

Abstract
Both dopamine D3 receptor (D3R) partial agonists and antagonists have been implicated as potential medications for substance use disorders. In contrast to antagonists, partial agonists may cause fewer side effects since they maintain some dopaminergic tone and may be less disruptive to normal neuronal functions. Here, we report three sets of 4-phenylpiperazine stereoisomers that differ considerably in efficacy: the (R)-enantiomers are antagonists/weak partial agonists whereas the (S)-enantiomers are much more efficacious. To investigate the structural basis of partial agonism, we performed comparative microsecond-scale molecular dynamics simulations starting from the inactive state of D3R in complex with these enantiomers. Analysis of the simulation results reveals common structural rearrangements near the ligand binding site induced by the bound (S)-enantiomers, but not by the (R)-enantiomers, that are features of partially activated receptor conformations. These receptor models bound with partial agonists may be useful for structure-based design of compounds with tailored efficacy profiles.
Keywords: Dopamine D3 receptor, orthosteric binding site, synthon, antagonist, partial agonist, phenylpiperazine, molecular dynamics
Graphical abstract
INTRODUCTION
The dopamine D3 receptor (D3R) belongs to the dopamine D2-like subgroup of G-protein-coupled receptors (GPCRs), and has been implicated as a promising therapeutic target for psychostimulant addiction.1 Early preclinical studies using the D3R-selective antagonist (N-{trans-4-[2-(6-cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl]cyclohexyl}quinoline-4-carboxamide; SB277011A)2–4 and weak partial agonist (N-(4-(4-(2-methoxyphenyl)piperazin-1-yl)butyl)-2-naphthamide; BP897)5, 6 prompted an intense effort to design novel drug-like molecules with D3R-selectivity as potential medications for substance use disorders. Our efforts on the 4-phenylpiperazine class of compounds have led to high-affinity D3R-selective compounds with varying efficacies.7, 8
Partial agonists may provide an improved therapeutic profile over antagonists by normalizing and stabilizing dopaminergic tone. Thus, unlike full dopamine receptor agonists and antagonists, partial agonists may have reduced abuse liability or disruptive effects on motivated behavior.9 In addition, compared to antagonists, partial agonists may have attenuated cardiovascular side effects. Recently, the D3R antagonist 1-(2-fluoro-4-trifluoromethyl-phenyl)-3-{3-[4-methyl-5-(4-methyl-oxazol-5-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-propyl}-3-aza-bicyclo[3.1.0]hexane (GSK598,809) was reported to potentiate cocaine-mediated increases in blood pressure and heart rate in dogs.10 In contrast, D3R partial agonists attenuated cocaine-mediated increases in blood pressure and heart rate in nonhuman primates implanted with indwelling telemetry devices (MA Nader, AH Newman, unpublished results).
Previously, we used molecular docking and simulations based on the D3R crystal structure11 to demonstrate that the 4-phenylpiperazine primary pharmacophore (PP) binds to the conserved orthosteric binding site (OBS) in which dopamine binds, whereas the arylamide secondary pharmacophore (SP) binds in a secondary binding pocket (SBP) that is topographically divergent between D2R and D3R.12, 13 These studies indicated that efficacy is primarily determined by the binding mode of the PP moiety within the OBS12.
However, the structural basis of partial agonism at D3R, and at GPCRs in general remains unclear. The conformational changes that occur during GPCR activation processes have only begun to be characterized in recent years based on the X-ray crystal structures of several GPCRs in their active- and inactive-states (reviewed in Katritch et al.14). The crystal structures of a receptor bound to a G protein or G protein mimetic indicate that the activation mechanism involves large conformational changes on the intracellular side, with a >10 Å outward movement of the intracellular end of TM6, and smaller conformational changes around the OBS, characterized by various degrees of contraction.15–17 The crystal structures and molecular dynamics (MD) simulations of the β2-adrenergic receptor further provide evidence that the motion of several residues in the connector region – the so called Pro5.50-Ile3.40-Phe6.44 motif (the residue indices are in Ballesteros-Weinstein numbering18) – may allosterically couple the OBS and the intracellular side.19, 20 In the β1-adrenergic receptor, the partial agonists salbutamol and dobutamine were unable to stabilize the fully-active-state structure of the OBS in these crystal structures, due to the lack of interaction with Ser5.46, in contrast to full agonists that form hydrogen bonds with both Ser5.42 and Ser5.46.17 Fluorescence spectroscopy experiments detecting conformational changes in the β2-adrenergic receptor suggested a sequential binding model in which the activation process occurs by kinetically distinguishable steps through discrete intermediate conformational states,21 which are likely differentially stabilized by ligands with different efficacies.
In the current study, we performed long-timescale MD simulations with a novel set of stereoisomers from the 4-phenylpiperazine class that switch their efficacy from antagonism/weak partial agonism to substantially more efficacious partial agonism, to further evaluate the key receptor-ligand interactions formed, and the corresponding receptor conformational changes. By simulating the binding modes of compounds of high similarity, we focused on detecting subtle but consistent conformational changes in the OBS that differentiate the partial agonist-stabilized versus the inactive state of the receptor.
RESULTS
Synthesis of novel piperazine derivatives
Scheme 1 shows the synthesis of the full-length ligands (3, (±)-, (R)- and (S)-4) in which the quinoline-3-carboxylic acid is coupled to the corresponding 4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-1-amine (1) or 4-amino-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol (2). The chiral analogues (R)- and (S)-4 were prepared with (R)- or (S)-4-amino-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol amine ((R)- or (S)-5), respectively,22, 23 via an in situ 1,1′-carbonyldiimidazole (CDI) coupling reaction. HPLC analysis and optical rotations confirmed the enantiopurity of the final compounds. Scheme 2 shows the synthesis of the synthons (R)- or (S)-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)butan-2-ol ((R)- and (S)-6) and (R)- or (S)-1-(4-(2,3-dichlorophenyl)piperazin-1-yl)propan-2-ol ((R)- and (S)-7). These enantiomers were obtained by reacting the commercially available enantiomers (R)- or (S)-2-ethyloxiranes or (R)- or (S)-2-methyloxiranes with 1-(2,3-dichlorophenyl)piperazine (8) under reflux conditions to form the ring-opened synthons. HPLC (Figures S6–S11, S.I.) and X-ray analysis of compounds (R)- and (S)-7 (Tables S3 and S4, Figures S12 and S13, S.I.) confirmed the structures assigned and enantiomeric purity.
Scheme 1. Synthesis of full-length D3R ligandsa.

aReagents and conditions: (a) (i) quinoline-3-carboxylic acid, CDI, THF, room temperature, 2 h; (ii) appropriate 4-arylpiperazine amine, THF, 0 °C to room temperature, overnight.
Scheme 2. Synthesis of (R)- and (S)-synthonsa.

aReagents and conditions: (a) appropriate epoxide, isopropanol, reflux, overnight.
Binding characterization of novel piperazine derivatives
The binding affinities of the synthesized compounds were measured by radioligand binding assays using HEK 293 cells expressing human D2Rs or D3Rs (Table 1). Similar to the compounds reported previously,12 the arylpiperazine synthons were non-selective or weakly selective for D3R over D2R as they bind to the OBS of both D2R and D3R, which are highly homologous.11 In contrast, the full-length compounds have significantly higher D3R selectivity, presumably by taking advantage of the divergence in the SBP between D3R and D2R as we have previously proposed.12, 13
Table 1.
