

Abstract
Aminoglycoside antibiotics represent a classical group of antimicrobials first discovered in the 1940s. Due to their ototoxic and nephrotoxic side effects, they are typically only used against Gram negative bacteria which have become resistant to other therapeutics. One family of aminoglycosides includes such compounds as butirosin, ribostamycin, neomycin, and kanamycin, amongst others. The common theme in these antibiotics is that they are constructed around a chemically stable aminocyclitol unit referred to as 2‐deoxystreptamine (2‐DOS). Four enzymes are required for the in vivo production of 2‐DOS. Here, we report the structure of RbmB from Streptomyces ribosidificus, which is a pyridoxal 5′‐phosphate dependent enzyme that catalyzes two of the required steps in 2‐DOS formation by functioning on distinct substrates. For this analysis, the structure of the external aldimine form of RbmB with 2‐DOS was determined to 2.1 Å resolution. In addition, the structure of a similar enzyme, BtrR from Bacillus circulans, was also determined to 2.1 Å resolution in the same external aldimine form. These two structures represent the first detailed molecular descriptions of the active sites for those aminotransferases involved in 2‐DOS production. Given the fact that the 2‐DOS unit is widespread amongst aminoglycoside antibiotics, the data presented herein provide new molecular insight into the biosynthesis of these sugar‐based drugs.
Keywords: aminoglycoside–aminocyclitol antibiotics, aminotransferase, Bacillus circulans, butirosin, 2‐deoxystreptamine, protein structure, ribostamycin, Streptomyces ribosidificus
Short abstract
Abbreviations
- IPTG
isopropyl β‐d‐1‐thiogalactopyranoside
- MOPS
3‐(N‐morpholino)propanesulfonic acid
- Ni‐NTA
nickel nitrilotriacetic acid
- PCR
polymerase chain reaction
- PLP
pyridoxal 5'‐phosphate
- PMP
pyridoxamine 5'‐phosphate
- Tris
tris‐(hydroxymethyl)aminomethane.
Introduction
According to Dr. Thomas R. Frieden, former director of the US Centers for Disease Control and Prevention, the alarming rise of antibiotic resistance could bring about the “next pandemic.” Without question, antibiotic‐resistant strains of bacteria are a significant threat to human existence. The main methods that bacteria utilize to confer resistance to antibiotics include modifying the drug, physically removing it from the cell, or altering the target site so that it is no longer recognized by the antimicrobial agent.1 Many antibiotics, such as penicillin, cephalosporin, vancomycin, and erythromycin A, amongst others, are derived from natural products. Indeed, these compounds, isolated from plants, bacteria, fungi, and many marine organisms have dramatically changed the course of human history. The “mining” of unusual organisms such as those that grow at high temperatures, in sewers, or in jungles, for example, has led to a dazzling array of important therapeutic compounds
As with any use of antibiotics, however, bacterial resistant strains arise. It is thus imperative to stay “ahead of the game” by developing new therapeutics. Within recent years it has become apparent that microbial genome mining is playing a key role in the discovery of novel compounds with antibiotic and antitumor activities.2, 3, 4, 5 In particular, bacteria belonging to the genus Streptomyces are a known prolific source of natural compounds. On average, a Streptomyces genome contains ∼30 secondary metabolite gene clusters.5
The focus of this investigation is on the pyridoxal 5'‐phosphate (PLP)‐dependent aminotransferase, RbmB, involved in the biosynthesis of ribostamycin derived from Streptomyces ribosidificus.6 As shown in Scheme 1, ribostamycin, a broad spectrum aminoglycoside antibiotic, consists of three groups: 2‐deoxystreptamine (2‐DOS), d‐neosamine, and d‐ribose. The 2‐DOS core is a common aglycone observed in various antibiotics including kanamycin, neomycin, gentamicin, and butirosin, amongst others.7
Scheme 1.
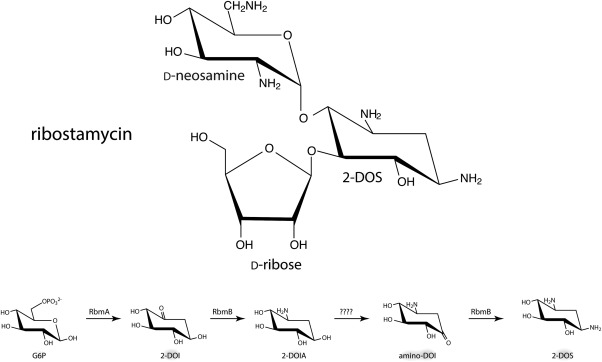
The structure of ribostamycin and the biochemical steps leading to the formation of 2‐DOS.
