

Summary
Yellow fever 17D vaccine is one of the oldest live-attenuated vaccines in current use that is recognized for historically immunogenic and safe properties. These unique properties of 17D are presently exploited in rationally designed recombinant vaccines targeting not only flaviviral antigens but also other pathogens of public health concern. Several candidate vaccines based on 17D have advanced to human trials, and a chimeric recombinant Japanese encephalitis vaccine utilizing the 17D backbone has been licensed. The mechanism(s) of attenuation for 17D are poorly understood; however, recent insights from large in silico studies have indicated particular host genetic determinants contributing to the immune response to the vaccine, which presumably influences the considerable durability of protection, now in many cases considered to be life-long. The very rare occurrence of severe adverse events for 17D is discussed, including a recent fatal case of vaccine-associated viscerotropic disease.
Keywords: Yellow Fever Virus, Flavivirus, Vaccine, Empiric, Primary Seed, Secondary Seed, Live-Attenuated, Recombinant Vaccine, Empiric Vaccine, Rational Vaccine, Viral Diversity
Historical Significance
Live-attenuated vaccines to prevent yellow fever (YF) were among the first to be successfully developed by empiric serial passage, and as such occupy a place of historical significance in the study and control of tropical diseases. YF is noted in colonial-era literature as a febrile disease of the tropics, from which patients would suffer fever, and jaundice, eventually succumbing to hemorrhage disease[1]. YF outbreaks were closely recorded in the late 1700s through early 1900s, in which the disease phenotype and eventually the complex mosquito-primate transmission cycle became described. Notably, losses to the disease stemming from American military deployments to Cuba during the Spanish-American War and construction of the Panama Canal prompted close study of the disease in the late nineteenth century [2]. The World Health Organization (WHO) recommended case definition for suspected YF disease specifies the observation of jaundice within two weeks of acute fever [3]. After a resolution phase in which symptoms decrease in severity, a minority of cases (12%) may advance to a toxic phase, featuring liver damage and hemorrhage, for which the mortality rate has been recently estimated to be 47 percent [4].
YFV is the Prototype Member of the Family Flaviviridae
Yellow fever virus (YFV) is the causative agent of YF. The virus is a member of the family Flaviviridae, genus Flavivirus, and as such bears similarities of size and genome organization to the related dengue, Japanese encephalitis, and West Nile viruses, and others of public health significance. The particle is enveloped, measuring between 50 and 60nm by transmission electron microscopy[5]. The YFV 17D genome is organized as a single-stranded, positive-sense strand of RNA, comprising in read order, a 5′ untranslated region (UTR) (118 bases), three structural proteins C(capsid), prM/M(premembrane/membrane), E(envelope), 7 nonstructural proteins NS(nonstructural)1, NS2A, NS3, NS4A, NS4B, NS5, and a 3′ UTR (507 bases)[Figure 1]. Following clathrin-dependent endocytosis and subsequent low pH endosomal fusion, the genome is immediately translated upon entry to the host cytoplasm, producing a single 3411 codon polyprotein that is co and post-translationally cleaved by a combination of host and viral proteases. Nonstructural genome products contribute to the replication complex and serve various roles in genome replication, which occurs on endoplasmic reticular membrane structures; the assembled virus is transited, and finally exocytosed through the trans-golgi network. Molecular aspects of replication for flaviviruses with respect to YFV vaccines has been extensively reviewed [6]. Wild-type YFV is principally vectored by Aedes spp. mosquitoes in Africa and Haemogogus and Sabethes spp. in South America and the non-human primate hosts differ by geographic region. The taxonomy of YFV implies considerable ecological and geographic restriction for the virus; transmission has only been observed in tropical and subtropical belts of South America, Africa, and certain Caribbean islands [7].
Figure 1.
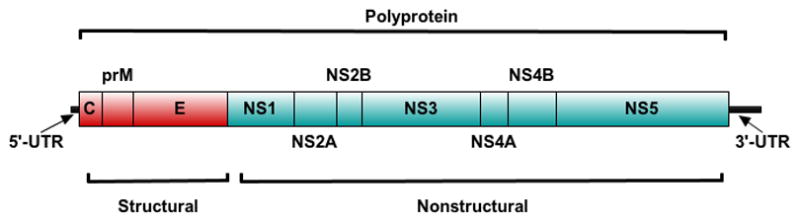
Canonical genome organization of YFV and members of the genus Flavivirus. The virus particle carries a single-stranded, positive sense strand of RNA, which is immediately translated. Structural proteins (red) are placed upstream and occur sequentially, and similarly for the downstream nonstructural proteins. 17D vaccine genomes are identically organized.
Early Development of YFV Vaccines
The development pathway of early YFV vaccines was notable for several technical advances that rendered the virus amenable to laboratory manipulation. The discovery that Indian crown (then identified as Macaca sinica) and rhesus (Macaca mulatta) macaques were susceptible to YFV infection permitted the isolation of wild-type strains in the absence of a cold chain [8]. Max Theiler first demonstrated that the virus could be propagated in the brains of mice, also showing that phenotype of the virus would change with serial passage [9]. The susceptibility of mice to intracerebral challenge by YFV was then usefully employed in serological protection assays, in which sera were titrated against lethality of a YFV strain with neurotropic phenotype [10]. These methods were used to successfully isolate two wild-type strains in 1927, these forming the basis of two concurrently developed, and then successfully deployed live-attenuated YFV vaccines. The first of these wild-type strains, called Asibi, was isolated from a Ghanaian of the same name, who suffered a mild case of YF and fully recovered [8]. The second was isolated from the Syrian Francois Mayali, which became known as the French viscerotropic virus (FVV), and is referred to in literature of the time as the “French” strain, whether as wild-type or in passaged, attenuated strain forms [11].
Development of the French Strain
FVV was adapted to growth in mouse brain (formerly also called as “fixed”) by 128 passages by Theiler before transfer to the Institut Pasteur at Dakar, Senegal [12, 13]. Neurotropism in monkeys was enhanced, a property reasonably attributed to fixation of the virus to mouse brain [14, 15]. At attenuated passage levels, the vaccine strain is referred to as the “neurotropic” strain, and later the “French neurotropic virus” (FNV). FNV served as the platform by which to investigate YFV vaccine delivery methods, including the co-administration with the virus of human immune serum [16]. The attendant impracticality of serovaccination, in conjunction with both the limited supply of serum and risk of pathogen contamination precluded widespread adoption of this technique. Through the 1940’s onward, FNV was used extensively in French west Africa, with 84 million doses produced between 1939 and 1954, which were administered by scarification from an Arabic gum suspension of infected and desiccated mouse brain [13]. FNV was noted for unacceptable incidence of post-vaccinal encephalitis in children, and in 1961 administration of FNV to children under 14 years was prohibited [17]. In 1981, FNV was discontinued by the Institut Pasteur in Senegal. Reference material for FNV is exceedingly rare, however a number of FNV collection strains were partially sequenced in reference to FVV and compared for neurotropism in vivo [18].
Development of the 17D Strain
The Asibi strain was passaged 53 times in monkeys before attempting to attenuate by passage in non-native tissues. From this juncture in handling of the wild-type strain, it was referred to as the “pantropic” or Asibi strain, meaning that the virus possessed both neurotropic and viscerotropic affinities. Under supervision of Max Theiler at the Rockefeller Foundation laboratories in New York, the Asibi strain was passaged in successive tissue preparations; a passage series called 17D was performed in minced mouse embryo (18x), minced whole chick embryo (58x) and finally minced whole chick embryo without brain and spinal cord (100x) [19]. At stages along the passage series, the virus was assayed for neurotropism in mice by intracerebral injection and pathogenicity in monkeys by both intracerebral and extraneural administration. At the 114th passage (in reference to the parental strain Asibi), the virus was observed to have lost pathogenicity for both mice and monkeys. For monkeys challenged by intracerebral or subcutaneous routes with the 114th passage of 17D, the strain produced a survivable encephalitis, with transient fever. 17D did not cause significant viremia in monkeys, and as such the virus was considered to have lost wild-type viscerotropism. The 176th passage of the virus was less lethal by average survival time in mice than a comparator strain at the 114th passage, when delivered intracerebrally and was selected for use as a vaccine. 17D infects Aedes spp. mosquitoes, however the dose required is greater than that required of for wild-type strains [20]. The attenuated strain does not disseminate to the tissues of the mosquito, preventing transmission [21]. The simultaneous loss of viscerotropism, neurotropism, and infectivity to mosquitoes ultimately rendered 17D to be safer than FNV, although immunogenicity of 17D was lower than that of the French strain [22, 23]. As with FNV, field immunogenicity trials of 17D were first undertaken using co-administration of normal human serum with the virus (non-aqueous 17D). 17D preparations without added serum (aqueous-base) were standardized in 1945, and is the current form of the vaccine; administration is by the subcutaneous route [24]. There have been few changes in the seed-lot system since 1945 and over 600 million doses of 17D have been distributed [25].