Binding data for full-length compounds and synthons
| Ligand | 2D structure | D2R | D3R | D2/D3 |
|---|---|---|---|---|
| Ki nM) ± SEM | ||||
| 3 |
|
10.0 ± 1.86 | 0.172 ± 0.00913 | 58 |
| (±)-4 |
|
268 ± 60.0 | 4.26 ± 0.76 | 63 |
| (R)-4 |
|
202 ± 48.6 | 4.12 ± 0.49 | 49 |
| (S)-4 |
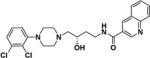
|
316 ± 8.72 | 23.4 ± 2.56 | 14 |
| (R)-6 |
|
588 ± 98.5 | 119 ± 25.9 | 5 |
| (S)-6 |
|
93.1 ± 23.2 | 34.3 ± 9.38 | 3 |
| (R)-7 |
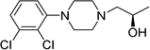
|
1400 ± 250 | 373 ± 88.5 | 4 |
| (S)-7 |
|
317 ± 18.7 | 75.2 ± 15.2 | 4 |
| (R)-9a |
|
433 ± 29.5 | 1.12 ± 0.21 | 394 |
| (S)-9a |
|
715 ± 6.23 | 16.6 ± 2.31 | 43 |
| 10b |
|
103 ± 20.8 | 1.4 ± 0.4 | 73 |
| 11b |
|
3.4 ± 0.4 | 1.9 ± 0.2 | 2 |
(R)- and (S)-enantiomers of full-length 4-phenylpiperazine derivatives have differential efficacies
A lead molecule from the 4-phenylpiperazine class of compounds, PG648 (9) was previously reported to be highly D3R-selective and enantioselective in binding at D3R ((R)-9: Ki = 1.12 nM; (S)-9: Ki = 16.6 nM).24 Compound 9 has a 2,3-diCl-phenylpiperazine PP, a terminal indole SP, and the butyl linker functionalized with a 3-OH group. Using a synthon-approach to deconstruct the full-length compounds into their pharmacophoric components, we previously found that the unsubstituted 2,3-diCl-phenylpiperazine PP by itself elicited partial agonism (Emax = 57.3% of dopamine efficacy by the BRET assay) whereas the addition of the linker and/or SP to the PP generally reduced efficacy.12
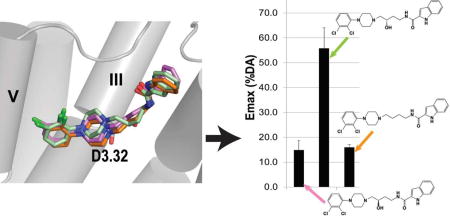
In this study, we first used the Go bioluminescence resonance energy transfer (BRET) assay to evaluate the functional activity of the full-length compound 9, and found its enantiomers showed dramatically different efficacies: whereas the (R)-enantiomer behaved as a weak partial agonist, the (S)-enantiomer behaved as a more efficacious partial agonist ((R)-9: Emax = 14.9%; (S)-9: Emax = 55.7%, see Figure 1), in agreement with the previous mitogenesis results.24 Motivated by this finding and the molecular modeling results described below, we synthesized and separated the (R)- and (S)-enantiomers of an analogue of compound 9 with the indole SP modified to a quinoline SP (compound 4), and found that the differential efficacies of the (R)- and (S)-enantiomers were retained ((R)-4: Emax = 19.9%; (S)-4: Emax = 53.9%, see Figure 1). Interestingly, the analogues without the 3-OH in the linker region showed similar profiles to the (R)-enantiomers (10: Emax = 16.0%; and 3: Emax = 20.9%, see Figure 1). Thus, the partial agonism of the (S)-enantiomers is not dependent on the specific aryl amide SP moiety but requires the (S)-chirality at the C3 position of the linker.
Figure 1. Functional efficacies measured by the Go BRET activation assay.

The (R)-enantiomers ((R)-9, -4, -6) and the analogues without the linker 3-OH group (10, 3, 11) are antagonists, whereas the (S)-enantiomers ((S)-9, -4, -6) are partial agonists. Emax is the % of dopamine efficacy. The efficacies of (S)-enantiomers are significantly different from their corresponding (R)-enantiomers and analogues without a linker (in each case, P < 0.005 using one-way ANOVA followed by post hoc Tukey test).
Similarly, differential efficacies of the (R)- and (S)-enantiomers of compounds 9 and 4 were also observed in a β-arrestin recruitment assay (Figure 2).
Figure 2. Functional efficacies measured by the β-arrestin recruitment assay.

The (R)-enantiomers ((R)-9, -4, -6) are antagonists, whereas the (S)-enantiomers ((S)-9, -4, -6) are partial agonists. Emax is the % of dopamine efficacy. NA, no activity. The efficacies of (S)-enantiomers are significantly different from their corresponding (R)-enantiomers; (S)-9 efficacy is significant different from (S)-4 or (S)-6 efficacy (in each case, P < 0.0001 using one-way ANOVA followed by post hoc Tukey test).
Overview of molecular dynamics (MD) simulations
To better understand the structural basis of efficacy from both the ligand and receptor perspectives, we performed more than 28 µs-long MD simulations of D3R in complex with six full-length and two synthon ligands, with the objective of characterizing their differential ligand binding modes and the corresponding receptor conformations that correlate with varied efficacies. All simulations were started from previously equilibrated inactive state models based on the antagonist-bound D3R crystal structure (PDB: 3PBL).11–13, 25 We carried out microsecond-scale MD simulations in order to induce a transition in the ligand binding site to accommodate the bound (S)-enantiomers, which are strong partial agonists. For each receptor-ligand complex, at least two trajectories of lengths longer than 300 ns were obtained. The summary of the MD trajectories is in Table S1.
Differential binding modes of full-length (R)- and (S)-enantiomers
The results of our molecular docking and MD simulations show that the overall binding mode of (S)-9 is similar to (R)-9: the 4-phenylpiperazine PP binds in the OBS, the indole SP binds in the SBP formed by TMs 2, 3, EL1, and EL2, and the linker 3-OH forms a hydrogen bond with the side chain of Asp1103.32 (Figure 3a). However, unlike in (R)-9, the four-carbon linking chain (C1-C4 in Figure 3b) of (S)-9 is in a non-extended conformation that consequently impacts the position of PP within the OBS, causing the PP of the (S)-enantiomer to shift slightly upward compared to that of the (R)-enantiomer (Figure 3c,f). Such differential binding modes of the enantiomers may underlie the substantial difference in their efficacies. Similarly to (R)- and (S)-9, the predicted binding modes for (R)- and (S)-4 differ at the linker region (Figure 3d,g). For analogues without the 3-OH in the linker region (3 and 10), the predicted binding modes showed extended linker conformations, similar to that of the (R)-enantiomers (Figure 3c,d,f,g). Thus, the 3-OH substitution may impose strain on the linker and thereby impact the PP binding mode to elicit partial agonism.
Figure 3. Differential binding modes of (R)- vs (S)-enantiomers.

(a) The overall binding mode of the full-length 2,3-diCl phenylpiperazine compound. (b) The dihedral angle in the linker region. Panels (c–e) show the representative binding modes of (R)-, (S)-9, and 10 (C), (R)-, (S)-4 and 3 (d), and (R)- and (S)-6 (e). The distribution of linker dihedral angle (C1-C2-C3-C4) differs substantially between the (R)- and (S)-enantiomers for the full-length compounds (f,g), whereas they are comparable for the synthons (h). Ligands are shown as sticks. TMs 6 and 7 are not shown for clarity.