Formation of the 2‐DOS core initiates with RbmA catalyzing the conversion of glucose‐6‐phosphate to 2‐deoxy‐scyllo‐inosose (2‐DOI) as indicated in Scheme 1. RbmB catalyzes the next step in the pathway leading to the formation of 2‐deoxy‐scyllo‐inosamine (2‐DOIA). The enzyme responsible for the conversion of 2‐DOIA to 3‐amino‐2‐deoxy‐scyllo‐inosose (amino‐DOI) has not yet been identified in S. ribosidificus, but has been studied in other aminoglycoside‐producing bacteria.7, 8 Finally, RbmB catalyzes the second amination reaction in the pathway to yield the 2‐DOS core. It thus functions on both 2‐DOI and amino‐DOI. As noted above, RbmB is a PLP‐dependent enzyme. Typically, these enzymes bind the PLP cofactor via a Schiff base with a conserved lysine residue, which is referred to as the internal aldimine. A transimination reaction occurs when an amino‐containing substrate displaces the conserved lysine thereby leading to the so‐called external aldimine.
Here, we describe the three‐dimensional structure of RbmB in its external aldimine form with 2‐DOS. In addition, we report the structure of a similar enzyme, BtrR from Bacillus circulans, also in complex with its external aldimine. The results reported herein provide new details into substrate binding amongst these “dual functional” aminotransferases.
Results and Discussion
The structure of RbmB in its external aldimine form was solved to 2.1 Å resolution and refined to an overall R‐factor of 18.5%. Shown in Figure 1(a) is a ribbon representation of the RbmB dimer. The total buried surface area is extensive at ∼5600 Å2. Each subunit consists of 12 α‐helices and 16 β‐strands, and the active sites are separated by ∼28 Å. The RbmB architecture places it into the well characterized fold type I or aspartate aminotransferase family.9 The α‐carbons for the two subunits in the dimer superimpose with a root‐mean‐square deviation of 0.2 Å for 386 target pairs.
Figure 1.

Structure of RbmB from S. ribosidificus. Shown in (a) is a ribbon representation of the dimer. The external aldimines are displayed in space‐filling representations. The electron density corresponding to the bound external aldimine in subunit 2 is presented in (b). The electron density map was calculated with (F o – F c) coefficients and contoured at 4σ. The ligands were not included in the X‐ray coordinate file used to calculate the omit map, and thus there is no model bias. A close‐up view of the active site with the bound external aldimine is depicted in stereo in (c). Possible hydrogen bonding interactions within 3.2 Å are indicated by the dashed lines. Ordered water molecules are displayed as red spheres. Those residues highlighted in yellow belong to subunit 1 of the dimer (labels are marked by asterisks).
The electron densities corresponding to the external aldimines in both subunits were unambiguous. Shown in Figure 1(b) is the observed electron density found in subunit 2. As indicated in Figure 1(c), the 2‐DOS entity is flanked on one side by the indole ring of Trp 102 and on the other side by Lys 202. The aromatic side chains of Tyr 311 and Tyr 346 project toward the 2‐DOS moiety but they do not interact with it. The side chain of Lys 202 lies within 3.2 Å of the C‐6 hydroxyl whereas the side chain of Arg 231 from the other subunit bridges the C‐4 and C‐5 hydroxyls. In the internal aldimine state, the side chain of Lys 202 forms a Schiff base with the PLP cofactor. The C‐3 amino group is surrounded only by two water molecules. Those side chains that lie within hydrogen bonding distance to the PLP group include Thr 77, Asp 173, and Ser 197 from subunit 2 and Asn 255 from subunit 1. Hydrogen bonds also occur between the backbone amide nitrogens of Gly 76 and Thr 77 and two phosphoryl oxygens of the cofactor. Three water molecules complete the hydrogen bonding pattern around the phosphoryl group.