17D vaccines are considered an essential component of childhood vaccination schedules in YF endemic countries, although the early 21st century has brought increased attention on programmatic lapses in YFV vaccination as a matter of economic development. Though surveillance of YFV is incomplete across the endemic range, recent estimates of YF disease burden modeled from African serosurveys and case data indicate that 1.3 million cases (95%CI = 850,000–1.8 million) would have occurred in 2013, resulting in 78,000 deaths (95%CI = 19,000–180,000) [26]. Vaccination coverage was estimated to have prevented 450,000 cases (95%CI = 340,000–560,000) and 28,000 deaths (95%CI = 7,200–62,000) in the same year, attesting in silico to a quantified, reduced burden of disease as a consequence of YF vaccination campaigns. Widespread mass vaccination campaigns in a number of African countries with FNV and 17D vaccines between the 1940s and 1960s had resulted in the almost-complete disappearance of yellow fever. However, immunization campaigns waned in the mid-1960s. Consequently, since the mid-2000s the nongovernmental Global Alliance for Vaccines and Immunizations (GAVI) has committed considerable resources to support of childhood YFV vaccination in endemic countries, including efforts supporting increased production of 17D vaccines from WHO-prequalified manufacturers [27]. Since 2006, 12 countries have completed preventive yellow fever vaccination campaigns involving over 70 million doses of vaccine with financial support from the GAVI Alliance, and its partners including the European Community Humanitarian Office (ECHO) and the Central Emergency Response Fund (CERF). These programs are ongoing, including a major program of vaccination in Nigeria. Currently, over 35 of the 44 countries at risk for yellow fever in Africa and the Americas have routine infant immunization programs. Large scale vaccination campaigns require enormous amount of vaccine. To meet increasing demand for vaccine, intradermal (ID) delivery of 17D has been investigated as an alternative to subcutaneous administration under a hypothesis that less virus would be needed to induce protection; the strategy represents both a dose-saving approach to vaccination and a potential administration route for recipients with egg allergy. In healthy adult recipients, immunogenicity and safety measurements were equivalent in a comparison of ID delivery (1/5 typical dose) of 17D to the same lot of vaccine administered conventionally [28]. Additionally, a retrospective cohort (n=7) of individuals who failed the 17D skin allergy test (ID administration of 1/5 typical dose) were found to have seroconverted subsequent to the test [29]. Results are promising, however more studies are needed to ascertain the true dose-response relationship of ID vaccine load with adverse events, immunogenicity, and viremia.
Lineage of 17D, and The Current Vaccine Standard Genotype
The lineage of 17D has been extensively described, consisting of seeds and vaccine lots from the three “substrains” 17D-204, 17D-213, and 17DD[6, 30]. Briefly, Theiler’s originating 17D strain at passage 176 was taken and the basal substrains 17D-204 and 17DD originate from subculture levels 204 and 195, respectively [Figure 2]. The 17D-213 substrain was produced by the Robert Koch Institute in 1977 as a derivative of 17D-204, to be free from contamination by avian leukosis virus (ALV), a retrovirus producing sarcomas in chickens, but with no known pathogenic effects in humans[31]. All 17D seeds in current use are free of ALV, and standard production methods require use of eggs from ALV-free flocks[6, 30]. Since the 1940s there have been many vaccine producers from over 10 countries but currently there are only six producers: United States (17D-204, YF-Vax®, Sanofi-Pasteur), France (17D-204, Stamaril®, Sanofi-Pasteur), Senegal (17D-204, Institut Pasteur, Dakar), the People’s Republic of China (17D-204, Tiantan®, Wuhan Institute of Biological Products), the Russian Federation (17D-213, Chumakov Institute of Poliomyelitis and Viral Encephalitides), and Brazil (17DD, Bio-Manguinhos/FIOCRUZ). The French, Russian, Senegalese and Brazilian producers are prequalified by the World Health Organization, serve international markets, and are used for mass vaccination campaigns. The U.S. and Chinese vaccines are used in domestic markets only.
Figure 2.

Lineage of 17D and derivative substrains, considering only present production of 17D vaccines. Substrains 17DD, 17D-204, and 17D-213 are shown in blue, green, and tan respectively. S1: Primary seed. S2: Secondary seed. V: Final vaccine lot. Adapted from [30] [6].
Initial genomic sequence comparison of 17D in reference to the wild-type parental Asibi strain was performed using a 17D-204 isolate obtained from the ATCC[32]. Similar information was later reported for the 17DD and 17D-213 substrains of the vaccine, which permitted elucidation of the conserved sequence features between all substrains[33]. Current data indicates that the three vaccine substrains share 20 common amino acid substitutions, and four nucleotide substitutions in the 3′ untranslated region [Table 1]. The contribution of the various amino acid substitutions and 3′UTR nucleotide changes to the attenuated phenotype is poorly understood (see below). Minimal consensus sequence divergence has been noted between substrains of 17D. Bayesian phylogenetic analysis for a set of 17D substrain consensus sequences was unable to resolve taxonomic groupings for 17D-204 and 17D-213 isolates, indicating very limited sequence divergence between these substrains for the clustering model employed [34]. These data are consistent with the very effective seed-lot system developed in the 1940s and probably contribute to the excellent safety record of 17D vaccine.
Table 1.
Amino acid changes from parental Asibi strain (AY640589) to representative vaccines from each of three substrains 17D-204 (Stamaril®, X15062), 17D-213(YFU17067) and 17DD(YFU1066). Substitutions indicated in grey are conserved between all substrains of 17D and are adapted from the current WHO standard for YFV vaccine production [30].
| Position | Gene | Codon | Amino Acid | 17-204(X15062) | 17D-213(YFU17067) | 17DD(YFU17066) |
|---|---|---|---|---|---|---|
| 854 | M | 36 | L | F | F | F |
| 1127 | E | 52 | G | R | R | R |
| 1140 | E | 56 | A | V | V | |
| 1431 | E | 153 | N | T | ||
| 1436 | E | 155 | D | S* | ||
| 1437 | E | 155 | D | S* | ||
| 1482 | E | 170 | A | V | V | V |
| 1491 | E | 173 | T | I | I | I |
| 1572 | E | 200 | K | T | T | T |
| 1870 | E | 299 | M | I | I | I |
| 1887 | E | 305 | S | F | F | F |
| 1946 | E | 325 | P | S | S | |
| 1965 | E | 331 | K | R | R | R |
| 2112 | E | 380 | T | R | R | R |
| 2193 | E | 407 | A | V | V | V |
| 2219 | E | 416 | A | T | T | |
| 2220 | E | 416 | A | V | ||
| 2687 | NS1 | 79 | L | F | F | F |
| 3371 | NS1 | 307 | I | V | V | V |
| 3860 | NS2A | 118 | M | V | V | V |
| 4007 | NS2A | 167 | T | A | A | A |
| 4013 | NS2A | 169 | L | F | F | |
| 4022 | NS2A | 172 | T | A | A | A |
| 4056 | NS2A | 183 | S | F | F | F |
| 4289 | NS2B | 37 | I | L | L | L |
| 4505 | NS2B | 109 | I | L | L | L |
| 5115 | NS3 | 182 | Q | R | ||
| 5153 | NS3 | 195 | I | V | V | |
| 6023 | NS3 | 485 | D | N | N | N |
| 6876 | NS4A | 146 | V | A | A | A |
| 7171 | NS4B | 95 | I | M | M | M |
| 7497 | NS4B | 204 | L | S | ||
| 7580 | NS4B | 231 | Y | H | H | H |
| 7701 | NS5 | 22 | Q | R | R | |
| 8808 | NS5 | 391 | N | S | ||
| 10142 | NS5 | 836 | E | K | K | K |
| 10338 | NS5 | 901 | P | L | L | L |
| 10367 | 3′UTR | - | T | C | C | C |
| 10418 | 3′UTR | - | T | C | C | C |
| 10550 | 3′UTR | - | T | C | C | |
| 10722 | 3′UTR | - | G | A | ||
| 10800 | 3′UTR | - | G | A | A | A |
| 10847 | 3′UTR | - | A | C | C | C |
Substitution is encoded at two positions at the indicated codon.
Little is known about the molecular basis of attenuation for the 17D vaccine. The 17D and FNV vaccines share two common attenuated mutations in reference to their parental strains (M-L36F and NS4B-I95M), however the influence of these residues on attenuation is unknown. The residue M-36 (L in Asibi, F in 17D) is contained in a pro-apoptotic sequence described for dengue virus [35]. Using a series of 17D/Asibi point mutants localized in the envelope protein, Lee and colleagues observed that the capacity of 17D to bind to heparin sulfate was reduced for viruses containing the wild-type residues E-380T and E-325P (the latter only found in 17D-204 and 17D-213 substrain vaccines); the substitutions also produced a low-neurovirulence phenotype in mice [36]. In summary, determinants of attenuation have been explored to a limited extent. Findings thus far have indicated the influence of alterations in binding of virus to cells, but the effects are likely multigenic. The wild-type and vaccine strain may differ in capacity to antagonize innate immune responses in the host, although this has not been definitively shown for any 17D genotype [37].