The aryl amide secondary pharmacophore is not required for the efficacy switch
In order to evaluate whether the SP moiety is required for the partial agonism of the full-length (S)-enantiomer compounds, we synthesized synthons consisting of the 2,3-diCl-phenylpiperazine PP and 3-OH four-carbon linking chain without the SP ((R)-6 and (S)-6)). Results of the Go BRET assay indicated that the differential efficacies of the (R)- and (S)-enantiomers are retained in the synthons ((R)-6: Emax = 11.6%; (S)-6: Emax = 58.6%, see Figure 1) and that the partial agonism of the (S)-enantiomers does not require the SP moiety. In the β-arrestin recruitment assay, (S)-6 is more efficacious than (R)-6 as well (Figure 2).
In our MD simulations of D3R in complex with (R)- or (S)-6, we found that the predicted binding mode for (R)-6 overlaps with that of the full-length R-enantiomers, (R)-9 and (R)-4, i.e. the four-carbon linker of the (R)-enantiomer synthon is in an extended conformation. In contrast, the linker of (S)-6, like the full-length S-enantiomers, is not fully extended (Figure 3e,h). However, the binding modes of (S)-6 from all the parallel simulated trajectories deviate from that of the full-length (S)-enantiomers, (S)-9 and (S)-4, and converge to a pose in which the 4-phenylpiperazine PP is oriented more parallel to the membrane within the OBS. Unlike for the full-length (S)-enantiomers, the 2,3-diCl substituents of the bound (S)-6 are oriented toward the interface of TMs 5 and 6 (Fig. 2e). Despite the divergence in the overall binding modes of the synthon and full-length (S)-compounds, there is spatial overlap between the tip of the synthon linker and C4 of the full-length compound linkers.
Development of a comparative analysis tool to detect subtle conformational changes in GPCRs
To further characterize the conformational changes in the ligand binding site of D3R that are induced by the binding of the (S)-enantiomer partial agonists, compared to those by the (R)-enantiomers, we performed pairwise comparisons of the resulting D3R conformations from our MD simulations of the protein in complex with the three enantiomer pairs, i.e., (R)-9 vs. (S)-9, (R)-4 vs (S)-4, and (R)-6 vs (S)-6. Such comparisons of the binding modes of enantiomers of the same chemical structure allowed us to identify relevant conformational divergences that may be responsible for the different efficacies of these compounds. In addition, the availability of three such pairs allows us to identify common trends, further strengthening the conclusions.
Specifically, we assumed that the two compounds in an enantiomeric pair have subtle but significant differences in binding modes. Differential conformational effects initiated by such differences from the ligand binding sites, including both OBS and SBP, are expected to propagate to other parts of the receptor. In particular, conformational differences near the ligand binding sites should be detectable even if MD simulations do not reveal the entire activation process. Thus, in order to detect relatively subtle changes near the binding sites and to identify a common trend with regard to ligand induced-effects on receptor conformation, we developed a comparative receptor conformational analysis tool (termed Protein Interaction Analyzer for GPCR (PIA-GPCR)) that quantifies the extent of the conformational changes at both residue and sub-segment levels. Based on the clustering protocol and division of the sub-segments of the transmembrane domain in the PIA program described previously,26 PIA-GPCR systematically measures structural features such as the distances among the centers of mass of transmembrane sub-segments as well as the distances among Cα atoms of residues in ligand binding site, and filters those that differ between two compared conditions (see Methods).
To validate PIA-GPCR, which was designed for Class-A GPCRs, we applied it to compare the available inactive- and active-state crystal structures of β2-adrenergic receptor (β2AR) and muscarinic M2 receptor (M2R). In both β2AR and M2R, the analysis recapitulated the previously well-characterized conformational changes associated with the activation process:15, 27 we observed larger distances and changes in angles between intracellular sub-segment of TM6 (TM6i, see Methods for the divisions of “e”, “m”, and “i” sub-segments for each TM) and other TM sub-segments in the active-state, indicating an outward movement of TM6i; and smaller distances between TM7i and TM3i, indicating an inward movement of TM7i (Figure S1). On the extracellular side, there is an inward rearrangement of TM6e to move toward TM3e and TM4e, which is more prominent in M2R (Figure S1).
PIA-GPCR also detects other changes that are not easily identifiable by visual inspection or simple superposition-based examination. For example, although the majority of the inter-residue Cα-Cα distances in the ligand binding site were smaller in the active-state, as expected from the agonist-induced contraction of the binding site, we detected a number of distances involving residues Phe5.47 and Phe6.44 that increased in the active-state, indicating that the change in the size of the ligand binding site during activation is not a uniform contraction. Phe6.44 is part of the “Pro5.50-Ile3.40-Phe6.44” connector motif, which has been proposed to play a critical role in connecting the conformational changes in the ligand binding site to the intracellular side.19
In addition, our results revealed some different rearrangements during the activation process in M2R compared to β2AR: for example, TM5e moves away from TM7e in M2R but moves towards TM7e in β2AR during activation (Figure S1).
To further demonstrate the strength of PIA-GPCR in detecting subtle but significant conformational changes that are often not obvious from visual inspection, we applied the method to compare the partial agonist-bound β1AR structures against the antagonist-bound β1AR structures. Interestingly, we detected significant rearrangements of TM6i in a direction similar to those observed in M2R and β2AR; however, the induced changes of TM2e appear to be unique to the partial agonists of β1AR; in addition, the contraction of the OBS near TM5e is similar to β2AR but not M2R, suggesting that some features of the conformational changes induced in the (partially) activated state may be specific to individual receptors (Figure S2).
Common conformational changes induced by partial agonists
We carried out comparative conformational analysis using PIA-GPCR on the simulations results of D3R in complex with either the full-length or synthon compounds. Our analysis indicates that compared to the (R)-9 bound state, TM6e moves inward in the presence of bound (S)-9, with the distances between TM6e and all other extracellular and middle sub-segments except for TM5m on average smaller in the (S)-9-bound state (the red pixels along the “TM6e” row on Figure 4a). Similarly, the analysis of (R)-4 vs (S)-4-bound state indicates that TM6e moves inward in the (S)-4-bound state (Figure 4b). For the synthons, even though the differences in the binding modes between (S)-6 and (R)-6 are larger than those between the full-length (S)- and (R)-enantiomers, we observed similar effects on receptor conformation, i.e., inward movement of TM6e in the presence of (S)-6 (Figure 4c). Correspondingly, the Cα-Cα distances in the binding site between TM6e residues and those in TM2e, TM3e, EL2, and TM7e/m, but not TM5e/m, are smaller in the (S)-enantiomer-bound states in all three comparisons (Figure 4). Interestingly, we observed that TM5e moves away from TM7e consistently in these comparisons (though to different extents), which is similar to M2R but not β2AR (see above) (Figure S1).
Figure 4. Comparative receptor conformational analysis using PIA-GPCR.

Heatmaps show the differences in the distances among TM sub-segments (left panels), and the distances among Cα atoms of ligand binding site residues (right panels), comparing between (R)- and (S)-9-bound frames (a); (R)- and (S)-4-bound frames (b); (R)- and (S)-6-bound frames (c). The color is scaled from blue to red, corresponding to the increase and decrease, respectively, of the metric values in the (S)-enantiomers bound state compared to the (R)-enantiomers bound state.
In order to evaluate whether or not the induced conformational changes by the (S)-enantiomers are directed toward full activation of the receptor, we carried out MD simulations of D3R in complex with the endogenous full-agonist dopamine, as a control (Table S1). We then performed the analysis using PIA-GPCR to compare the resulting D3R conformation with that stabilized by the inverse-agonist eticlopride. Our results indicate that dopamine induced a significant inward movement of TM6e, with a slightly larger magnitude than observed with the (S)-enantiomers (Figure S3).