In 2006, the crystal structures of the PLP‐ and pyridoxamine 5'‐phosphate (PMP)‐bound forms of BtrR from B. circulans were reported.10 This particular enzyme is involved in the biosynthesis of butirosin, and its substrates are identical to those of RbmB. Curious as to whether BtrR also binds the external aldimine in a similar manner, we determined its structure in such a complex. The model for the BtrR/external aldimine complex was refined at 2.1 Å resolution to an overall R‐factor of 19.2%. Shown in Figure 2(a) is the electron density corresponding to the bound external aldimine in subunit 2. Only PLP was observed binding in subunit 1. The α‐carbons for BtrR and RbmB superimpose with a root‐mean‐square deviation of 0.8 Å for 358 target pairs. A superposition of the BtrR and RbmB active sites is presented in Figure 2(b), and as can be seen they are remarkably similar. The only minor difference is the replacement of Ser 67 in BtrR with Thr 77 in RbmB. It was suggested in the original study of BtrR that the side chain of Trp 92 would have to flip in order to accommodate substrate binding. The authors were absolutely correct as shown in Figure 2(c). Indeed, the Cη2 carbon of the Trp 92 indole ring moves by ∼5 Å upon external aldimine binding.
Figure 2.
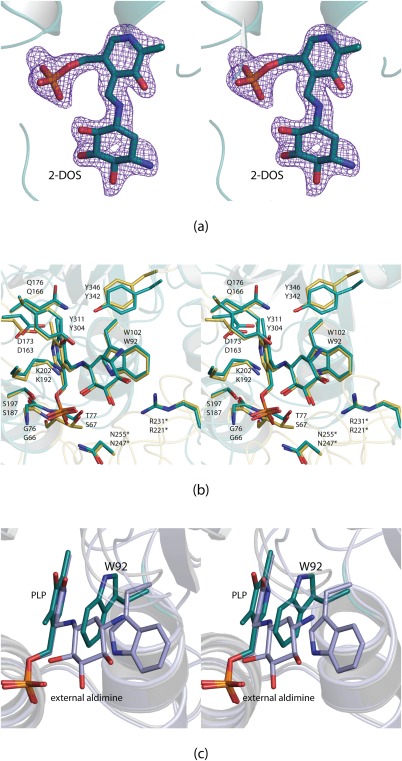
Structure of BtrR from B. circulans. Electron density corresponding to the bound external aldimine is shown in stereo in (a). It was calculated as described in the legend to Figure 1 and contoured at 4σ. A superposition of the BtrR and RbmB active sites is presented in (b). The RbmB and BtrR models are colored in yellow and teal, respectively. The top labels correspond to those amino acids in RbmB whereas the bottom labels refer to those found in BtrR. In the first structural analysis of BtrR, it was predicted that Trp 92 would have to swing away from the PLP cofactor.10 The hypothesis was, indeed, correct as shown in stereo in (c). The structure of BtrR with bound PLP is highlighted in teal whereas the structure of BtrR determined in this investigation is displayed in light blue.
The manner in which both BtrR and RbmB can accommodate the external aldimine form with 2‐DOS has now been defined. However, how these enzymes bind 2‐DOIA (Scheme 1) in the external aldimine form is still unknown. Clearly, the structure analysis of either of these enzymes in an external aldimine form with 2DOIA would address this essential question. This work is in progress as we attempt to produce a soluble form of RbmA (Scheme 1) that can ultimately be utilized to produce the required reagents for this aspect of the investigation. Regardless, the research presented herein reports the first three‐dimensional models for aminotransferases in their external aldimine forms that are involved in 2‐DOS biosynthesis. Given the number of 2‐DOS‐containing aminoglycoside antibiotics that have been discovered to date, the data provide new insight into 2‐DOS biosynthesis on a molecular level.11
Materials and Methods
Cloning of the genes encoding RbmB and BtrR
Genomic DNA from S. ribosidificus (NRRL B‐11466) served as the polymerase chain reaction (PCR) template for gene amplification of rbmB. For the analysis of the B. circulans BtrR, a synthetic gene was constructed on the basis of its amino acid sequence. The gene was prepared for Escherichia coli optimized codon usage by Integrated DNA Technologies. The genes encoding RbmB or BtrR were ligated into pET28t vectors that ultimately generated proteins with N‐terminal His6‐tags.12
Protein expression and purification
The pET28t‐rbmB or pET28t‐btrR plasmids were used to transform Rosetta2(DE3) E. coli cells (Novagen). The cultures were grown in lysogeny broth supplemented with kanamycin (50 mg/L) and chloramphenicol (50 mg/L) at 37°C with shaking until an optical density of 0.8 was reached at 600 nm. The flasks were cooled in an ice bath, and the cells were induced with 1 mM isopropyl β‐d‐1‐thiogalactopyranoside and allowed to express protein at 21°C for 24 h.
