional predictions based on binding site analysis suggested other possible ligands. To investigate, chemical shift perturbation experiments were repeated with cyanocobalamin (vitamin B12), zinc (II) ions, EDTA, and peptides. Again, no changes in the spectrum were observed, suggesting other likely functions for HKU9 C.
Discussion
Conservation of the SARS‐unique region in betacoronaviruses
The structure determination of HKU9 C revealed unexpected structural similarity with the corresponding SARS‐unique domain in the human SARS‐CoV. These two sequences share only 18% sequence similarity. An area of strong conservation is present around the residues Arg 588–Trp 590 in the loop joining α1 to β1, where the residues are conserved [indicated by stars, Fig. 3(C)] and the protein surfaces have similar polar character [Fig. 3(A,B)]. Additional similarity is present around the residues Lys 609–Gly 611. In particular, the residues Arg 588–Asp 589 –Trp 590 in HKU9 and the residues Arg 670–Asp 671–Trp 672 in SARS adopt nearly identical side chain orientations (Fig. 4). This suggests a possible conserved function between the two viruses. We describe this surface as the conserved face (CF) of the protein. This surface is defined by the loop connecting α1 and β1 and the beta turn between β3 and β4, near the C‐terminus of the protein.
Figure 4.
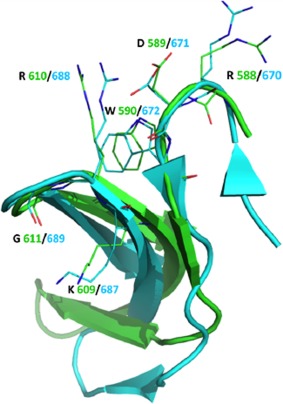
Overlay of HKU9‐C (green) and SARS‐C (cyan) backbone with secondary structures shown. Sidechains from Arg 588‐Asp 589‐Trp 590, and Lys 609‐Arg 610‐Gly 611 (HKU9 C) and Arg 670‐Asp 671‐Trp 672, and Lys 687‐Arg 688‐Gly 689 (SARS C) are shown with the corresponding one‐letter amino acid code
In contrast, the corresponding region of the MHV C domain has acidic and hydrophobic character [Fig. 3(A,B)]. This is a consequence of the substitution of the sequence Arg 588–Asp 589–Trp 590 by Thr–Asp–Trp and Lys 609–Arg 610–Gly 611 by Glu‐Cys‐Pro. Since the three proteins share a low level of overall sequence identity (15–18%), this difference would not have been apparent without a structural comparison.
This structure represents clear evidence that the SARS‐unique domain is also conserved in bat CoVs. The overall structural similarity between the HKU9 C domain and the SARS C domain, from betacoronavirus lineage B, was assessed by the program DALI.51 The resulting RMSD value was 1.66 Å with a DALI score of 8.2, indicating a strong match. The RMSD value for the MHV C domain from the betacoronavirus lineage A was 2.16 Å, with a DALI score of 8.7. Since the β‐CoV HKU9 belongs to lineage D, this analysis reinforces the hypothesis advanced by us and others that the unique region of SARS nsp3 is actually conserved across multiple β‐CoVs.30, 32, 43, 60, 61 A structure‐based sequence alignment of the C domains and related proteins is shown in Figure 3(C). Residues such as Phe 578, Val 581, and Trp 590 are conserved when compared to the sequence and structure of the SARS and MHV orthologues. In contrast, Trp 590 is replaced by other aromatic or hydrophobic residues in the frataxins. Based on their low solvent accessibility, we conclude that these residues are likely to be important in stabilizing the fold, rather than for intermolecular interactions. However, other conserved residues that contribute to the surface potential such as the side chains of Arg 588, Asp 589, Lys 609, Arg 610, and Gly 611, described above, that are oriented to the same face of the protein, are likely to be responsible for a shared function between the CoV groups 2b and 2d (Fig. 3). Correspondingly, these residues are not conserved throughout the protein family.