Thus, the receptor conformations induced by these partial agonists, either the full-length molecules or the synthons, consistently showed an inward movement of TM6e, which is similar to the TM6e rearrangements observed in the fully-active-state crystal structures described above. To understand how this movement may have been triggered by the re-positioning of the 4-phenylpiperazine PP moiety of the ligand within the OBS, we analyzed other correlative conformational changes within the OBS. We observed that the side chain of Phe6.52 is oriented toward the interface of TMs 5 and 6 in all the (S)-enantiomer and dopamine-bound receptor conformations, leading to an increased distance between TM5m and TM6e, whereas Phe6.52 pointed into the center of OBS in the (R)-enantiomer and eticlopride-bound receptor conformations (Figure 5). This rearrangement affects the orientation of Phe5.47 in TM5m that is sandwiched between Phe6.52 and Phe6.44.
Figure 5. Common rearrangements at the TM5-TM6 interface.

Scatterplots of X1-dihedral angle of Phe3466.52 and the distance between TMs 5 and 6 compared between (R)- and (S)-9, 4, 6. The common trend of the differences between the (R)- and (S)-enantiomer bound conditions suggest that the rotamer change of Phe6.52 is correlated with the observed movements of TMs 5 and 6 during partial activation.
Taken together, the presence of the (S)-3-OH on the butyl linker results in a non-extended linker conformation that affects the binding mode of the 4-phenylpiperazine PP in the OBS. Compared to the (R)-enantiomers, the PP of the (S)-enantiomers is shifted toward the interface of TMs 5 and 6, and consequently induces TM5 to rotate clockwise, leading to an inward movement of TM6e, and also moving EL2 closer to TM6e. These coordinated rearrangements ultimately destabilize the inactive conformation of the receptor, and the common trends in all three pair-wise (R)- versus (S)-enantiomer comparisons as well as between dopamine and eticlopride suggest that these reconfigurations are a hallmark of the initial step of receptor activation (Figure 6).
Figure 6. Molecular mechanism of D3R partial activation.

Schematic representation shows the common trend of movements of TM helices and the extracellular loop regions based on the comparisons between the antagonist- and partial agonist-bound D3R MD simulations.
DISCUSSION AND CONCLUSIONS
In this study, we synthesized and characterized enantiomeric pairs of full-length 2,3-diCl-phenylpiperazine D3R-selective ligands and their synthons. Using long MD simulations, we compared the binding modes of three pairs of the enantiomers in D3R – in each pair, the (S)-enantiomer demonstrated substantially higher efficacy than the (R)-enantiomer, regardless of the presence or absence of the SP. The molecular models of the receptor-ligand complexes showed differential binding modes of the enantiomers in the OBS: for the full-length compounds, (R)- and (S)-chirality at the 3-OH substitution in the linker region rendered the four-carbon linking chain to be in an extended or non-extended conformation, respectively, which is associated with distinct positions/orientations of the 2,3-diCl-phenyl moieties. This was also the case for the linkers of the (R)- and (S)-synthon enantiomers that lack an arylamide SP moiety. However, there was a greater difference in the binding mode of synthon PPs within the OBS, whereby the 2,3-diCl-phenyl moiety shifted more towards the interface of TMs 5 and 6 in the (S)-enantiomer.
The binding modes of the PPs of the (S)-enantiomers are reminiscent of our previous finding for the unsubstituted 2,3-diCl-phenylpiperazine synthon that is also a strong partial agonist.12 In that study we focused only on the ligand binding modes and found that the 3-Cl of the 2,3-diCl-phenylpiperazine, when bound to the OBS of D3R, is oriented similarly towards the interface of TMs 5 and 6.12 In the current study, our analysis using greatly extended MD simulations shows common conformational changes in and around the OBS that are induced by the three (S)-enantiomer partial agonists. Compared to the (R)-enantiomer, the binding of (S)-enantiomer induces an inward movement of TM6e, a rotation of TM5e/m, and the movement of EL2 toward TM6e. In particular, we predict that the rearrangements near Phe5.47 in the presence of (S)-enantiomer will eventually propagate to the Pro5.50-Ile3.40-Phe6.44 motif, the reconfiguration of which has been proposed to promote the conformational changes on the intracellular side necessary for receptor activation19 – however, we have not detected such changes in our simulations at the current timescales. Thus although the TM sub-segment rearrangements in the presence of the (S)-enantiomers or dopamine showed similar features to those in the fully-active-state crystal structures of other receptors (see Results), the conformational transition we have revealed so far are likely to represent the initial or intermediate steps of the activation process. Nevertheless, the common trend we observed in the binding site from four sets of comparative simulations and analysis points to the structural basis for the switch in efficacy from the inactive to the partially activated state.
It is intriguing that subtle changes in ligand stereochemistry can result in such a profound difference in the functional activation of the receptor. Similar observations have been reported in other studies: stereoisomers of an aminotetralin-derived D2R agonist have been shown to elicit differential biased signaling properties;28 differential pharmacological properties of the stereoisomers of a novel D3R agonist, 8-hydroxy-3-(n-propyl)1,2,3a,4,5,9b-hexahydro-1H-benz[e]indole, were found to be related to the different interactions with the extracellular portion of TM6.29 These are in line with our previous findings that small modifications of ligands can lead to different efficacies.12 Furthermore, this phenomenon seems to be prevalent in other GPCRs as well: close analogs of naltrindole, 5′-guanidinonaltrindole and 6′-guanidinonaltrindole that differ only in the substitution site of the guanidinium group were shown to act as a biased agonist and an antagonist respectively for the κ opioid receptor.30, 31 For allosteric ligands of metabotropic glutamate receptors, minor structural changes to the ligand scaffold have been shown to lead to opposing modes of pharmacology.32 Thus, small modifications of the ligand not only can optimize binding affinity, but also provide an opportunity to modulate functional efficacy.
Indeed, in the context of fragment-based drug design, the linker region has typically been considered to serve only the role to connect the fragments within the binding pockets.33 Yet, we have found that the stereochemistry within the linker region in our series of compounds has a dramatic impact on their functional efficacy by altering the binding mode of the PP and the subsequently induced receptor conformational changes. Thus, given the enantio-specific efficacy profiles of ligands studied herein, the design of the linker should be evaluated not only for optimizing the distance between the two primary and secondary pharmacophores, but also for the potential to modulate pharmacological properties such as efficacy.
In summary, the results of this study provide a hypothesis, at the molecular level, for the partial agonism of the PP and the (S)-enantiomers in our 2,3-diCl-phenylpiperazine series of compounds. In our future synthesis of D3R partial agonists, we will design compounds that induce similar conformational changes in the ligand binding site (Fig. 6), and expect that the iterations between computational analysis and experimental validation will render refined insights for more efficient detection of structural features necessary for partial agonism. In addition, the continuing advancement of computational hardware allows extensive and relatively long-timescale of MD simulations described in this study to be more routinely performed. Importantly, even though MD-based rational drug design requires significantly more resources than those based on individual snapshots of protein conformations, adequate consideration of the receptor conformational changes in response to binding of specific ligands appears to be the key to more challenging drug discovery tasks.34 Thus, in combination with quantitative tools to identify, analyze, and classify the conformational changes, such as the PIA-GPCR described herein, MD-based molecular mechanistic studies establish practical platforms for structure-based drug design by taking functionally relevant conformational spectra into account.