The cells were harvested by centrifugation and disrupted by sonication on ice. The lysate was cleared by centrifugation, and the recombinant proteins were purified utilizing nickel nitrilotriacetic acid resin (Qiagen) according to the manufacturer's instructions. The purified proteins were dialyzed against 10 mM Tris‐HCl (pH 8.0) and 200 mM NaCl and concentrated to 15 mg/mL based on an extinction coefficient of 1.14 (mg/mL)−1 cm−1 for RbmB and 25 mg/mL based on an extinction coefficient of 0.73 (mg/mL)−1 cm−1 for BtrR.
Crystallization
Crystallization conditions were surveyed by the hanging drop method of vapor diffusion using a laboratory‐based sparse matrix screen. The enzymes were tested for crystallization properties in the presence of 1 mM PLP and 5 mM 2‐DOS. X‐ray diffraction quality crystals of RbmB were grown from precipitant solutions composed of 24%–26% poly(ethylene glycol) 3350, 100 mM MgCl2, and 100 mM 3‐(N‐morpholino)propanesulfonic acid (MOPS) (pH 7.0). The crystals belonged to the tetragonal space group P41212 with unit cell dimensions of a = b = 101.1 Å, and c = 160.0 Å. The asymmetric unit contained one dimer. The crystals were prepared for X‐ray data collection by serially transferring them to a cryoprotectant solution composed of 30% poly(ethylene glycol) 3350, 200 mM NaCl, 100 mM MgCl2, 1 mM PLP, 5 mM 2‐DOS, 10% ethylene glycol, and 100 mM MOPS (pH 7.0).
X‐ray diffraction quality crystals of BtrR were grown from precipitant solutions composed of 15–17% poly(ethylene glycol) 5000, 200 mM NaCl, and 100 mM MOPS (pH 7.0). The crystals belonged to the orthorhombic space group P212121 with unit cell dimensions of a = 69.4 Å, b = 74.8 Å, and c = 180.1 Å. The asymmetric unit contained one dimer. The crystals were prepared for X‐ray data collection by serially transferring them to a cryoprotectant solution composed of 22% poly(ethylene glycol) 5000, 300 mM NaCl, 1 mM PLP, 5 mM 2‐DOS, 16% ethylene glycol, and 100 mM MOPS (pH 7.0).
X‐ray data collection and processing
All X‐ray data sets were collected in house using a Bruker AXS Platinum 135 CCD detector controlled with the PROTEUM software suite (Bruker AXS Inc.) The X‐ray source was Cu Kα radiation from a Rigaku RU200 X‐ray generator equipped with Montel optics and operated at 50 kV and 90 mA. The data sets were processed with SAINT and scaled with SADABS (Bruker AXS Inc.). Relevant X‐ray data collection statistics are listed in Table 1.
Table 1.
X‐ray Data Collection and Model Refinement Statistics
| RbmB from S. ribosidificus | BtrR from B. circulans | |
|---|---|---|
| Resolution limits (Å) | 50.0–2.1 (2.2–2.1)a | 50.0–2.1 (2.2–2.1)a |
| Number of independent reflections | 48736 (6185) | 54841 (6774) |
| Completeness (%) | 99.2 (98.2) | 98.7 (95.0) |
| Redundancy | 8.3 (4.1) | 6.3 (3.1) |
| avg I/avg σ(I) | 10.3 (2.8) | 7.4 (2.1) |
| R sym (%)b | 9.2 (37.5) | 9.6 (38.2) |
| c R‐factor (overall)%/no. reflections | 18.5/45736 | 19.2/54841 |
| R‐factor (working)%/no. reflections | 18.2/46279 | 19.0/52130 |
| R‐factor (free)%/no. reflections | 23.3/2457 | 23.9/2711 |
| Number of protein atoms | 6174 | 6504 |
| Number of heteroatoms | 398 | 427 |
| Average B values | ||
| Protein atoms (Å2) | 20.7 | 23.2 |
| Ligand (Å2) | 15.2 | 18.6 |
| Solvent (Å2) | 20.3 | 23.7 |
| Weighted RMS deviations from ideality | ||
| Bond lengths (Å) | 0.012 | 0.014 |
| Bond angles (°) | 2.1 | 1.7 |
| Planar groups (Å) | 0.006 | 0.008 |
| Ramachandran regions (%)d | ||
| Most favored | 97.3 | 96.9 |
| Additionally allowed | 2.5 | 2.8 |
| Generously allowed | 0.1 | 0.2 |
Statistics for the highest resolution bin.