The sequence alignment of Figure 3(C) reveals the conserved topology in the frataxin fold family. It also reveals differences between the viral domains and more distantly related proteins. The viral proteins retain a similar sequence length, have conserved residues in both helices and the β‐sheet, and align with high DALI scores of 8.2 and above, indicating a strong match. These features are not conserved in the distantly related frataxin‐like folds. For example, the adenylate‐binding AcsD domain (PDB ID: 2W04)55 has an extended loop with an alpha turn insertion between β1 and β2 and another long loop between β3 and β4. DALI scores for the alignment of the HKU9 C domain to the human frataxin (PDB: 3T3X) and to the bacterial frataxin (PDB: 4HS5) are 3.8 and 3.5, respectively. These scores are also significant (>2.0) and indicate a conserved fold, but with some structural variability.51 This is underscored by the presence of structural insertions relative to the viral proteins.
Possible functions of the SARS‐unique region in BatCoV HKU9
We employed bioinformatics analysis with the COACH56 meta‐server to predict possible functions for the bat CoV HKU9. Several possible functions emerged from this analysis. One possible function is as a nucleic acid‐binding protein, predicted by the COACH and COFACTOR62 servers (Table 2) with low confidence score values of 0.02 and 0.01. This is also highlighted by the sequence and structure alignment of the SARS and HKU9 C domains. Several residues involved in the binding of SARS C to RNA are conserved in HKU9‐C.35 The RNA‐binding residues from the SARS‐CoV protein, such as His 695 (β5−β6 loop), Gly 707 (β6−β7 loop) and Val 709 (β7strand), align to Phe 617 (β4−β5 loop), Gly 627 (β5−β6 loop), and Val 630 (β6) in HKU9 C [Fig. 3(C)]. Additionally, a distantly related viral frataxin, the C terminal domain of the T4 activator MotA, (PDB ID: 1KAF),53 binds an E. coli DNA promoter sequence. The MotACTD double‐wing β‐sheet utilizes asparagine residues to bind DNA. These residues are not conserved; for instance, one (Asn 187) aligns to Gly 627 in HKU9 C [Fig. 3(C)]. Consistent with this lack of conservation, no nucleic acid binding was observed for HKU9 C. However, it is possible that this function is present but requires the presence of neighboring nsp3 domains.
A second possible function for the HKU9 C domain is protein/protein interaction. In the SARS‐CoV, the SUD region interacts with host cell proteins to enhance p53 degradation.41 The frataxins also have protein binding partners, where the interaction is mediated by side chains that are exposed on the planar face of the β‐sheet. It is notable that in the viral proteins, the β‐sheet face is smaller and less planar than that of the frataxins. The latter proteins have the β1−β2 hairpin in the same plane as the β‐sheet [Fig. 3(A)]. However, in the β‐CoV domains, the β1−β2 hairpin wraps over the β‐sheet, obscuring the side chains in β2−β6 from the protein surface. In addition, the β‐sheet side chains that are important to frataxin binding and catalysis such as Trp 155 and Arg 165 (human) or Arg 53 and Trp 61 (Psychromonas ingrahamii) are not conserved in the β‐CoV proteins.52, 54
Protein or peptide binding is another function that is predicted by the bioinformatic analysis of HKU9 C (Table 2). A potential protein‐binding site is predicted to be present on the conserved face (CF) of the protein. The site shows structural and chemical similarity to that of the AAA+ delivery protein63 and the Nsl1 protein.64 The surface identified by this prediction includes the residues Arg 588–Asp 589–Trp 590 and Lys 609 – Arg 610 – Gly 611 that we have identified as a conserved functional site.
Analysis of the bioinformatics results displays a theme with respect to HKU9 C surface regions. The conserved face of the fold [Fig. 3(B)] is the only region of the protein that was predicted to have protein‐protein interactions, while the other surface regions predicted metal ion and small molecule recognition [Table 2(B)]. The β‐sheet sidechains are not solvent exposed, but side chains from the β2−β3 and β4−β5 loops and from the β6 strand could potentially bind small molecules. Interestingly, the metal ion ligands such as Ca2+ and Zn2+ were predicted to bind to the α‐helices. To date, we were not able to experimentally confirm any metal ion binding activity or nucleic acid binding activity for HKU9 C. The prediction of a possible protein/protein interaction function is intriguing and awaits further experiment.