EXPERIMENTAL METHODS
Refinement of D3R model
Of note, in the D3R crystal structure the N-terminus and a large stretch of residues in intracellular loop 3 (ICL3) were not resolved.11 Starting with our previous equilibrated D3R models,11–13 we improved the model further by building the missing N-terminus, as well as a small intracellular loop connecting TM5 and TM6. We built the N-terminus de novo using Rosetta.35 For the large stretch of missing residues in ICL3 (222–318), we used Modeller 36 to extend two helical turns at the TM5 C-terminus and three residues at the TM6 N-terminus based on the sequence of D3R, and connected these two ends with a poly-Gly chain. By varying the length of the poly-Gly chain from 6 to 16 residues, the optimal length was determined to prevent distortion of the TM5 C-terminus. The criterion was that the helical axis was not significantly bent in the top 40% of the scored ensemble of homology models based on each linker length. The final construct had 9 Gly and was found to be fully functional in a Go BRET assay (Prashant Donthamsetti and Jonathan Javitch, unpublished). An ICL3 deletion construct of similar length (4 residues shorter than our truncated ICL3) has been crystallized for the β1-adrenergic receptor, and this construct was also found to couple to G proteins17.
Computational characterization of the ligand binding modes
Docking was performed using a core-restrained induced-fit docking (IFD) protocol in the Schrödinger software (release 2014–3; Schrödinger, LLC: New York, NY),13 and representative poses were selected for simulations.
MD simulations of the receptor-ligand complexes were performed in the explicit water-POPC lipid bilayer solvent environment using Desmond Molecular Dynamics System (version 3.8–4.3; D. E. Shaw Research, New York, NY) with the CHARMM36 protein force field,37–39 CHARMM36 lipid force field,40 and TIP3P water model. The ligand poses in the starting model of the simulations was derived by molecular docking, and/or by superimposing and mutating a pose from a structurally similar ligand – for example, (S)-4 poses were derived from both molecular docking of this compound in the D3R model, and mutating from a (S)-9 pose), in order to generate different starting points – however, we have assumed that the PP, 4-phenylpiperazine, is bound in the OBS.11 The ligand parameters were obtained from the GAAMP server,41 with the initial force field based on CGenFF assigned by ParamChem.42 The protonation state of ligands at pH 7.0 was predicted by the Epik program in the Schrödinger software (release 2014–3, Schrödinger, LLC: New York, NY, 2013). The system charges were neutralized and a solvent concentration of 0.15 M NaCl was added. The total system consisted of ~107,000 atoms. The system was initially minimized and equilibrated with restraints on the ligand heavy atoms and protein backbone atoms, followed by an isothermal-isobaric MD production at 310 K with all atoms unrestrained, as described previously.25 For each ligand, we ran multiple trajectories; total simulation lengths of more than 28 µs were collected (Table S1).
Conformational Clustering Analysis
For each MD trajectory, the frames were clustered by the clustering protocol in our previously described program Protein Interaction Analyzer (PIA) to identify the segment of the trajectory that has equilibrated (in the cases of the trajectories for the (S)-enantiomers, they have evolved significantly from the starting point of the simulation).25, 26, 43 Briefly, the clustering protocol is based on a dissimilarity matrix of pairwise Cα-ifRMSDs (i.e., iterative fit RMSD of all the Cα atoms 44) for all frames, using the agglomerative nesting algorithm and the average cluster linkage criterion. Such a protocol ensures the same clustering criteria applied consistently for all the trajectories. The conformational analysis was applied to the equilibrated segments of the MD trajectories.
Comparative conformational analysis
The TM helices in D3R were divided into extracellular, middle, and intracellular sub-segments for the coarse-grained conformational analysis. We defined the sub-segments by taking into considerations of a few general rules: i) the residues forming one helical turn before and after the highly conserved proline residues within TMs, e.g. Pro2005.50, should be assigned within one sub-segment, ii) the OBS residues in a TM should be assigned to the extracellular and/or middle sub-segments, iii) the boundaries between adjacent sub-segments at extracellular-middle and middle-intracellular should be aligned at similar z-coordinates (z-axis is the membrane normal) across all seven TMs. The sub-segments in D3R were defined as follows: NT (N-terminus, residues 1–26), TM1e (extracellular (e) sub-segment of TM1, residues 271.29−331.36), TM1m (middle (m) sub-segment of TM1, residues 341.37−421.45), TM1i (intracellular (i) sub-segment of TM1, residues 431.46−571.60), IL1 (intracellular loop, residues 58–62), TM2i (residues 632.38−762.51), TM2m (residues 772.52−862.61), TM2e (residues 872.62−912.66), EL1 (residues 92–99), TM3e (residues 1003.22−1093.31), TM3m (residues 1103.32−1163.38), TM3i (residues 1173.39−1343.56), IL2 (residues 135–144), TM4i (residues 1454.37−1574.49), TM4m (residues 1584.50−1634.55), TM4e (residues 1644.56−1704.62), EL2 (residues 171–185), TM5e (residues 1865.36−1945.44), TM5m (residues 1955.45−2025.52), TM5i (residues 2035.53−2185.68), IL3 (residues 219–321), TM6i (residues 3226.28−3406.46), TM6m (residues 3416.47−3486.54), TM6e (residues 3496.55−3546.60), EL3 (residues 355–361), TM7e (residues 3627.32−3687.38), TM7m (residues 3697.39−3767.46), TM7i (residues 3777.47−3857.55), H8 (residues 386–400).
We developed an automated conformational analysis tool (PIA-GPCR) to quantify the complexity of conformational transitions in the partial activation mechanism by systematically measuring four structural features: 1) distance between centers of mass of TM sub-segments, 2) angle between the axes of TM sub-segments, 3) distance between Cα atoms of ligand binding site residues, and 4) X1 dihedral angle of ligand binding site residues (Fig. S1). The analysis workflow consists of the following steps: i) measurement of the four metrics across all models in the compared states; ii) generation of the difference matrices for each measure. To compare crystal structures, the measurements are compared for each pair of models across the two compared states; to compare MD simulation results, the measurements from the same cluster of frames are averaged, then the difference between the averages of the compared states are calculated; iii) cancellation of the noise using comparisons within the same state; iv) visualization of the difference matrices between compared states as heat maps; and v) identification and visualization of the most significantly different measurements. The most significantly different measurements are found by ranking the absolute values of the differences, as well as the signal to noise ratios of the difference over the variation.
PIA-GPCR is implemented in Python, and depends on MDTraj45 in managing MD trajectories. The input for PIA-GPCR consists of: 1) definition of the TM sub-segments, 2) binding site residues, and 3) Ballesteros-Weinstein numbering for the binding site residues. Current version of PIA-GPCR comes equipped with these user-defined parameters for β1AR, β2AR, D3R, D2R, M2R, and Rhodopsin.