Genetic determinants of mosquito infectivity for the vaccine were investigated by Higgs and coworkers, in which Asibi-17D chimeric infectious clones were constructed to contain swapped residues in NS2A (four amino acids), NS4B (one amino acid), and the 3′ untranslated region (four nucleotides) [38]. Dissemination of the chimeric viruses to salivary glands of orally infected Aedes aegypti was reduced for wild-type (Asibi) backbone constructs bearing 17D residues. In Ae. aegypti the 17D envelope residue E-380R was paradoxically observed to increase titers of the Asibi strain in mosquito salivary glands [39]. Although not explaining attenuation in the vaccinee, reduced mosquito infectivity of 17D is hypothesized to contribute to the safety profile of the vaccine by preventing transmission of the attenuated virus post-administration.
Nonclinical comparison of 17D substrain vaccines is infrequently reported. Vaccine seeds are evaluated using a standard WHO monkey neurovirulence assay, in which 10 rhesus or cynomolgous (Macaca fascicularis) macaques are administered a known quantity of seed virus intracerebrally, and monitored for 30 days. Clinical and histological outcomes of neurotropism are compared to an equivalently sized group challenged with a reference strain of known acceptable properties; viscerotropism (serum viremia) and immunogenicity (PRNT50) are assessed by numeric limits. Minor and coworkers used this method to compare three 17D seeds of differing substrain origin [40]. The WHO reference virus strain 168–73 (17D-213 substrain) was of equivalent immunogenicity to secondary seeds originating from Senegal and the United Kingdom (both 17D-204), however greater viremias and lower histological scores were observed for 168–73 when compared to the seeds.
Very little clinical information exists on the comparative performance of vaccines derived from the 17D substrains in human subjects, as modern efficacy trials would be considered unethical due to known historical properties of the vaccine and the severity of YF disease. Some noninferiority trials have been reported for 17D-derived products, both within and between substrains. A comparison of two 17D-204 substrain products Stamaril® and Arilvax® (Chiron; originally produced by Wellcome labs in the U.K. and no longer manufactured) was performed in a healthy adult cohort (n=211)[41]. Both vaccines were found to be immunogenic, observing similar rates of local and systemic adverse reactions; the test group receiving Stamaril® developed moderately but significantly higher neutralizing antibody titers at days 10 and 28 post-administration. Again for two products within the 17D-204 substrain, ARILVAX® and YF-Vax® were compared in a cohort of Peruvian children of ages ranging from 9 to 10 years, observing similar rates of adverse events for both products; seroconversion rates differed significantly and were 94.9% and 90.6%, respectively [42].
For studies comparing 17D vaccines produced from different substrains, a series of trials were reported by Fiocruz comparing the immunogenicity and reactogenicity of two serially generated Brazilian 17DD substrain products with that of a 17D-213 substrain derivative vaccine [43] [44]. In a healthy adult cohort (n=1087), neutralizing antibody titers were equivalent for the 17DD vaccines investigated, while moderately higher for the vaccine derived from the 17D-213 substrain, although immunogenicity results for all groups would be considered highly protective. In seronegative recipients, seroconversion rates for the three test groups were equivalent (>= 98%), and no significant differences in rates of post-vaccinal adverse events were reported. Comparison of the 17D-204 substrain Stamaril® with 17D-213 substrain products RKI-YF® (Robert Koch Institute, Germany) and Berna-YF(Flavimun®) (neither vaccine produced currently) was reported in a cohort of healthy adults (n=304), from which was observed statistical dissimilarity of neutralizing titers between the groups receiving Berna-YF® and Stamaril® [45]. Statistically significant difference of neutralizing titers was not observed between groups receiving RKI-YF and Berna-YF, and all subjects seroconverted (titer >=1:10); the result is of particular interest for the reason that the two vaccines are identically related on the 17D lineage, arising from passage 228 relative to the parental strain and from the same secondary seed. For a cohort of healthy Argentinian adults and children (n=2514, ages=1–70 years), the Fiocruz 17DD vaccine was analyzed for comparative safety to a vaccine derived from 17D-213 substrain; no statistical differences in either local or systemic adverse events were reported, immunogenicity data were not analyzed [46]. In summary, there is little evidence to suggest divergence of phenotype across the 17D substrain lineage. Differences of immunogenicity observed in clinical trials exist at very high ranges of neutralizing antibody titer, and as such would not likely represent any meaningful change in protection. Trials comparing outcomes for 17D vaccines are summarized in Table 2.
Table 2.
Summary of trials discussed comparing 17D substrain vaccines, including endpoints, cohort size, and results.
| Trial Type and Reference | Cohort | Endpoints | Strain, (Substrain, National Origin) | Results |
|---|---|---|---|---|
| Nonclinical [40] | Cynomolgous Macaques | Viremia (Viscerotropism), Immunogenicity | 1. 168–73 (17D-213, WHO Reference Strain) 2. (17D-204 Secondary Seed, UK), 3. (17D-204 Secondary Seed, Senegal) | 1. All groups of equivalent immunogenicity. 2. Greater viremias observed for group receiving 168–73. 3. Lower histological scores for group receiving 168–73. |
| Clinical [41] | Healthy Adults (n=211) | Immunogenicity, Adverse reactions | 1. Stamaril® (17D-204, France) 2. Arilvax® (17D-204, UK) | 1. Similar rates of adverse reactions and seroconversion for both vaccines. 2. Higher immunogenicity for group receiving Stamaril®. |
| Clinical [42] | Children, 9mos.-10yrs. (n=1,107) | Immunogenicity, Adverse reactions | 1. YF-Vax ® (17D-204, USA) 2. Arilvax® (17D-204, UK) | 1. Greater seroconversioon rates for Arilvax®. 2. Similar rates of adverse events. 3. Similar immunogenicity for both vaccines. |
| Clinical [43, 44] | Healthy Adults (n=1087) | Immunogenicity, Adverse Reactions | 1. 17D-WHO (17D-213, Brazil) 2. 17DD/013Z (17DD, Brazil) 3. 17DD/102/84 (17DD, Brazil) | 1. Greater immunogenicity for 17D-213 substrain vaccine. 2. Similar rates of serovonversion for all vaccines considered. 3. Slightly higher rates of minor adverse reactions and detectable viremia for 17D-213 substrain. |
| Clinical[45] | Healthy Adults (n=304) | Immunogenicity, Adverse Reactions | 1. Stamaril® (17D-204, France) 2. RKI-YF® (17D-213, Germany) 3. Berna-YF® (17D-213, Netherlands) | 1. All groups immunogenic (GMT >1:10), 2. Higher ab titers for males receiving 17D-213 substrain products. 3. No difference in immunogenicities between 17D-213 products administered. 4. Higher rate of injection site erythema and pain for RKI-YF®, compared to Berna-YF®. |
| Clinical [46] | Healthy Adults and Children (n=2514) | Adverse Reactions | 1. 17DD/Fiocruz (17DD, Brazil) 2. Stamaril® (17D-204, France) | No significant differences in rates of local or systemic adverse events. |
Use YFV as a Recombinant Backbone of other Vaccines
Owing to the considerable safety profile of 17D, the nonstructural genes of the vaccine has been used as a backbone in a number of vaccine candidates designed to deliver both flaviviral and non-related antigens. Specifically, infectious clones of 17D virus have been generated with the 17D genome cDNA in one or more plasmids; using reverse genetics the cDNA can be mutated to swap or insert foreign sequences. The cDNA is transcribed to RNA in vitro (which is equivalent to YFV genomes), transfected into cells, and is able to replicate in cells to make a recombinant virus carrying desired mutations [47]. Thus, the 17D genome is variably amenable to insertion of heterologous antigenic elements; the strategy is used in a number of flavivirus vaccine candidates at various preclinical and clinical development stages. Recombinant 17D virus bearing inserts of known Plasmodium epitopes showed moderate immunogenicity to the insert peptides in monkeys, while both immunogenicity to the 17D backbone and viremia were reduced in comparison to the parental 17D vaccine, which was hypothesized to result from partially abrogated endosomal fusion in the recombinant virus [48]. A recombinant 17DD construct expressing from the amastigote surface protein (ASP-2) of Trypanasoma cruzi was partiality protective in a susceptible mouse model, a significant result considering the dearth of preventatives and treatments for Chagas’ disease [49]. A recombinant 17D was investigated as a backbone to stimulate cellular immunity against human immunodeficiency virus (HIV) by delivering a known CD8+ T-cell epitopes [50]. The epitope was derived from a known immunodominant section of the simian immunodeficiency virus(SIV) Gag polyprotein, ligated to be expressed by a nonstructural region of the 17D genome. With this construct, CD8+ responses to the 17D vector were stimulated in a nonhuman primate model, with specific cellular immunogenicity to the SIV/gag polyprotein. Recombinant 17D virus with Lassa virus(LAV) glycoproteins were generated by insertion of the LAV GP1 and GP2 genes into the C-terminal region of 17D E protein gene; the construct was immunogenic in a guinea pig model of LAV infection, although less protective (80%) by comparison to a live-attenuated Mopeia virus/LAV reassortant [51].