We hypothesize that the conserved face of the HKU9 C domain is a likely interface for HKU9 C binding partners. Based on our FFAS (Fold and Function Assignment System) analysis,65 and on the experimental results reported here, we predict structural conservation between the nsp3 proteins of the human SARS‐CoV and bat HKU9. We used this structural alignment to predict the linker regions that would join the HKU9 C domain to the neighboring domains in nsp3. At the N‐terminus of HKU9 C, a short, three‐residue linker is predicted to join the domain to the neighboring M domain; while at the C‐terminus, on the “conserved face” of the protein, a seven‐residue linker joins the C domain to the papain‐like protease of the virus.36, 44, 66 A longer linker would provide flexibility to accommodate binding partners and interactions. This would coincide with our hypothesis that the conserved face of the protein near the C‐terminus may harbor a potential functional site for reactivity or binding to other biomolecules.
Conclusions
The frataxin or double‐wing fold of the bat HKU9 nsp3 C domain reported here has high structural similarity to the human SARS‐CoV C domain. Although there is low sequence similarity to the other CoV nsp3 proteins, some residues are structurally conserved. The conservation of specific surface polar residues relative to the human SARS virus may indicate a conserved function among certain betacoronaviruses.
Materials and Methods
Protein expression and purification
The DNA sequence encoding the central region of nsp3 (37 − 1037) was obtained as a codon‐optimized synthetic gene from Genscript (Piscataway, NJ). The residues 573 − 646 of nsp3, corresponding to the recombinant HKU9 C domain, were cloned into the vector pET‐15b‐TEV67 vector from the Northeast Structural Genomics Consortium (DNASU). The construct was expressed in E. coli strain BL21 (DE3) with a 6xHis tag. The Ser‐His‐Met sequence at the N‐terminus of the proteins remained after tag cleavage with the tobacco etch virus protease. The sample was prepared in both LB medium for natural isotopic abundance and in minimal medium for uniform 15N‐ and 13C‐labeling. These samples were used for functional analysis and structure determination, respectively. Sample conditions such as buffer, pH, and salt concentration were optimized based on peak intensity and linewidth in the [15N,1H]‐HSQC spectrum, leading to the selection of 20 mM sodium phosphate (pH 6.0), 150 mM NaCl, and 5 mM DTT. The protein was monomeric as assessed by size‐exclusion chromatography on a GE Healthcare HiLoad 26/600 SuperdexTM 200 pg column.
NMR spectroscopy
The C domain structure was determined based on multidimensional NMR experiments using uniformly 15N‐ or [15N, 13C]‐labeled protein solution with 97% H2O/3% D2O (v/v). All experiments were conducted on Bruker Avance III HD spectrometers (600 and 850 MHz) equipped with Bruker 5 mm TCI cryoprobes and on a Bruker Avance II 700 MHz spectrometer equipped with a CP TCI H‐C/N‐D cryoprobe. The sequence‐specific backbone assignments were based on 3D HNCACB, CBCA(CO)NH, HNCA, HNHA, and HNCO experiments. Aliphatic and aromatic side chain assignments were determined using the ASCAN68 protocol in the J‐UNIO47 suite of programs followed by interactive correction and completion using 3D CC(CO)NH, HBHA(CO)NH, (HB)CB(CGCD)HD‐COSY, (HB)CB(CGCDCE)HE‐COSY, 3D HC(C)H‐TOCSY, 15N‐resolved [1H,1H]‐NOESY (τ m =150 ms), 13C‐resolved aliphatic [1H,1H]‐NOESY (τ m =150 ms), and 13C‐resolved aromatic [1H,1H]‐NOESY (τ m =150 ms) experiments.69 All assignments were verified manually using the CARA and CCPNmr Analysis programs.70, 71 1H chemical shifts were calibrated from internal 3‐(trimethylsilyl)propane‐1‐sulfonic acid (DSS) and 15N and 13C shifts were referenced indirectly.72
The ATNOS and CANDID73, 74 algorithms in the J‐UNIO suite were used to pick the NOESY spectra and to calculate the structure of the C domain. A globular fold obtained after the first cycle remained consistent throughout the calculation with a steady decrease in RMSD of the ensemble. A set of 132 tight dihedral angle restraints were obtained from the program Talos+.75 A set of loose ϕ, ψ, and χ1 restraints produced by the HABAS algorithm in CYANA 2.076 based on intraresidual and sequential NOEs provided an additional 347 dihedral angle restraints for the structure calculation.73, 74 The set of unambiguous NOE assignments obtained in the final cycle of calculation included 1828 restraints or 24 restraints/residue (Table 1). The 20 structures with the lowest CYANA target function values in cycle 7 were further refined by explicit solvent minimization using the AMBER03 force field in explicit solvent (TIP3PBOX) with a 10 Å box geometry using the web‐based AMBER interface AMPS‐NMR in the WeNMR portal.77
Structure validation of the final ensemble employed the Protein Data Bank validation suite, MolMol 2K.2, and ProCheck 3.5.4 from the PSVS suite 1.5.78, 79, 80, 81 Validation also employed input and output of the Unio calculation and agreement of the structure with NMR observables (Supporting Information Table S2).