Synthesis
Anhydrous solvents were purchased from Aldrich and were used without further purification except for tetrahydrofuran, which was freshly distilled from sodium-benzophenone ketyl. All other chemicals and reagents were purchased from Sigma-Aldrich Co. LLC, Combi-Blocks, TCI America, OChem Incorporation, Acros Organics, Maybridge, and Alfa Aesar. Reaction conditions and yields were not optimized. All amine final products were converted into either the oxalate or hydrochloride salt. Spectroscopic data and yields refer to the free base form of the compounds. Teledyne ISCO CombiFlash Rf or glass flash column chromatography was performed using silica gel (EMD Chemicals, Inc.; 230–400 mesh, 60 Å). 1H and 13C NMR spectra were acquired using a Varian Mercury Plus 400 spectrometer at 400 MHz and 100 MHz, respectively. Chemical shifts are reported in parts-per-million (ppm) and referenced according to deuterated solvent for 1H spectra (CDCl3, 7.26 or CD3OD, 3.31) and 13C spectra (CDCl3, 77.2, CD3OD, 49.0). Infrared (IR) spectra were obtained (neat) on a Perkin Elmer Spectra Two FTIR spectrometer. Gas chromatography-mass spectrometry (GC/MS) data were acquired (where obtainable) using an Agilent Technologies (Santa Clara, CA) 6890N GC equipped with an HP-5MS column (cross-linked 5% PH ME siloxane, 30 m × 0.25 mm i.d. × 0.25 µm film thickness) and a 5973 mass-selective ion detector in electron-impact mode. Ultrapure grade helium was used as the carrier gas at a flow rate of 1.2 mL/min. The injection port and transfer line temperatures were 250 and 280 °C, respectively, and the oven temperature gradient used was as follows: the initial temperature (100 °C) was held for 3 min and then increased to 295 °C at 15 °C/min over 13 min, and finally maintained at 295 °C for 10 min. In some cases, HPLC (see details below) and HRMS were additionally used to demonstrate compound identification and purity. To acquire high resolution mass spectrometry (HRMS) data, 1 µL of sample was mixed with 1 µL of matrix (saturated solution of 2,4,6-trihydroxyacetophenone/2,5-dihydroxybenzoic acid in 50/50 ethanol/water), then 1 µL was deposited on a stainless steel plate and analyzed in positive ion mode in a MALDI LTQ-XL-Orbitrap (Thermo-Scientific, San Jose, CA) using a laser energy fixed at 6 µJ and a mass resolution of 60,000 at an m/z of 400 Th. Combustion analysis was performed by Atlantic Microlab, Inc. (Norcross, GA) and the results agree within ±0.4% of calculated values\Melting point determination was conducted using a Thomas-Hoover melting point apparatus or SRS OptiMelt MPA100-Automated melting point system and are uncorrected. On the basis of NMR and combustion data, all final compounds are ≥95% pure.
General amidation procedure
1,1’-Carbonyldiimidazole (CDI), (1 equiv) was added to a solution of carboxylic acid (1 equiv) in THF (10 mL/mmol). The resulting mixture was stirred at room temperature for 2 h. The solution was cooled to 0 °C and the amine (1 equiv) in THF (3 mL/mmol) was added drop wise. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was concentrated, and the crude product was diluted with CHCl3 (20 mL/mmol) and washed with saturated aqueous NaHCO3 solution (2 × 10 mL). The organic layer was dried over MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography (8% MeOH:CHCl3) or as indicated.
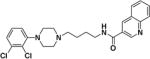
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butyl)quinoline-3-carboxamide (3)
Compound 3 was synthesized according to General Method A using 122, 46 (990 mg, 3.27 mmol) and quinoline-3-carboxylic acid (562 mg, 3.24 mmol). The crude product was purified by column chromatography as described for General Method A to give 3 (701 mg, 47% yield) as an off-white solid, which was converted to the oxalate salt in 2-propanol. Mp. 222–224 °C (oxalate salt); 1H NMR (CDCl3) δ 9.26 (sd, J = 2.4 Hz, 1H), 8.58 (sd, J = 2.4 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.81 (td, J = 8.2, 1.5 Hz, 1H), 7.61 (td, J = 8.4, 1.0 Hz, 1H), 7.20 (br s, 1H), 7.13 (dd, J = 8.0, 1.6 Hz, 1H), 7.06 (t, J = 8.0 Hz, 1H), 6.95 (dd, J = 8.0, 1.6 Hz, 1H), 3.57 (q, J = 12.0 Hz, 2H), 2.97 (br s, 4H), 2.63 (br s, 4H), 2.50 (t, J = 6.8 Hz, 2H), 1.82-1.67 (m, 4H); 13C NMR (CDCl3) δ 165.9, 151.0, 149.2, 148.3, 135.5, 133.97, 131.1, 129.4, 128.7, 127.7, 127.5, 127.47, 127.40, 126.9, 124.6, 118.5, 58.0, 53.3, 51.1, 40.3, 27.5, 24.6; Anal. (C24H26Cl2N4O· C2H2O4·¼H2O) C, H, N.
N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)quinoline-3-carboxamide ((±)4).47
Compound (±)-4 was synthesized according to General Method A using 248 (740 mg, 2.32 mmol) and quinoline-3-carboxylic acid (400 mg, 2.31 mmol). The crude product was purified by column chromatography as described for General Method A to give (±)-4 (300 mg, 27% yield) as a light brown solid, which was converted to the oxalate salt in 2-propanol/acetone. Mp. 193–194.5 °C (oxalate salt); 1H NMR (CDCl3) δ 9.30 (sd, J = 2.4 Hz, 1H), 8.62 (sd, J = 2.0 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.79 (td, J = 8.2, 1.6 Hz, 1H), 7.71 (br s, 1H), 7.60 (td, J = 8.2, 1.2 Hz, 1H), 7.19-7.13 (m, 2H), 6.95 (dd, J = 7.0, 2.6 Hz, 1H), 4.02–3.93 (m, 3H), 3.56-3.48 (m, 1H), 3.08 (br s, 4H), 2.91-2.87 (m, 2H), 2.63-2.62 (m, 2H), 2.53-2.43 (m, 2H), 1.91-1.84 (m, 1H), 1.71-1.62 (m, 1H); 13C NMR (CDCl3) δ 165.3, 150.9, 149.2, 148.3, 135.6, 134.1, 131.1, 129.4, 128.8, 127.6, 127.5, 127.4, 127.2, 127.0, 124.8, 118.6, 66.9, 63.6, 53.2, 51.4, 38.9, 32.9; Anal. (C24H26Cl2N4O2· ½C2H2O4·½H2O) C, H, N.
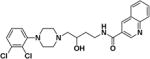
(R)-N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)quinoline-3-carboxamide ((R)-4)
Compound (R)-4 was synthesized according to General Method A using (R)-549 (74 mg, 0.23 mmol) and quinoline-3-carboxylic acid (40 mg, 0.23 mmol). The crude product was purified by column chromatography as described for General Method A to give (R)-4 (33 mg, 30% yield) as a brown solid. Mp. 159–160 °C (free base); [α]25D + 2.07° (c 0.82, MeOH); 1H NMR (CDCl3) δ 9.30 (d, J = 2.0 Hz, 1H), 8.61 (d, J = 2.1 Hz, 1H), 8.13 (d, J = 8.5 Hz, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.78 (tt, J = 6.9, 1.8 Hz, 2H), 7.59 (t, J = 7.5 Hz, 1H), 7.16 – 7.14 (m, 2H), 6.94 (dt, J = 7.0, 2.1 Hz, 1H), 3.96 (dq, J = 16.3, 6.0, 5.2 Hz, 2H), 3.78 (s, 1H), 3.59 – 3.45 (m, 1H), 3.07 (s, 4H), 2.88 (dd, J = 10.9, 5.3 Hz, 2H), 2.62 (d, J = 8.6 Hz, 2H), 2.53 – 2.41 (m, 2H), 1.94 – 1.80 (m, 1H), 1.66 (dtd, J = 13.9, 9.2, 4.1 Hz, 1H); 13C NMR (CDCl3) δ 165.3, 150.9, 149.1, 148.4, 135.6, 134.1, 131.1, 129.3, 128.8, 127.5, 127.4, 127.3, 127.2, 127.0, 124.8, 118.6, 66.8, 63.6, 53.2, 51.3, 38.8, 33.0; IR: 3329.1 (s, br) cm−1; HRMS (MALDI-TOF) m/z 473 (M+H)+. HRMS (MALDI-TOF) Calcd for: m/z 473.1506 (M+H)+ Found: m/z 473.1503 (M+H)+, Fig. S2, S.I.).