The ChimeriVax® (Sanofi-Pasteur, Lyon, France) platform is perhaps the most advanced in development of this class of vaccines, and is derived by swap chimeras of heterologous flavivirus prM/M-E structural cassettes into the nonstructural backbone of the 17D genome [52]. Resulting recombinant vaccines are efficiently delivered in a live-attenuated context with a safety profile afforded by the 17D nonstructural genes [53]. To date, one chimera, ChimeriVax-JE (Imojev®), has progressed to licensure and is registered in nine countries [54]. The most advanced candidate in development using this technology is the tetravalent dengue virus (DENV) preparation. Two phase III, multi-center trials of the ChimeriVax® DENV candidate were recently reported, testing efficacy to prevent hospitalized dengue disease in Asian and Latin American pediatric cohorts, for which combined-serotype efficacy was 56.5% and 60.8%, respectively [55, 56]. Similarly, the ChimeriVax® West Nile virus candidate WNV02 was recently assessed in a dose-ranging, phase II safety trial enrolling 208 healthy adults for three age cohorts, revealing 96 percent seroconversion at 28 days that was correlated with vaccine dose [57, 58]. While these studies with mosquito-borne flaviviruses suggest that the nonstructural protein genes of 17D encode the attenuated phenotype, utilization of the ChimeriVax® technology for a tick-borne encephalitis virus prM/M-E chimera did not produce the desired attenuated phenotype in preclinical studies [59].
New Report of A Serious Adverse Viscerotropic Reaction to 17D
Serious adverse events (SAE) following vaccination with 17D are rare, and generally are categorized as either neurotropic, referring to infection of the central nervous system (yellow fever vaccine-associated neurotropic disease: YEL-AND) or viscerotropic (yellow fever vaccine associated viscerotropic disease: YEL-AVD), referring to multi organ disease with infection of the liver as is similarly observed in wild-type YF. Clinical data on SAE are consequently rare, but support a paradigm of SAE occurrence that is considerably influenced by host factors rather than reversion of the vaccine virus to a virulent phenotype.
A recent case of fatal YEL-AVD, the 65th reported case of this type, occurred in 2014, in a female recipient receiving the vaccine for travel to South America [60]. The patient developed signs consistent with the WHO case definition of YEL-AVD, eventually succumbing to systemic disease [61]. YFV antigen was observed in recovered tissues by immunostain, with positive IgM titer of 1:640. On autopsy, a thymoma was found in association with serology indicating myasthenia gravis, which if previously identified would potentially have contraindicated receipt of the vaccine. The directly precedent reported cases of YEL-AVD occurred in 2009, which was not reported in open literature. Preceding this in 2007, in Peru, five individuals were treated for systemic viscerotropic disease following 17D vaccination in a large campaign, with four fatalities [62]. The consensus genomic sequence of the originating vaccine lot and three of the fatal cases were determined, and there was no divergence from the secondary seed lot used in manufacture. Single-nucleotide polymorphism content from the 2009 cluster and previous YFV-AVD case tissues were sequenced and compared at loci hypothesized to influence innate immune responses, but the results did not describe association of particular SNPs with the cases. Since the case descriptions include evidence of altered immune status for the vaccinees in association with viscerotropic disease, host factors are likely contributors.
Post-vaccinal SAE have been extensively reviewed [25]. Risk factors for YEL-AVD are estimated from the limited clinical data available and include age (>60), systemic lupus erythematosus, thymectomy/thymoma, or other autoimmune condition [63] [64]. A hypothesis that competent innate immune responses are required to prevent dissemination of 17D is supported by animal models of YFV disease, in which 17D vaccine virus is lethal in mice lacking alpha/beta and gamma interferon receptors (AG129) [65]. Similarly, widespread dissemination of genetically tagged 17D strains was observed in a mouse model lacking alpha/beta interferon receptors [66]. In a study comparing naive 17D recipients in young and elderly age groups, viremia in the older subjects was found to be relatively greater [67]. Mutations localized to CCR5/RANTES genes or their regulatory elements are associated with variable response to viral infection, and thus have been hypothesized to influence development of YEL-AVD, however the association of known alleles with the adverse syndrome is inconclusive [68].
New YFV Vaccine Candidate Technologies and Monitoring of Safety
As aforementioned, the occurrence of adverse events is rare, however the use of inactivated YFV vaccines has been proposed to ameliorate concerns adverse reactions for recipients for whom administration of live 17D strains would be contraindicated, including the elderly, immunosuppressed, or those with egg allergy. Design of inactivated YFV vaccines have typically been frustrated by necessity to deliver multiple doses to achieve protection in typical neurovirulent mouse models of YFV infection.
Nonclinical data for two inactivated, whole-virion 17D candidates has been reported. Characterization of the β-propriolactone-inactivated 17D-204 candidate XRX-001 (Xcellerex/GE Healthcare, Marlborough, MA) was performed, showing that the vaccine was protective in Syrian golden hamsters against challenge by the hamster-adapted Jimenez strain of YFV [69]. Immunogenicity of XRX-001 was assessed in cynomolgous macaques for two and three-dose administration schedules, from which sera variably neutralized the heterologous 14-FA (Angola) strain. Results of a phase I safety and dose-ranging trial of XRX-001 in 60 recipients were reported in 2011, observing seroconversion (endpoint/baseline PRNT50 >4) for all participants receiving a booster dose at 21 days [70]. Although neutralizing antibody titers for the two-dose administration of XRX-001 did not exceed those typically found in clinical studies of live-attenuated 17D vaccines, the seropositivity criterion used (PRNT50 >10) is thought to confer protection in humans [43]. Two inactivated 17D candidates have been reported by the Oswaldo Cruz Foundation (Brazil). The first of these to be characterized was a pressure-inactivated, 17DD-substrain candidate; immunogenicity data in mice revealed that three doses were necessary to produce equivalent neutralization titers to those subjects receiving live 17DD vaccine [71]. The second, a β-propriolactone-inactivated 17DD substrain derivative was tested for immunogenicity in outbred mice (subcutaneous route), using a live-attenuated 17DD vaccine as comparison. Greatest immunogenicity (GMT = 922, 95%C.I. = 666–1274, 43.75% seroconversion) was observed in a group receiving three doses, adjuvanted with aluminum hydroxide [72]. No information is available on the durability of protection offered by any inactivated 17D vaccine, however under reported test conditions, immunogenicities for these exceed typical standards for seroconversion, and as such would be expected to be protective.
Recombinant YFV vaccine candidates have been investigated using diverse vectoring strategies. Very commonly, constructs are designed to deliver the 17D prM/M-E structural cassette, which contains the predominant surface-exposed neutralizing epitopes of YFV and all flaviviruses. Delivery of YFV antigens by live-attenuated, recombinant vaccinia constructs was shown to be feasible in the late 1990s[73]. A recent effort used the replication-deficient modified vaccinia virus Ankara (MVA) and d4R defective vaccinia virus (dVV) to vector the 17D prM/M-E antigen protected mice from intracerebral challenge with 17D when administered in a single dose (17D substrains, though highly attenuated in humans, are virulent for mice when directly introduced to the brain) [74]. The study not only demonstrated protection and immunogenicity of the constructs, but also high apparent safety in mice, as the vaccines themselves were not neurotropic when administered at doses exceeding the that required to induce protection. A plasmid-vectored DNA vaccine was constructed by fusion-ligation of the 17DD substrain prM/M-E onto that of human LAMP-1, a strategy designed to target the antigen structural subunit for efficient trafficking to MHC-II compartments and consequent presentation and stimulation of cellular immune responses [75]. A 45 day, three-dose course of the LAMP-1 plasmid construct was protective when assayed in mice against live 17DD virus delivered intracerebrally.
17D is manufactured in embryonated chicken eggs, a legacy technology that remains unchanged since standardization of the vaccine in the 1940s. The adaptation of the vaccine to other cell based systems has been investigated, with the goal of efficiently producing the vaccine in vitro. The adaptive capacity of YFV has been investigated since the early development of all YFV vaccines, with multiple studies recognizing that passage history could reverse the attenuation or immunogenicity of the vaccine [76, 77]. A 17DD substrain seed was adapted to chick embryo fibroblasts (CEF), to permit growth of the vaccine lot by microcarrier bead culture technology [78]. The CEF-prepared vaccine lot was tested using the standard WHO monkey neurovirulence test; the authors noted elevated neurotropism scores in the monkeys tested with respect to a conventionality prepared seed, although these results were not statistically significant. Adaptation of the inactivated 17D-204 candidate XRX-001 to Vero cell culture was performed, revealing some instability in the vaccine genome localized to the envelope, NS2A, and NS4B genes, although differences in phenotype conferred by mutations have not been tested in an infectious clone system [79]. The substitutions were stably observed between three independently conducted passage series, associated with greater yields of the virus from Vero cell cultivation. In both of these cases, the vaccines were safe and immunogenic in nonclinical studies involving nonhuman primates, however the known potential for instability of live virus phenotype under in vitro passage adaptation warrants close attention.