The atomic coordinates of the ensemble of conformers of Figure 2(B) have been deposited in the Protein Data Bank with accession number 5UTV. The sequence‐specific resonance assignments have been deposited in the BioMagResBank with accession number 30247.
Chemical shift perturbation experiments with BatCoV C domain
NMR titrations were conducted by diluting the protein sample with NMR buffer consisting of 20 mM sodium phosphate (pH 6.0), 150 mM NaCl, 3% D2O (v/v), 5 mM DTT‐d10, and 0.02% NaN3, to a final concentration of 50 μM. The potential ligands were dissolved in NMR buffer at a 20 mM final concentration. Titrations were conducted by measuring NMR [15N,1H]‐HSQC spectra at increasing ligand:protein concentration ratios. An initial measurement without ligand present and with 2048 (1H) × 256 (15N) points was taken as a baseline. The ratios of ligand to protein concentration were 0.25:1, 0.5:1, 1.0:1, 5.0:1, 10.0:1, 20.0:1 for cyanocobalamin, ZnCl2, ADP, ADP‐ribose, and 10:1 and 20:1 for EDTA.
Electrophoretic mobility shift assays
Binding assays were conducted by incubating purified HKU9 C protein with a set of DNA and RNA oligonucleotides available in our laboratory for studying protein‐nucleic acid interactions. Protein‐oligonucleotide mixtures were incubated at 25°C for 1 h in EMSA buffer: 20 mM sodium phosphate (pH 6.0), 75 mM NaCl, and 3% glycerol. G‐quadruplex oligomers were annealed by heating to 95°C for 5 min and slowly cooling to 18°C overnight in buffer: 20 mM sodium phosphate (pH 6.0), 75 mM NaCl. The mixtures were resolved by native electrophoresis on 10% TBE gels (Invitrogen) for 1 h at 4°C. Gels were stained with SYBR Gold stain (Invitrogen) and visualized by the Safe Imager 2.0 Blue‐Light Transilluminator (Invitrogen).
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
Supporting Information Figure 1.
Supporting Information Table 1.
Supporting Information Table 2.
Acknowledgments
We thank Chong Tian, Pamlea N. Brady and members of the Johnson laboratory for technical assistance.
Significance: The three‐dimensional structure of a protein in the SARS‐unique region of the bat coronavirus HKU9 (Hong Kong University 9) was solved by NMR. The structure is highly similar to that of the human severe acute respiratory syndrome (SARS) coronavirus. This may indicate conserved functions among animal and human viruses. The fold reveals a potential functional site. This represents the first structure of a domain from a bat coronavirus HKU9 nonstructural protein.