R sym = (∑|I – |/∑I)⋅× 100.
R‐factor = (Σ|F o – F c|/Σ|F o|) × 100 where F o is the observed structure‐factor amplitude and F c is the calculated structure‐factor amplitude.
Distribution of Ramachandran angles according to PROCHECK.17
The structure of RbmB was solved by molecular replacement with PHASER13 using as the search probe the coordinates of a previously determined version of BtrR (PDB 2C7T).10 Iterative cycles of model building with COOT14, 15 and refinement with REFMAC16 reduced the R work and R free to 18.2% and 23.3%, respectively, from 50 to 2.1 Å resolution.
The BtrR structure was determined by molecular replacement with PHASER. Iterative cycles of model building with COOT and refinement with REFMAC reduced the R work and R free to 19.0% and 23.9%, respectively, from 50 to 2.1 Å resolution.
Model refinement statistics for both structures are listed in Table 1.
The authors have no competing financial interests.
X‐ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, N. J. (accession nos. 5W70 and 5W71).
References
- 1. Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ (2015) Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol 13:42–51. [DOI] [PubMed] [Google Scholar]
- 2. Bachmann BO, Van Lanen SG, Baltz RH (2014) Microbial genome mining for accelerated natural products discovery: is a renaissance in the making? J Ind Microbiol Biotechnol 41:175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Monciardini P, Iorio M, Maffioli S, Sosio M, Donadio S (2014) Discovering new bioactive molecules from microbial sources. Microb Biotechnol 7:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rutledge PJ, Challis GL (2015) Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol 13:509–523. [DOI] [PubMed] [Google Scholar]
- 5. Ziemert N, Alanjary M, Weber T (2016) The evolution of genome mining in microbes—a review. Nat Prod Rep 33:988–1005. [DOI] [PubMed] [Google Scholar]
- 6. Subba B, Kharel MK, Lee HC, Liou K, Kim BG, Sohng JK (2005) The ribostamycin biosynthetic gene cluster in Streptomyces ribosidificus: comparison with butirosin biosynthesis. Mol Cells 20:90–96. [PubMed] [Google Scholar]
- 7. Kudo F, Eguchi T (2016) Aminoglycoside antibiotics: new insights into the biosynthetic machinery of old drugs. Chem Rec 16:4–18. [DOI] [PubMed] [Google Scholar]
- 8. Kudo F, Eguchi T (2009) Biosynthetic enzymes for the aminoglycosides butirosin and neomycin. Methods Enzymol 459:493–519. [DOI] [PubMed] [Google Scholar]
- 9. Schneider G, Kack H, Lindqvist Y (2000) The manifold of vitamin B6 dependent enzymes. Structure 8:R1–R6. [DOI] [PubMed] [Google Scholar]
- 10. Popovic B, Tang X, Chirgadze DY, Huang F, Blundell TL, Spencer JB (2006) Crystal structures of the PLP‐ and PMP‐bound forms of BtrR, a dual functional aminotransferase involved in butirosin biosynthesis. Proteins 65:220–230. [DOI] [PubMed] [Google Scholar]
- 11. Kudo F, Eguchi T (2009) Biosynthetic genes for aminoglycoside antibiotics. J Antibiot (Tokyo) 62:471–481. [DOI] [PubMed] [Google Scholar]
- 12. Thoden JB, Holden HM (2005) The molecular architecture of human N‐acetylgalactosamine kinase. J Biol Chem 280:32784–32791. [DOI] [PubMed] [Google Scholar]
- 13. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 15. Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum‐likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. [DOI] [PubMed] [Google Scholar]
- 17. Laskowski RA, Moss DS, Thornton JM (1993) Main‐chain bond lengths and bond angles in protein structures. J Mol Biol 231:1049–1067. [DOI] [PubMed] [Google Scholar]