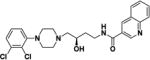
(S)-N-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)-3-hydroxybutyl)quinoline-3-carboxamide ((S)-4)
Compound (S)-4 was synthesized according to General Method A using (S)-549 (82 mg, 0.26 mmol) and quinoline-3-carboxylic acid (44 mg, 0.26 mmol). The crude product was purified by column chromatography as described for General Method A to give (S)-4 (42 mg, 34% yield) as a light brown solid. Mp. 160–161 °C (free base); [α]25D –4.40° (c 0.84, MeOH); 1H NMR (CDCl3) δ 9.30 (d, J = 2.2 Hz, 1H), 8.66 – 8.56 (m, 1H), 8.13 (dd, J = 8.5, 1.0 Hz, 1H), 7.90 (dd, J = 8.2, 1.4 Hz, 1H), 7.80–7.76 (m, 1H), 7.74 (s, 1H), 7.59 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.21 – 7.08 (m, 2H), 6.94 (dd, J = 7.0, 2.5 Hz, 1H), 4.06 – 3.90 (m, 2H), 3.59 – 3.46 (m, 1H), 3.17 – 2.99 (m, 4H), 2.88 (dt, J = 10.1, 4.5 Hz, 2H), 2.62 (d, J = 7.4 Hz, 2H), 2.53 – 2.40 (m, 2H), 1.91-1.87 (m, 1H), 1.71-1.63 (m, 1H); 13C NMR (CDCl3) δ 165.3, 150.9, 149.2, 148.3, 135.5, 134.1, 131.0, 129.4, 128.8, 127.5, 127.5, 127.3, 127.2, 127.0, 124.8, 118.6, 66.9, 63.6, 53.2, 51.4, 38.9, 32.9; IR: 3321.17 (s, br) cm−1; HRMS (MALDI-TOF) m/z 473 (M+H)+. HRMS (MALDI-TOF) Calcd for: m/z 473.1506 (M+H)+ Found: m/z 473.1497 (M+H)+ (Fig. S3, S.I.).
(R)-1-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butan-2-ol ((R)-6)
To a solution of 8 (1.60 g, 6.93 mmol) in 2-propanol (30 mL) was added (R)-2-ethyloxirane (500 mg, 6.93 mmol). The reaction mixture was stirred at reflux (82 °C) overnight under an argon atmosphere. After cooling the mixture to room temperature, the solvent was removed in vacuo, and the crude product was purified by flash column chromatography (50-50% EtOAC:Hexanes) to give the product (R)-6. (215 mg, 10% yield) as an off-white solid. Mp. 54–56 °C (free base)); [α]23D – 21.95° (c 0.92, MeOH); 1H NMR (CDCl3) δ 7.17 – 7.10 (m, 2H), 6.94 (dd, J = 6.7, 2.8 Hz, 1H), 3.67-3.61 (m, 1H), 3.06 (s, 4H), 2.85 (dd, J = 10.8, 5.5 Hz, 2H), 2.57 (d, J = 9.2 Hz, 2H), 2.43 (dd, J = 12.3, 3.1 Hz, 1H), 2.32 (dd, J = 12.3, 10.5 Hz, 1H), 1.59 – 1.36 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3) δ 151.1, 134.0, 127.5, 127.4, 124.6, 118.6, 67.4, 63.8, 53.3, 51.4, 27.7, 9.9; IR: 3447.78 (s, br) cm−1; GC-MS (EI) m/z 302 (M+); Anal. (C14H20Cl2N2O) C, H, N.
(S)-1-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butan-2-ol ((S)-6)
Compound (S)-6 was synthesized as described for (R)-6 using 8 (1.60 g, 6.93 mmol) and (S)-2-ethyloxirane (500 mg, 6.93 mmol) in 2-propanol (30 mL). The solvent was removed under reduced pressure, and the crude product was purified by column chromatography (50-50% EtOAC:Hexanes) to give (S)-6 (215 mg, 12% yield) as a brown solid. Mp. 55–57 °C (free base); [α]23D + 22.27° (c 1.01, MeOH); 1H NMR (CDCl3) δ 7.17 – 7.10 (m, 2H), 6.94 (dd, J = 6.7, 2.8 Hz, 1H), 3.67-3.61 (m, 1H), 3.06 (s, 4H), 2.86 (dd, J = 10.7, 5.6 Hz, 2H), 2.57 (d, J = 8.2 Hz, 2H), 2.43 (dd, J = 12.3, 3.1 Hz, 1H), 2.32 (dd, J = 12.3, 10.5 Hz, 1H), 1.55 – 1.35 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3) δ 151.1, 134.0, 127.5, 127.4, 124.6, 118.6, 67.4, 63.8, 53.3, 51.4, 27.7, 9.9; IR: 3451.13 (s, br) cm−1; GC-MS (EI) m/z 302 (M+); Anal. (C14H20Cl2N2O) C, H, N.
(R)-1-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propan-2-ol ((R)-7)
Compound (R)-7 was synthesized as described for (R)-6 using 8 (3.98 g, 17.2 mmol) and (R)-2-methyloxirane (1.21 mL, 17.2 mmol) in 2-propanol (35 mL). The solvent was removed under reduced pressure and the crude product was purified by column chromatography (50-50% EtOAC:Hexanes) to give (R)-7 (1.15 g, 23% yield) as a white solid. Mp. 101–102 °C (free base); [α]23D – 20.09° (c 1, MeOH); 1H NMR (CDCl3) δ 7.16 – 7.09 (m, 2H), 6.93 (dd, J = 6.7, 2.9 Hz, 1H), 3.92 – 3.80 (m, 1H), 3.44 (s, 1H), 3.05 (q, J = 6.1, 5.4 Hz, 4H), 2.83 (dt, J = 10.0, 4.6 Hz, 2H), 2.64 – 2.49 (m, 2H), 2.39 (dd, J = 12.3, 3.0 Hz, 1H), 2.28 (dd, J = 12.4, 10.4 Hz, 1H), 1.14 (d, J = 6.1 Hz, 3H); 13C NMR (CDCl3) δ 151, 134.0, 127.5, 127.4, 124.6, 118.6, 65.6, 62.3, 53.3, 51.4, 19.9; IR: 3438.79 (s, br) cm−1; GC-MS (EI) m/z 288 (M+) Anal. (C13H18Cl2N2O) C, H, N.
(S)-1-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propan-2-ol ((S)-7)
Compound (S)-7 was synthesized as described for (R)-6 using 8 (3.98 g, 17.22 mmol) and (S)-2-methyloxirane (1.00 g, 17.2 mmol) in 2-propanol (35 mL). The solvent was removed under reduced pressure and the crude product was purified by column chromatography (50-50% EtOAC:Hexanes) to give (S)-7 (1.06 g, 21% yield) as an off-white solid. Mp. 100–102 °C (free base); [α]23D + 20.98° (c 1.02, MeOH); 1H NMR (CDCl3) δ 7.16 – 7.10 (m, 2H), 6.93 (dd, J = 6.7, 2.9 Hz, 1H), 3.92 – 3.79 (m, 1H), 3.05 (d, J = 7.5 Hz, 4H), 2.83 (dt, J = 9.9, 4.5 Hz, 2H), 2.56 (d, J = 8.3 Hz, 2H), 2.39 (dd, J = 12.3, 3.1 Hz, 1H), 2.28 (dd, J = 12.4, 10.4 Hz, 1H), 1.14 (d, J = 6.1 Hz, 3H); 13C NMR (CDCl3) δ 151, 134.0, 127.5, 127.4, 124.6, 118.6, 65.6, 62.3, 53.3, 51.4, 19.9; IR: 3437.14 (s, br) cm−1; GC-MS (EI) m/z 288 (M+); Anal. (C13H18Cl2N2O) C, H, N.