References
- 1. Gralinski LE, Baric RS (2015) Molecular pathology of emerging coronavirus infections. J Pathol 235:185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fehr AR, Perlman S (2015) Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol 1282:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peiris JS, Guan Y, Yuen KY (2004) Severe acute respiratory syndrome. Nat Med 10:S88–S97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Wit E, van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: Recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(ICTV) ICoToV. Available from: https://talk.ictvonline.org/taxonomy/
- 6. Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, Wong BH, Gao K, Tsoi HW, Huang Y, Li KS, Lam CS, Chan KH, Zheng BJ, Yuen KY (2007) Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol 81:1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF (2005) Bats are natural reservoirs of SARS‐like coronaviruses. Science 310:676–679. [DOI] [PubMed] [Google Scholar]
- 8. Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang YJ, Luo CM, Tan B, Wang N, Zhu Y, Crameri G, Zhang SY, Wang LF, Daszak P, Shi ZL (2013) Isolation and characterization of a bat SARS‐like coronavirus that uses the ACE2 receptor. Nature 503:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang XL, Hu B, Wang B, Wang MN, Zhang Q, Zhang W, Wu LJ, Ge XY, Zhang YZ, Daszak P, Wang LF, Shi ZL (2015) Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J Virol 90:3253–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anthony SJ, Gilardi K, Menachery VD, Goldstein T, Ssebide B, Mbabazi R, Navarrete‐Macias I, Liang E, Wells H, Hicks A, Petrosov A, Byarugaba DK, Debbink K, Dinnon KH, Scobey T, Randell SH, Yount BL, Cranfield M, Johnson CK, Baric RS, Lipkin WI, Mazet JA (2017) Further evidence for bats as the evolutionary source of Middle East respiratory syndrome coronavirus. MBio 8:e00373–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Anthony SJ, Ojeda‐Flores R, Rico‐Chavez O, Navarrete‐Macias I, Zambrana‐Torrelio CM, Rostal MK, Epstein JH, Tipps T, Liang E, Sanchez‐Leon M, Sotomayor‐Bonilla J, Aguirre AA, A, Flores R, Medellin RA, Goldstein T, Suzan G, Daszak P, Lipkin WI (2013) Coronaviruses in bats from Mexico. J Gen Virol 94:1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lau SK, Li KS, Tsang AK, Lam CS, Ahmed S, Chen H, Chan KH, Woo PC, Yuen KY (2013) Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J Virol 87:8638–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lau SK, Poon RW, Wong BH, Wang M, Huang Y, Xu H, Guo R, Li KS, Gao K, Chan KH, Zheng BJ, Woo PC, Yuen KY (2010) Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel betacoronavirus subgroup. J Virol 84:11385–11394. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Tao Y, Tang K, Shi M, Conrardy C, Li KS, Lau SK, Anderson LJ, Tong S (2012) Genomic characterization of seven distinct bat coronaviruses in Kenya. Virus Res 167:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwanda B, Breiman RF, Anderson LJ, Rupprecht CE (2009) Detection of novel SARS‐like and other coronaviruses in bats from Kenya. Emerg Infect Dis 15:482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ge X, Li Y, Yang X, Zhang H, Zhou P, Zhang Y, Shi Z (2012) Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J Virol 86:4620–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang C, Qi J, Lu G, Wang Q, Yuan Y, Wu Y, Zhang Y, Yan J, Gao GF (2016) Putative receptor binding domain of bat‐derived coronavirus HKU9 spike protein: evolution of betacoronavirus receptor binding motifs. Biochemistry 55:5977–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hurst KR, Koetzner CA, Masters PS (2013) Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase‐transcriptase complex. J Virol 87:9159–9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hurst KR, Ye R, Goebel SJ, Jayaraman P, Masters PS (2010) An interaction between the nucleocapsid protein and a component of the replicase‐transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J Virol 84:10276–10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fehr AR, Channappanavar R, Jankevicius G, Fett C, Zhao J, Athmer J, Meyerholz DK, Ahel I, Perlman S (2016) The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome coronavirus infection. MBio 7:e01721–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fehr AR, Athmer J, Channappanavar R, Phillips JM, Meyerholz DK, Perlman S (2015) The nsp3 macrodomain promotes virulence in mice with coronavirus‐induced encephalitis. J Virol 89:1523=1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang R, Li Y, Cowley TJ, Steinbrenner