To this end, massively parallel sequencing (next-generation sequencing or NGS) approaches have been deployed to interrogate the fine population structure of vaccines, including the presence of contaminants[80]. These methods partially derive from theoretical insight of the “quasispecies” paradigm, in which viral diversity is considered to influence fitness in vivo[81]. Recently, a commercial lot of YF-Vax® 17D-204 vaccine (Sanofi-Pasteur, Swiftwater, USA) was sequenced by massively parallel methods recovering evidence of very low ALV contamination, confirmed by RT-PCR [82]. Low coverages for YFV (n=<100) prevented complete assessment of population structure for the targeted viruses. Comparison of a 17D-204 commercial lot with a collection lot of the parental Asibi strain was performed at ultra deep coverage (>5000), revealing that the wild-type virus population structure differs from that of the vaccine by virtue of relative homogeneity suggesting that the lack of diversity in the 17D vaccine virus population may contribute to the attenuated phenotype [83]. Resolution of population structure for viral vaccines by NGS or other methods is a potential avenue by which to measure the presence of virulent subpopulations which naturally occur in RNA viruses, and in doing so predict safety profiles of the vaccines before use.
New Insights to Immunogenicity and Durability of Protection for 17D
Production of YFV vaccines is regulated using a seed-lot system, developed in 1945 in response to evidence that phenotypic instability of the vaccine could be introduced by improper handling resulting in excessive passaging of the vaccine virus [24]. Among other contributing findings, an early Brazilian trial cohort observed absence of immunogenicity for a group receiving vaccine from a seed at extended passage levels [77]. Presently, the protection conferred by 17D lots of 17D production seeds is characterized for immunogenicity by neutralization antibody titers during performance of the WHO monkey neurovirulence test (criterion: PRNT50 >1:10), and by comparison trials in humans for lots of vaccine originating from newly generated seeds [30]. Presently, a neutralization titer of 1 in 10 or a fourfold increase of neutralizing antibody from baseline is considered evidence of seroconversion. International Health Regulations (IHR) require that, for certain travelers in areas of YFV endemicity, a certificate of prophylaxis (“yellow card”) be issued by the vaccinator, which is considered valid for ten years following the tenth day post-administration[84].
The WHO Strategic Advisory Group of Experts (SAGE) working group for yellow fever vaccination evaluated open and closed-literature findings on the durability of immunogenicity conferred by 17D [85]. Specifically, the working group recommended revision to the 2003 WHO position on 17D booster schedules, recommending that both the WHO position and IHR be revised to reflect an expectation that 17D confers lifetime protection without the necessity of 10-year booster intervals. The amendment to the IHR was approved by the World Health Assembly, to be included in a 2016 revision. In February 2015, the Advisory Committee on Immunization Practices (ACIP) working group on yellow fever vaccines presented these findings to the ACIP, which were adopted by unanimous vote of the committee [86]. The 2013 SAGE findings were assembled into an extensive review of immunogenicity for 17D, which in numerous cases of seropositivity have been observed in vaccinees at intervals greater than 10 years post-administration [87]. Briefly, of 8 open-literature studies considered by the working group, duration of 17D immunity was maintained for 10 years in all datasets, with an upper reported boundary of 38 years [88]. Some countries have questioned the proposed removal of booster doses and it remains to seen whether or not all will elect to adopt the WHO recommendation.
Mechanisms underlying the long-lasting immunogenicity of 17D are poorly understood, but are attributed in part to the combined stimulation of innate, humoral and cellular immunities from the presentation of endogenous antigen expressed by the live, replicating virus [89]. In a passive serum transfer experiment using the hamster adapted challenge model, neutralizing PRNT50 titers of <1:40 were sufficient to confer protection, supporting the canonical model for protective humoral seroconversion in humans [90]. Systems biology approaches have recently been deployed to interrogate these mechanisms, in order to more closely elucidate determinants of immunogenicity for the vaccine in the host. Significantly, studies of this type have identified key cytokines and innate response elements that determine both the diversity and longevity of the immune response to 17D. A series of in silico studies reported associations of networked antiviral response elements that were predictive of CD8+ cell responses to the vaccine [91]. In a comparative study of several vaccines, humoral responses to 17D are partially correlated to cellular stress responses, specifically that of transcription factor ATF2, and translation factor kinase EIF2AK4, both markers for stress response and amino-acid starvation, which contributes to programming of DCs to present antigens to stimulate CD8+T cell immunity [92]. The cellular response to 17D is highly diverse, involving activation of multiple TLR response elements and lineages of dendritic cells [93]. This results in the induction of various pro-inflammatory cytokines, including IL-12p40, IL-6, and interferon-α, and consequently the stimulation of both T and B cell responses. In a cohort of healthy adults receiving 17D vaccine, the viral load at day 14 post-administration correlated with effector CD8 T-cell responses; a saturation effect for CD8+ T-cell response is observed above a certain viremia sustained in the vaccinee, above which no changes were observed [94]. The study is especially significant to understanding the immunological responses of 17D recipients under conditions of dose-sparing. In a comparison of 17DD and 17D-213 substrains in a pediatric cohort, the cytokine responses of the vaccines were similar, however revealing an enhanced inflammatory component for the group receiving the 17D-213 substrain vaccine [95]. IFN-gamma responses are observed to correlate with humoral, CD4+, and CD8+ responses [96, 97]. It is hoped that, by interrogation of specific immune response elements, predictive models of response to live-attenuated vaccines could be constructed, and in such a manner offer personalized insight to the safety and eventual protection available to the recipient.
Expert Commentary
The yellow fever 17D vaccine was a milestone in the development of live attenuated vaccines and has proved enormously successful at controlling a highly pathogenic virus. Over 600 million doses have been distributed since the development of the vaccine in 1937 with an excellent record of safety and immunogenicity. Serious adverse events are rare and the vaccine can be co-administered with 10 other vaccines. In many ways, this is an ideal vaccine for use in developing countries where YF disease is found. Somewhat surprisingly, the vaccine is still produced in embryonated chicken eggs, a legacy technology that has changed little since the introduction of the seed-lot system in 1945. Attempts to transfer the manufacturing process to cell culture have not proven successful.
Although the vaccine has proved very successful, our understanding of the molecular basis of attenuation of the vaccine is very limited, although recent studies indicate this may be due to a lack of diversity in the quasispecies population of the virus, limiting the potential for virulent reversion. Similarly, our understanding of how one dose of vaccine can induce protection for at least 10 years, and probably life-long, is rudimentary. However, recent studies of the innate immune response and systems biology are starting to reveal the mechanisms of protective immunity. Finally, reverse genetics of the 17D vaccine virus genome that was developed over 25 years ago is being applied to generate recombinant vaccines utilizing the 17D genome as a backbone, that have found success in clinical trial settings.
Five-Year View
The use of YFV vaccines encompasses the entire history of empiric viral vaccination, and as such represents a rich historical dataset by which to understand newer perspectives on the rational design of live-attenuated vaccines. The ChimeriVax® 17D-Japanese encephalitis vaccine has been recently licensed and it is likely that the same technology may yield a licensed dengue vaccine in the near future. The 17D lineage is broadly recognized to possess an exemplary safety profile, however, the increasing use of 17D genomic components in rationally designed vaccine candidates should prompt closer investigation of the discrete mechanisms of attenuation for the virus. The attenuation is certainly complex and multigenic, as 17D is attenuated for viscerotropism, neurotropism and mosquito vector competence. In particular, understanding the molecular basis of loss of vector competence will have potentially important applications to the development of live attenuated vaccines for other mosquito-borne pathogens.
One major gap in our understanding of 17D vaccine are the mechanisms influencing the occurrence of severe adverse events. Yellow fever vaccine-associated viscerotropic disease (YEL-AVD) and neurotropic disease (YEL-AND) are rare conditions, but unfortunately can be fatal, especially YEL-AVD. Most studies of YEL-AVD and YEL-AND are clinical case studies and so our understanding of these conditions is limited and results suggest they are due to host genetic factors and not to virulent reversion of the virus, Continuing advances in systems biology and molecular biology of the virus will likely lead to a better understanding of the mechanism(s) of these rare conditions, and improved approaches to determining contraindications for 17D and other live-attenuated vaccines.
Similarly, systems biology studies of 17D vaccine have identified critical response pathways in the induction of protective immunity. Nonetheless, the studies to date are still rudimentary and continued interrogation of the host immune response will likely lead to a comprehensive understanding of how a proactive and durable immune response is induced. Overall, 17D has taught us much about live attenuated vaccines and continues to offer important insights in to how live attenuated vaccines work. This will have important applications to other vaccine systems and will provide critical information for the eventual design of “personalized” vaccines, although the latter is unlikely to take place in the next 5 years.