AD, Phillips JM, Yount BL, Baric RS, Weiss SR (2015) The nsp1, nsp13, and M proteins contribute to the hepatotropism of murine coronavirus JHM.WU. J Virol 89:3598–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zust R, Cervantes‐Barragan L, Kuri T, Blakqori G, Weber F, Ludewig B, Thiel V (2007) Coronavirus non‐structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog 3:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Case JB, Ashbrook AW, Dermody TS, Denison MR (2016) Mutagenesis of S‐adenosyl‐l‐methionine‐binding residues in coronavirus nsp14 N7‐methyltransferase demonstrates differing requirements for genome translation and resistance to innate immunity. J Virol 90:7248–7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mielech AM, Deng X, Chen Y, Kindler E, Wheeler DL, Mesecar AD, Thiel V, Perlman S, Baker SC (2015) Murine coronavirus ubiquitin‐like domain is important for papain‐like protease stability and viral pathogenesis. J Virol 89:4907–4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ (2013) Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double‐membrane vesicles. MBio 4:e00524–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oostra M, Hagemeijer MC, van Gent M, Bekker CP, te Lintelo EG, Rottier PJ, de Haan CA (2008) Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J Virol 82:12392–12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P (2005) Structural basis of severe acute respiratory syndrome coronavirus ADP‐ribose‐1″‐phosphate dephosphorylation by a conserved domain of nsP3. Structure 13:1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bailey‐Elkin BA, Knaap RC, Johnson GG, Dalebout TJ, Ninaber DK, van Kasteren PB, Bredenbeek PJ, Snijder EJ, Kikkert M, Mark BL (2014) Crystal structure of the Middle East respiratory syndrome coronavirus (MERS‐CoV) papain‐like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J Biol Chem 289:34667–34682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Y, Savinov SN, Mielech AM, Cao T, Baker SC, Mesecar AD (2015) X‐ray structural and functional studies of the three tandemly linked domains of non‐structural protein 3 (nsp3) from murine hepatitis virus reveal conserved functions. J Biol Chem 290:25293–25306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hurst‐Hess KR, Kuo L, Masters PS (2015) Dissection of amino‐terminal functional domains of murine coronavirus nonstructural protein 3. J Virol 89:6033–6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neuman BW, Joseph JS, Saikatendu KS, Serrano P, Chatterjee A, Johnson MA, Liao L, Klaus JP, Yates JR, 3rd , Wuthrich K, Stevens RC, Buchmeier MJ, Kuhn P (2008) Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J Virol 82:5279–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perlman S, Holmes K (2007) The Nidoviruses: Toward Control of SARS and Other Nidovirus Diseases. Springer Science & Business Media, New York. [Google Scholar]
- 34. Sulea T, Lindner HA, Purisima EO, Menard R (2005) Deubiquitination, a new function of the severe acute respiratory syndrome coronavirus papain‐like protease?. J Virol 79:4550–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnson MA, Chatterjee A, Neuman BW, Wuthrich K (2010) SARS coronavirus unique domain: three‐domain molecular architecture in solution and RNA binding. J Mol Biol 400:724–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Masters PS (2006) The molecular biology of coronaviruses. Adv Virus Res 66:193–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Graham RL, Denison MR (2006) Replication of murine hepatitis virus is regulated by papain‐like proteinase 1 processing of nonstructural proteins 1, 2, and 3. J Virol 80:11610–11620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mielech AM, Chen Y, Mesecar AD, Baker SC (2014) Nidovirus papain‐like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res 194:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuri T, Eriksson KK, Putics A, Zust R, Snijder EJ, Davidson AD, Siddell SG, Thiel V, Ziebuhr J, Weber F (2011) The ADP‐ribose‐1″‐monophosphatase domains of severe acute respiratory syndrome coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J Gen Virol 92:1899–1905. [DOI] [PubMed] [Google Scholar]
- 40. Malet H, Coutard B, Jamal S, Dutartre H, Papageorgiou N, Neuvonen M, Ahola T, Forrester N, Gould EA, Lafitte D, Ferron F, Lescar J, Gorbalenya AE, de Lamballerie X, Canard B (2009) The crystal structures of Chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J Virol 83:6534–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ma‐Lauer Y, Carbajo‐Lozoya J, Hein MY, Muller MA, Deng W, Lei J, Meyer B, Kusov Y, von Brunn B, Bairad DR, Hunten S, Drosten C, Hermeking H, Leonhardt H, Mann M, Hilgenfeld R, von Brunn A (2016) p53 down‐regulates SARS coronavirus replication and is targeted by the SARS‐unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc Natl Acad Sci U S A 113:E5192–E5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chatterjee A, Johnson MA, Serrano P, Pedrini B, Joseph JS, Neuman BW, Saikatendu K, Buchmeier MJ, Kuhn P, Wuthrich K (2009) Nuclear magnetic resonance structure shows that the severe acute respiratory syndrome coronavirus‐unique domain contains a macrodomain fold. J Virol 83:1823–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kusov Y, Tan J, Alvarez E, Enjuanes L, Hilgenfeld R (2015) A G‐quadruplex‐binding macrodomain within the “SARS‐unique domain” is essential for the activity of the SARS‐coronavirus replication‐transcription complex. Virology 484:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prentice E, McAuliffe J, Lu X, Subbarao K, Denison MR (2004) Identification and characterization of severe acute respiratory syndrome coronavirus replicase proteins. J Virol 78:9977–9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang X, Chen X, Bian G, Tu J, Xing Y, Wang Y, Chen Z (2014) Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain‐like protease. J Gen Virol 95:614–626. [DOI] [PubMed] [Google Scholar]
- 46. Kilianski A, Mielech AM, Deng X, Baker SC (2013) Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain‐like and 3C‐like proteases using luciferase‐based biosensors. J Virol 87:11955–11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Serrano P, Pedrini B, Mohanty B, Geralt M, Herrmann T, Wuthrich K (2012) The J‐UNIO protocol for automated protein structure determination by NMR in solution. J Biomol NMR 53:341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bencze KZ, Kondapalli KC, Cook JD, McMahon S, Millan‐Pacheco C, Pastor N, Stemmler TL (2006) The structure and function of frataxin. Crit Rev Biochem Mol Biol 41:269–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cho SJ, Lee MG, Yang JK, Lee JY, Song HK, Suh SW (2000) Crystal structure of Escherichia coli CyaY protein reveals a previously unidentified fold for the evolutionarily conserved frataxin family. Proc Natl Acad Sci U S A 97:8932–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y, Skolnick J (2005) TM‐align: a protein structure alignment algorithm based on the TM‐score. Nucleic Acids Res 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bridwell‐Rabb J, Winn AM, Barondeau DP (2011) Structure‐function analysis of Friedreich's ataxia mutants reveals determinants of frataxin binding and activation of the Fe‐S assembly complex. Biochemistry 50:7265–7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li N, Sickmier EA, Zhang R, Joachimiak A, White S (2002) The MotA transcription factor from bacteriophage T4 contains a novel DNA‐binding domain: the ‘double wing’ motif. Mol Microbiol 43:1079–1088. [DOI] [PubMed] [Google Scholar]
- 54. di Maio D, Chandramouli B, Yan R, Brancato G, Pastore A (2017) Understanding the role of dynamics in the iron sulfur cluster molecular machine. Biochim Biophys Acta 1861:3154–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schmelz S, Kadi N, McMahon SA, Song L, Oves‐Costales D, Oke M, Liu H, Johnson KA, Carter LG, Botting CH, White MF, Challis GL, Naismith JH (2009) AcsD catalyzes enantioselective citrate desymmetrization in siderophore biosynthesis. Nat Chem Biol 5:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang J, Roy A, Zhang Y (2013) Protein‐ligand binding site recognition using complementary binding‐specific substructure comparison and sequence profile alignment. Bioinformatics 29:2588–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Karras GI, Kustatscher G, Buhecha HR, Allen MD, Pugieux C, Sait F, Bycroft M, Ladurner AG (2005) The macro domain is an ADP‐ribose binding module. EMBO J 24:1911–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Neuvonen M, Ahola T (2009) Differential activities of cellular and viral macro domain proteins in binding of ADP‐ribose metabolites. J Mol Biol 385:212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen D, Vollmar M, Rossi MN, Phillips C, Kraehenbuehl R, Slade D, Mehrotra PV, von Delft F, Crosthwaite SK, Gileadi O, Denu JM, Ahel I (2011) Identification of macrodomain proteins as novel O‐acetyl‐ADP‐ribose deacetylases. J Biol Chem 286:13261–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Johnson MA, Goel A, Chan M, Tan X, Hammond R (2015) Computational and experimental studies of mono‐ and poly‐ADP‐ribosylation of peptides. American Chemical Society. SERMACS‐SWRM‐719.