Continual use of 17D in large vaccination programs is ongoing, necessitating the periodic regeneration of production seeds and close attention to any changes in the safety profile of the vaccine, however slight. Many of the primary vaccine seeds were generated over 50 years ago and given the large numbers of doses being manufactured each year it can only be a matter of time before regeneration of primary vaccine seeds will need to be considered.
Key Issues.
YFV vaccines are historically significant as they were developed by empiric methods, and were among the first vaccines of this class.
The 17D strain is still manufactured by legacy technologies involving embryonated chicken eggs that have changed little since the 1940s; manufacture in alternative cell culture systems has been investigated but has not been implemented by any producer.
The World Health Organization and the GAVI vaccine alliance have undertaken to immunize all people in sub-Saharan Africa who are at risk for yellow fever disease. This is in excess of 250 million people.
All RNA viruses consist of a quasispecies structure consisting of a “cloud” of RNA species with “high diversity”. The low diversity of 17D vaccine virus is thought to contribute to the high level of attenuation and great safety record of the vaccine.
17D is considered to be highly safe and protective, however the increasing use of 17D nonstructural genomic components into rationally-designed vaccine products demands closer scrutiny into discrete mechanisms of attenuation for the vaccine strain.
International recommendations for 17D booster schedules have been reduced for healthy adults; the 10-year booster International Health Regulations requirement has been modified to reflect an expectation that protection conferred by the vaccine is lifelong.
Systems biology has been used successively to understand the molecular basis of immunogenicity of 17D vaccine.
YFV vaccines represent a lengthy history of deployment, and as such represent a large historical dataset for interrogation of flaviviral pathogenesis mechanisms, including those of interferon antagonism, cellular immunity, and viral quasispecies theory.
Footnotes
Financial and competing interests disclosure
The authors were supported by grants from the National Institutes of Health (R21 AI 113407) and the Gillson-Longenbaugh Foundation. AS Beck was supported in part by National Institutes of Health grant T32 AI 060549. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Reference annotations
* Of interest
** Of considerable interest
- 1.Vainio J, Cutts F. Yellow Fever. Geneva: World Health Organization; 1998. [Google Scholar]
- 2.Bazin H. Yellow Fever Vaccine. Vaccinations: A History: John Libbey Eurotext. 2011:407–54. [Google Scholar]
- 3.World Health Organization. WHO-recommended standards for surveillance of selected vaccine preventable diseases. 2003 [Google Scholar]
- 4.Johansson MA, Vasconcelos PFC, Staples JE. The whole iceberg: estimating the incidence of yellow fever virus infection from the number of severe cases. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2014;108:482–7. doi: 10.1093/trstmh/tru092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burlaud-Gaillard J, Sellin C, Georgeault S, et al. Correlative scanning-transmission electron microscopy reveals that a chimeric flavivirus is released as individual particles in secretory vesicles. PLoS ONE. 2014;9:e93573. doi: 10.1371/journal.pone.0093573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monath TP, Gershman M, Staples JE, Barrett ADT. Yellow fever vaccine. In: Plotkin S, editor. Vaccines. 6. Elsevier Inc; 2013. pp. 870–15. [Google Scholar]
- 7.Jentes ES, Poumerol G, Gershman MD, et al. The revised global yellow fever risk map and recommendations for vaccination, 2010: consensus of the Informal WHO Working Group on Geographic Risk for Yellow Fever. The Lancet Infectious diseases. 2011;11:622–32. doi: 10.1016/S1473-3099(11)70147-5. [DOI] [PubMed] [Google Scholar]
- 8.Stokes A, Bauer JH, Hudson NP. Experimental transmission of yellow fever to laboratory animals. The American Journal of Tropical Medicine and Hygiene. 1928;1:103–64. [Google Scholar]
- 9.Theiler M. Susceptibility of white mice to the virus of yellow fever. Science (New York, NY) 1930;71:367. doi: 10.1126/science.71.1840.367. [DOI] [PubMed] [Google Scholar]
- 10.Theiler M. Neutralization tests with immune yellow fever sera and a strain of yellow fever virus adapted to mice. Annals of tropical medicine and parasitology. 1931;25:69–77. [Google Scholar]
- 11.Sellards AW, Hindle E. The Preservation of Yellow Fever Virus. British medical journal. 1928;1:713–4. doi: 10.1136/bmj.1.3512.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theiler M. Studies on the Action of Yellow Fever Virus in Mice. Ann trop Med Parasit. 1930;24:249–72. [Google Scholar]
- 13.Durieux C. Preparation of yellow fever vaccine at the Institut Pasteur, Dakar. In: Smithburn KC, Duriex C, Koerber R, Penna HA, Dick GWA, Courtois G, de Sousa Manso C, Stuart G, Bonnel PHYF, editors. Vaccination Monograph Series. 1956. pp. 31–2. [Google Scholar]
- 14.Lloyd W, Penna HA. Studies on the pathogenesis of neurotropic yellow fever virus in Macacus rhesus. The American Journal of Tropical Medicine and Hygiene. 1933;8:1–45. [Google Scholar]
- 15.Ni H, Ryman KD, Wang H, et al. Interaction of yellow fever virus French neurotropic vaccine strain with monkey brain: characterization of monkey brain membrane receptor escape variants. Journal of Virology. 2000;74:2903–6. doi: 10.1128/jvi.74.6.2903-2906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawyer WA, Kitchen SF, Lloyd W. Vaccination Against Yellow Fever With Immune Serum and Virus Fixed for Mice. The Journal of experimental medicine. 1932;55:945–69. doi: 10.1084/jem.55.6.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stones PB, Macnamarra FN. Encephalitis following neurotropic yellow fever vaccine administered by scarification in Nigeria: epidemiological and laboratory studies. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1955;49:176–86. doi: 10.1016/0035-9203(55)90044-4. [DOI] [PubMed] [Google Scholar]
- 18.Wang E, Ryman KD, Jennings AD, et al. Comparison of the genomes of the wild-type French viscerotropic strain of yellow fever virus with its vaccine derivative French neurotropic vaccine. Journal of General Virology. 1995;76( Pt 11):2749–55. doi: 10.1099/0022-1317-76-11-2749. [DOI] [PubMed] [Google Scholar]
- 19.Theiler M, Smith HH. The effect of prolonged cultivation in vitro upon the pathogenicity of yellow fever virus. The Journal of Experimental Medicine. 1937;65:767–86. doi: 10.1084/jem.65.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitman L. Failure of Aedes Aegypti To Transmit Yellow Fever Cultured Virus (17D) American Journal of Tropical Medicine. 2015;19:19–26. [Google Scholar]
- 21.Miller BR, Adkins D. Biological characterization of plaque-size variants of yellow fever virus in mosquitoes and mice. Acta virologica. 1988;32:227–34. [PubMed] [Google Scholar]
- 22.United Nations Relief and Rehabilitation Administration. Report to the UNRAA Expert Commission on Quarantine: Dakar Yellow-Fever Vaccine. 2014:618–37. [Google Scholar]
- 23.Stuart G. The Problem of Mass Vaccination Against Yellow Fever. World Health Organization; 1953. pp. 1–13. [Google Scholar]
- 24.World Health Organization. World Health Organ Epidemiol Bull. Vol. 1. Geneva: World Health Organization; 1945. Standards for the Manufacture and Control of Yellow Fever Vaccine; pp. 365–70. [Google Scholar]
- 25.Barrett AD, Teuwen DE. Yellow fever vaccine—how does it work and why do rare cases of serious adverse events take place? Current opinion in immunology. 2009;21:308–13. doi: 10.1016/j.coi.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 26.Garske T, Van Kerkhove MD, Yactayo S, et al. Yellow Fever in Africa: Estimating the Burden of Disease and Impact of Mass Vaccination from Outbreak and Serological Data. PLoS Medicine. 2014;11:e1001638. doi: 10.1371/journal.pmed.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.GAVI. [Accessed June 18, 2015];Yellow Fever Supply and Procurement Roadmap. 2015 Available at: http://www.gavi.org/library/gavi-documents/supply-procurement/yellow-fever-roadmap-public-summary/
- 28.Roukens AH, Vossen AC, Bredenbeek PJ, van Dissel JT, Visser LG. Intradermally administered yellow fever vaccine at reduced dose induces a protective immune response: a randomized controlled non-inferiority trial. PLoS ONE. 2008;3:e1993. doi: 10.1371/journal.pone.0001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roukens AH, Vossen AC, van Dissel JT, Visser LG. Reduced intradermal test dose of yellow fever vaccine induces protective immunity in individuals with egg allergy. Vaccine. 2009;27:2408–9. doi: 10.1016/j.vaccine.2009.02.049. [DOI] [PubMed] [Google Scholar]