- 61. Chan M, Hammond R, Tian C, Tan X, Goel A, Johnson MA (2015) Characterization and biochemical analysis of noncanonical coronavirus macrodomains. American Chemical Society. SERMACS‐SWRM‐779.
- 62. Roy A, Yang J, Zhang Y (2012) COFACTOR: an accurate comparative algorithm for structure‐based protein function annotation. Nucleic Acids Res 40:W471–W477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Levchenko I, Grant RA, Wah DA, Sauer RT, Baker TA (2003) Structure of a delivery protein for an AAA+ protease in complex with a peptide degradation tag. Mol Cell 12:365–372. [DOI] [PubMed] [Google Scholar]
- 64. Petrovic A, Mosalaganti S, Keller J, Mattiuzzo M, Overlack K, Krenn V, De Antoni A, Wohlgemuth S, Cecatiello V, Pasqualato S, Raunser S, Musacchio A (2014) Modular assembly of RWD domains on the Mis12 complex underlies outer kinetochore organization. Mol Cell 53:591–605. [DOI] [PubMed] [Google Scholar]
- 65. Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A (2011) FFAS server: novel features and applications. Nucleic Acids Res 39:W38–W44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD (2006) Severe acute respiratory syndrome coronavirus papain‐like protease: structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci U S A 103:5717–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Acton TB, Gunsalus KC, Xiao R, Ma LC, Aramini J, Baran MC, Chiang Y‐W, Climent T, Cooper B, Denissova NG, Douglas SM, Everett JK, Ho CK, Macapagal D, Rajan PK, Shastry R, Shih L‐y, Swapna GVT, Wilson M, Wu M, Gerstein M, Inouye M, Hunt JF, Montelione GT (2005) Robotic cloning and protein production platform of the northeast structural genomics consortium. Methods Enzymol. 394:210–243. [DOI] [PubMed] [Google Scholar]
- 68. Fiorito F, Herrmann T, Damberger FF, Wuthrich K (2008) Automated amino acid side‐chain NMR assignment of proteins using (13)C‐ and (15)N‐resolved 3D [(1)H, (1)H]‐NOESY. J Biomol NMR 42:23–33. [DOI] [PubMed] [Google Scholar]
- 69. Sattler M (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Magn Reson Spectrosc 34:93–158. [Google Scholar]
- 70. Keller RLJ. (2005) Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment. [Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment]. [Zürich].
- 71. Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59:687–696. [DOI] [PubMed] [Google Scholar]
- 72. Wishart D, Bigam C, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley J, Sykes B (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 6:135–140. [DOI] [PubMed] [Google Scholar]
- 73. Herrmann T, Güntert P, Wüthrich K (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319:209–227. [DOI] [PubMed] [Google Scholar]
- 74. Herrmann T, Güntert P, Wüthrich K (2002) Protein NMR structure determination with automated NOE‐identification in the NOESY spectra using the new software ATNOS. J Biomol NMR 24:171–189. [DOI] [PubMed] [Google Scholar]
- 75. Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guentert P, Braun W, Billeter M, Wuethrich K (1989) Automated stereospecific proton NMR assignments and their impact on the precision of protein structure determinations in solution. J Am Chem Soc 111:3997–4004. [Google Scholar]
- 77. Bertini I, Case DA, Ferella L, Giachetti A, Rosato A (2011) A Grid‐enabled web portal for NMR structure refinement with AMBER. Bioinformatics 27:2384–2390. [DOI] [PubMed] [Google Scholar]
- 78. Bhattacharya A, Tejero R, Montelione GT (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66:778–795. [DOI] [PubMed] [Google Scholar]
- 79. Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14:51–5; 29–32. [DOI] [PubMed] [Google Scholar]
- 80. Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291. [Google Scholar]
- 81. Chen VB, Arendall WB, 3, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Cryst D66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Table 1.
Supporting Information Table 2.