- 30.WHO Expert Committee on Biological Standardization. World Health Organization Technical Report Series. Vol. 978. Geneva: 2013. Recommendations to Assure the Quality, Safety, and Efficacy of Live Attenuated Yellow Fever Vaccines; pp. 241–314. [Google Scholar]
- 31.Bres P, Koch M. Standardization WECoB, editor. World Health Organization Technical Report Series. Vol. 36. Geneva: World Health Organization; 1987. Production and Testing of the WHO Yellow Fever Virus Primary Seed Lot 213–77 and Reference Batch 168–73; pp. 113–41. [Google Scholar]
- 32.Hahn CS, Dalrymple JM, Strauss JH, Rice CM. Comparison of the virulent Asibi strain of yellow fever virus with the 17D vaccine strain derived from it. Proc Natl Acad Sci U S A. 1987;84:2019–23. doi: 10.1073/pnas.84.7.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.dos Santos CN, Post PR, Carvalho R, Ferreira II, Rice CM, Galler R. Complete nucleotide sequence of yellow fever virus vaccine strains 17DD and 17D-213. Virus research. 1995;35:35–41. doi: 10.1016/0168-1702(94)00076-o. [DOI] [PubMed] [Google Scholar]
- 34.Stock NK, Boschetti N, Herzog C, Appelhans MS, Niedrig M. The phylogeny of yellow fever virus 17D vaccines. Vaccine. 2012;30:989–94. doi: 10.1016/j.vaccine.2011.12.057. [DOI] [PubMed] [Google Scholar]
- 35.Catteau A. Dengue virus M protein contains a proapoptotic sequence referred to as ApoptoM. Journal of General Virology. 2003;84:2781–93. doi: 10.1099/vir.0.19163-0. [DOI] [PubMed] [Google Scholar]
- 36.Lee E, Lobigs M. E protein domain III determinants of yellow fever virus 17D vaccine strain enhance binding to glycosaminoglycans, impede virus spread, and attenuate virulence. Journal of Virology. 2008;82:6024–33. doi: 10.1128/JVI.02509-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muñoz-Jordán JL, Laurent-Rolle M, Ashour J, et al. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. Journal of Virology. 2005;79:8004–13. doi: 10.1128/JVI.79.13.8004-8013.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McElroy KL, Tsetsarkin KA, Vanlandingham DL, Higgs S. Manipulation of the yellow fever virus non-structural genes 2A and 4B and the 3′non-coding region to evaluate genetic determinants of viral dissemination from the Aedes aegypti midgut. Am J Trop Med Hyg. 2006;75:1158–64. [PubMed] [Google Scholar]
- 39.Huang Y-JS, Nuckols JT, Horne KM, Vanlandingham D, Lobigs M, Higgs S. Mutagenesis analysis of T380R mutation in the envelope protein of yellow fever virus. Virology Journal. 2014;11:1–4. doi: 10.1186/1743-422X-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minor PD. Neurovirulence tests of three 17D yellow fever vaccine strains. Biologicals : journal of the International Association of Biological Standardization. 2011;39:167–70. doi: 10.1016/j.biologicals.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Lang J, Zuckerman J, Clarke P, Barrett P, Kirkpatrick C, Blondeau C. Comparison of the immunogenicity and safety of two 17D yellow fever vaccines. American Journal of Tropical Medicine and Hygiene. 1999;60:1045–50. doi: 10.4269/ajtmh.1999.60.1045. [DOI] [PubMed] [Google Scholar]
- 42.Belmusto-Worn VE, Sanchez JL, McCarthy K, et al. Randomized, double-blind, phase III, pivotal field trial of the comparative immunogenicity, safety, and tolerability of two yellow fever 17D vaccines (Arilvax and YF-VAX) in healthy infants and children in Peru. American Journal of Tropical Medicine and Hygiene. 2005;72:189–97. [PubMed] [Google Scholar]
- 43.Camacho LAB, Freire MdS, Leal MdLF, et al. Immunogenicity of WHO-17D and Brazilian 17DD yellow fever vaccines: a randomized trial. Revista de saúde pública. 2004;38:671–8. doi: 10.1590/s0034-89102004000500009. [DOI] [PubMed] [Google Scholar]
- 44.Camacho LAB, Aguiar SGd, Freire MdS, et al. Reactogenicity of yellow fever vaccines in a randomized, placebo-controlled trial. Revista de saúde pública. 2005;39:413–20. doi: 10.1590/s0034-89102005000300012. [DOI] [PubMed] [Google Scholar]
- 45.Pfister M, Kürsteiner O, Hilfiker H, et al. Immunogenicity and safety of BERNA-YF compared with two other 17D yellow fever vaccines in a phase 3 clinical trial. American Journal of Tropical Medicine and Hygiene. 2005;72:339–46. [PubMed] [Google Scholar]
- 46.Ripoll C, Ponce A, Wilson MM, et al. Evaluation of two yellow fever vaccines for routine immunization programs in Argentina. Human vaccines. 2014;4:121–6. doi: 10.4161/hv.4.2.5216. [DOI] [PubMed] [Google Scholar]
- 47.Rice CM, Grakoui A, Galler R, Chambers TJ. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. The New biologist. 1989;1:285–96. [PubMed] [Google Scholar]
- 48.Bonaldo MC, Garratt RC, Marchevsky RS, et al. Attenuation of Recombinant Yellow Fever 17D Viruses Expressing Foreign Protein Epitopes at the Surface. Journal of Virology. 2005;79:8602–13. doi: 10.1128/JVI.79.13.8602-8613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nogueira RT, Nogueira AR, Pereira MCS, et al. Recombinant yellow fever viruses elicit CD8+ T cell responses and protective immunity against Trypanosoma cruzi. PLoS ONE. 2013;8:e59347. doi: 10.1371/journal.pone.0059347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonaldo MC, Martins MÂ, Rudersdorf R, et al. Recombinant Yellow Fever Vaccine Virus 17D Expressing Simian Immunodeficiency Virus SIVmac239 Gag Induces SIV-Specific CD8+ T-Cell Responses in Rhesus Macaques. Journal of Virology. 2010;84:3699–706. doi: 10.1128/JVI.02255-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bredenbeek PJ, Molenkamp R, Spaan WJM, et al. A recombinant Yellow Fever 17D vaccine expressing Lassa virus glycoproteins. Virology. 2006;345:299–304. doi: 10.1016/j.virol.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bray M, Lai CJ. Construction of intertypic chimeric dengue viruses by substitution of structural protein genes. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:10342–6. doi: 10.1073/pnas.88.22.10342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guy B, Barrere B, Malinowski C, Saville M, Teyssou R, Lang J. From research to phase III: preclinical, industrial and clinical development of the Sanofi Pasteur tetravalent dengue vaccine. Vaccine. 2011;29:7229–41. doi: 10.1016/j.vaccine.2011.06.094. [DOI] [PubMed] [Google Scholar]
- 54.Appaiahgari MB, Vrati S. IMOJEV ®: a Yellow fever virus-based novel Japanese encephalitis vaccine. Expert review of vaccines. 2010;9:1371–84. doi: 10.1586/erv.10.139. [DOI] [PubMed] [Google Scholar]
- 55.Capeding MR, Tran NH, Hadinegoro SRS, et al. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. Lancet. 2014;384:1358–65. doi: 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- 56**.Villar L, Dayan GH, Arredondo-García JL, et al. Efficacy of a Tetravalent Dengue Vaccine in Children in Latin America. The New England journal of medicine. 2015;372:113–23. doi: 10.1056/NEJMoa1411037. Most recent of two reports for phase III clinical trials measuring efficacy for the 17D-based recombinant tetravalent dengue vaccine. [DOI] [PubMed] [Google Scholar]
- 57.Monath TP, Liu J, Kanesa-Thasan N, et al. A live, attenuated recombinant West Nile virus vaccine. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6694–9. doi: 10.1073/pnas.0601932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Biedenbender R, Bevilacqua J, Gregg AM, Watson M, Dayan G. Phase II, randomized, double-blind, placebo-controlled, multicenter study to investigate the immunogenicity and safety of a West Nile virus vaccine in healthy adults. Journal of Infectious Diseases. 2011;203:75–84. doi: 10.1093/infdis/jiq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rumyantsev AA, Goncalvez AP, Giel-Moloney M, et al. Single-dose vaccine against tick-borne encephalitis. Proceedings of the National Academy of Sciences. 2013;110:13103–8. doi: 10.1073/pnas.1306245110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60**.DeSilva M, Sharma A, Staples E, et al. Notes from the field: fatal yellow fever vaccine-associated viscerotropic disease--Oregon, September 2014. MMWR Morbidity and mortality weekly report. 2015;64:279–81. Most recent case report of YF vaccine associated viscerotropic disease (YEL-AVD) [PMC free article] [PubMed] [Google Scholar]
- 61.Gershman MD, Staples JE, Bentsi-Enchill AD, et al. Viscerotropic disease: case definition and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2012;30:5038–58. doi: 10.1016/j.vaccine.2012.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whittembury A, Ramirez G, Hernández H, et al. Viscerotropic disease following yellow fever vaccination in Peru. Vaccine. 2009;27:5974–81. doi: 10.1016/j.vaccine.2009.07.082. [DOI] [PubMed] [Google Scholar]
- 63.Monath TP, Cetron MS, McCarthy K, et al. Yellow fever 17D vaccine safety and immunogenicity in the elderly. Human vaccines. 2005;1:207–14. doi: 10.4161/hv.1.5.2221. [DOI] [PubMed] [Google Scholar]
- 64.Staples JE, Gershman M, Fischer M. Yellow fever vaccine: recommendations of the Advisory Committee on Immunization Practices (ACIP) In: Shaw F, editor. MMWR Recommendations and reports : Morbidity and mortality weekly report Recommendations and reports/Centers for Disease Control. Vol. 59. Centers for Disease Control and Prevention (CDC); 2010. pp. 1–27. [PubMed] [Google Scholar]
- 65.Meier KC, Gardner CL, Khoretonenko MV, Klimstra WB, Ryman KD. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009;5:e1000614. doi: 10.1371/journal.ppat.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Erickson AK, Pfeiffer JK. Dynamic Viral Dissemination in Mice Infected with Yellow Fever Virus Strain 17D. Journal of Virology. 2013;87:12392–7. doi: 10.1128/JVI.02149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roukens AH, Soonawala D, Joosten SA, et al. Elderly Subjects Have a Delayed Antibody Response and Prolonged Viraemia following Yellow Fever Vaccination: A Prospective Controlled Cohort Study. PLoS ONE. 2011;6:e27753–6. doi: 10.1371/journal.pone.0027753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pulendran B, Miller J, Querec TD, et al. Case of Yellow Fever Vaccine–Associated Viscerotropic Disease with Prolonged Viremia, Robust Adaptive Immune Responses, and Polymorphisms in CCR5 and RANTES Genes. Journal of Infectious Diseases. 2008;198:500–7. doi: 10.1086/590187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monath TP, Lee CK, Julander JG, et al. Inactivated yellow fever 17D vaccine: development and nonclinical safety, immunogenicity and protective activity. Vaccine. 2010;28:3827–40. doi: 10.1016/j.vaccine.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 70**.Monath TP, Fowler E, Johnson CT, et al. An inactivated cell-culture vaccine against yellow fever. The New England journal of medicine. 2011;364:1326–33. doi: 10.1056/NEJMoa1009303. Development and trial of inactivated YFV vaccine based on the 17D-204 substrain. [DOI] [PubMed] [Google Scholar]
- 71.Gaspar LP, Mendes YS, Yamamura AMY, et al. Pressure-inactivated yellow fever 17DD virus: implications for vaccine development. Journal of virological methods. 2008;150:57–62. doi: 10.1016/j.jviromet.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 72.Pereira RC, Silva ANMR, Souza MCO, et al. An inactivated yellow fever 17DD vaccine cultivated in Vero cell cultures. Vaccine. 2015 doi: 10.1016/j.vaccine.2015.03.077. [DOI] [PubMed] [Google Scholar]
- 73.Pincus S, Mason PW, Konishi E, et al. Recombinant vaccinia virus producing the prM and E proteins of yellow fever virus protects mice from lethal yellow fever encephalitis. Virology. 1992;187:290–7. doi: 10.1016/0042-6822(92)90317-i. [DOI] [PubMed] [Google Scholar]
- 74.Schäfer B, Holzer GW, Joachimsthaler A, et al. Pre-Clinical Efficacy and Safety of Experimental Vaccines Based on Non-Replicating Vaccinia Vectors against Yellow Fever. PLoS ONE. 2011;6:e24505. doi: 10.1371/journal.pone.0024505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maciel M, Cruz FdSP, Cordeiro MT, et al. A DNA Vaccine against Yellow Fever Virus: Development and Evaluation. PLoS Neglected Tropical Diseases. 2015;9:e0003693. doi: 10.1371/journal.pntd.0003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Findlay GM, Clarke LP. Reconversion of the neurotropic into the viscerotropic strain of yellow fever virus in rhesus monkeys. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1935;28:579–600. [Google Scholar]
- 77.Soper FL. Vaccination with Virus 17D in the Control of Jungle Yellow Fever in Brazil. 1938:19. [Google Scholar]
- 78.Freire MS, Mann GF, Marchevsky RS, et al. Production of yellow fever 17DD vaccine virus in primary culture of chicken embryo fibroblasts: yields, thermo and genetic stability, attenuation and immunogenicity. Vaccine. 2005;23:2501–12. doi: 10.1016/j.vaccine.2004.10.035. [DOI] [PubMed] [Google Scholar]
- 79.Beasley DWC, Morin M, Lamb AR, et al. Adaptation of yellow fever virus 17D to Vero cells is associated with mutations in structural and non-structural protein genes. Virus research. 2013:1–5. doi: 10.1016/j.virusres.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 80.Neverov A, Chumakov K. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc Natl Acad Sci U S A. 2010;107:20063–8. doi: 10.1073/pnas.1012537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andino R, Domingo E. Viral quasispecies. Virology. 2015;479–480C:46–51. doi: 10.1016/j.virol.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Victoria JG, Wang C, Jones MS, et al. Viral nucleic acids in live-attenuated vaccines: detection of minority variants and an adventitious virus. Journal of Virology. 2010;84:6033–40. doi: 10.1128/JVI.02690-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83**.Beck A, Tesh RB, Wood TG, Widen SG, Ryman KD, Barrett ADT. Comparison of the live attenuated yellow fever vaccine 17D-204 strain to its virulent parental strain Asibi by deep sequencing. Journal of Infectious Diseases. 2014;209:334–44. doi: 10.1093/infdis/jit546. In silico deep sequencing study examining the population structure of 17D vaccine, observing considerable homogeneity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Assembly WH. International health regulations (2005) Geneva: World Health Organization; 2006. [Google Scholar]
- 85.World Health Organization. Meeting of the Strategic Advisory Group of Experts on immunization, April 2013 – conclusions and recommendations. Weekly Epidemiological Record. 2013;88:201–6. [Google Scholar]
- 86.Centers for Disease Control and Prevention(CDC) Advisory Committee on Immunization Practices (ACIP) -Summary Report February 26, 2015. Atlanta, GA: 2015. pp. 1–72. [Google Scholar]
- 87**.Gotuzzo E, Yactayo S, Córdova E. Efficacy and duration of immunity after yellow fever vaccination: systematic review on the need for a booster every 10 years. The American journal of tropical medicine and hygiene. 2013;89:434–44. doi: 10.4269/ajtmh.13-0264. Review of WHO SAGE findings on durability of protection for 17D, which contributed to the revised WHO position and revision of the international health regulations (IHR) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Niedrig M, Lademann M, Emmerich P, Lafrenz M. Assessment of IgG antibodies against yellow fever virus after vaccination with 17D by different assays: neutralization test, haemagglutination inhibition test, immunofluorescence assay and ELISA. Tropical medicine & international health : TM & IH. 1999;4:867–71. doi: 10.1046/j.1365-3156.1999.00496.x. [DOI] [PubMed] [Google Scholar]
- 89.Mudd PA, Piaskowski SM, Neves PCC, et al. The live-attenuated yellow fever vaccine 17D induces broad and potent T cell responses against several viral proteins in Indian rhesus macaques--implications for recombinant vaccine design. Immunogenetics. 2010;62:593–600. doi: 10.1007/s00251-010-0461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Julander JG, Trent DW, Monath TP. Immune correlates of protection against yellow fever determined by passive immunization and challenge in the hamster model. Vaccine. 2011;29:6008–16. doi: 10.1016/j.vaccine.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Querec TD, Akondy RS, Lee EK, et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nature Immunology. 2008;10:116–25. doi: 10.1038/ni.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li S, Rouphael N, Duraisingham S, et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nature Immunology. 2014;15:195–204. doi: 10.1038/ni.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gaucher D, Therrien R, Kettaf N, et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. Journal of Experimental Medicine. 2008;205:3119–31. doi: 10.1084/jem.20082292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94**.Akondy RS, Johnson PLF, Nakaya HI, et al. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proceedings of the National Academy of Sciences. 2015;112:3050–5. doi: 10.1073/pnas.1500475112. In silico study describing a continuous relationship of 17D vaccine dose to CD8+ T-cell response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Campi-Azevedo AC, de Araújo-Porto LP, Luiza-Silva M, et al. 17DD and 17D-213/77 yellow fever substrains trigger a balanced cytokine profile in primary vaccinated children. PLoS ONE. 2012;7:e49828. doi: 10.1371/journal.pone.0049828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neves PCC, Rudersdorf RA, Galler R, et al. CD8+ gamma-delta TCR+ and CD4+ T cells produce IFN-γ at 5–7 days after yellow fever vaccination in Indian rhesus macaques, before the induction of classical antigen-specific T cell responses. Vaccine. 2010;28:8183–8. doi: 10.1016/j.vaccine.2010.09.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Neves PCC, Santos JR, Tubarão LN, Bonaldo MC, Galler R. Early IFN-Gamma Production after YF 17D Vaccine Virus Immunization in Mice and Its Association with Adaptive Immune Responses. PLoS ONE. 2013;8:e81953. doi: 10.1371/journal.pone.0081953. [DOI] [PMC free article] [PubMed] [Google Scholar]