Chiral Chromatography analysis
The chiral purity of compounds (R)- and (S)-4 and (R)- and (S)-7 was determined by HPLC analysis using a Daicel CHIRALCEL OD-H 14325 semi-preparative column (I.D. × L = 10 × 250 mm). Elution \ was achieved using hexane/2-PrOH (70:30 v/v) for compound (±)-, (R)- and (S)-4 at a flow rate of 0.5 mL/min with ultraviolet absorption at 225 nM (Waters 2487 detector, Waters Corp.) Chromatographic data were acquired using the Empower 2 software. The retention times for compounds (R)- and (S)-4 were 21.98 and 27.39 min respectively. Elution was achieved using hexane/2-PrOH (99:1 v/v) for compound (±)-, (R)- and (S)-7; the retention times for compounds (R)- and (S)-7 were 45.08 and 49.34 min respectively.
X-ray crystal data on compounds (R)-7 and (S)-7
Single-crystal X-ray diffraction data on compounds (R)-7 and (S)-7 were collected using CuKα radiation (λ = 1.54178Å) and a Bruker Platinum-135 CCD area detector. Crystals were prepared for data collection by coating with high viscosity microscope oil. The oil-coated crystal was mounted on a micro-mesh mount (Mitergen, Inc.) and transferred to the cold stream on the diffractometer and a diffraction data collected at 150°K. The structures were solved by direct methods and refined by full-matrix least squares on F2 values using the programs found in the SHELXTL suite (Bruker, SHELXTL v6.10, 2000, Bruker AXS Inc., Madison, WI). Corrections were applied for Lorentz, polarization, and absorption effects. Parameters refined included atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms on carbons were included using a riding model [coordinate shifts of C applied to H atoms] with C-H distance set at 0.96 Å. The absolute configuration was determined from the diffraction data using the method of Parsons et al.50 Complete information on data collection and refinement is available in S.I. (Tables S3 and S4).
For (R)-7 the 0.445 × 0.161 × 0.084 mm3 data crystal was orthorhombic in space group P 212121, with unit cell dimensions a = 7.4149(3), b = 11.2516(4), and c = 16.6967(5) Å. The data was 96.1% complete to 68.87° θ (~ 0.83 Å) with an average redundancy of 5.13. The final anisotropic full matrix least-squares refinement on F2 with 96 variables and one constraint converged at R1 = 1.97%, for the observed data and wR2 = 5.36% for all data. The absolute structure parameter was 0.047(4).
For (S)-7 the 0.291 × 0.184 × 0.055 mm3 data crystal was orthorhombic in space group P 212121, with unit cell dimensions a = 7.4199(3), b = 11.2614(5), and c = 16.7031(7) Å. The data was 98.8% complete to 68.30° θ (~ 0.83 Å) with an average redundancy of 5.43. The final anisotropic full matrix least-squares refinement on F2 with 96 variables and one constraint converged at R1 = 1.99%, for the observed data and wR2 = 5.73% for all data. The absolute structure parameter was 0.047(5).
Radioligand binding assays
Binding at dopamine D2-like receptors was determined using previously described methods. 51 Membranes were prepared from HEK293 cells stably expressing human D2R or D3R, grown in a 50:50 mix of DMEM and Ham’s F12 culture media, supplemented with 20 mM HEPES, 2 mM L-glutamine, 0.1 mM non-essential amino acids, 1X antibiotic/antimycotic, 10% heat-inactivated fetal bovine serum, and 200 µg/mL hygromycin (Life Technologies, Grand Island, NY) and kept in an incubator at 37°C and 5% CO2. Upon reaching 80–90% confluence, cells were harvested using pre-mixed Earle’s Balanced Salt Solution (EBSS) with 5 mM EDTA (Life Technologies) and centrifuged at 3000 rpm for 10 min at 21 °C. The supernatant was removed and the pellet was resuspended in 10 mL hypotonic lysis buffer (5 mM MgCl2 · 6 H2O, 5 mM Tris, pH 7.4 at 4 °C) and centrifuged at 20,000 rpm for 30 min at 4 °C. The pellet was then resuspended in fresh EBSS buffer made from 8.7 g/L Earle’s Balanced Salts without phenol red (US Biological, Salem, MA), 2.2 g/L sodium bicarbonate, pH to 7.4. A Bradford protein assay (Bio-Rad, Hercules, CA) was used to determine the protein concentration and membranes were diluted to 500 µg/mL and stored in a −80 °C freezer for later use.
Radioligand competition binding experiments were conducted using thawed membranes. Test compounds were freshly dissolved in 30% DMSO and 70% H2O to a stock concentration of 100 µM. To assist the solubilization of free-base compounds, 10 µl of glacial acetic acid was added along with the DMSO. Each test compound was then diluted into 11 half-log serial dilutions using 30% DMSO vehicle; final test concentrations ranged from 10 µM to 10 pM. Previously frozen membranes were diluted in fresh EBSS to a 200 µg/mL (for hD2R or hD3R) stock for binding. Radioligand competition experiments were conducted in 96-well plates containing 300 µl fresh EBSS buffer with 0.2 mM sodium metabisulfite, 50 µl of diluted test compound, 100 µl of membranes (20 µg total protein for hD2R or hD3R), and 50 µl of [3H]N-methylspiperone (0.4 nM final concentration; Perkin Elmer). Nonspecific binding was determined using 10 µM butaclamol (Sigma-Aldrich, St. Louis, MO) and total binding was determined with 30% DMSO vehicle. The reaction was incubated for one hour at room temperature and then terminated by filtration through Perkin Elmer UniFilter-96 GF/B filters, presoaked for one hour in 0.5% polyethylenimine, using a Brandel 96-Well Plates Harvester Manifold (Brandel Instruments, Gaithersburg, MD). The filters were washed 3 times with 3 mL (3 × 1 mL/well) of ice cold EBSS buffer. 65 µL Perkin Elmer MicroScint 20 Scintillation Cocktail was added to each well and filters were counted using a Perkin Elmer MicroBeta Microplate Counter. IC50 values for each compound were determined from dose-response curves and Ki values were calculated using the Cheng-Prusoff equation;52 these analyses were performed using GraphPad Prism version 6.00(GraphPad Software, San Diego, CA). Reported Ki values were determined from least three independent experiments, each with duplicate determinations.
Functional assay
The BRET-based Go activation assay and β-arrestin recruitment assay were described previously.12, 53 Briefly, for Go activation assay, HEK293T cells were transiently transfected with pcDNA3.1 vectors carrying D2R or D3R, GαoA fused to Renilla luciferase 8 (Rluc8) within anα-helical domain, Gβ1 fused to V1 (the N-terminal split of mVenus; residues 1–155) at its N-terminus, and Gγ2 fused to V2 (the C-terminal split of mVenus; residues 156–240) and for β-arrestin recruitment assay the cells were transiently transfected with pcDNA3.1 vectors carrying D3R fused to Rluc8 at the C-terminus, β-arrestin 2 fused to mVenus at the N-terminus, and G protein receptor kinase 2 using polyethylenimine (Polysciences, Inc.). Transfected cells were maintained in culture with DMEM (GIBCO) supplemented with 10% FBS, and transfection media was replaced with fresh media after ~24 h. Experiments were performed ~48 h after transfection. Transfected cells were washed, harvested, and resuspended in PBS supplemented with 5 mM glucose and distributed in 96-well black/white plates (Wallac, PerkinElmer Life and Analytical Sciences). Cells were incubated with coelenterazine H (5 µM) (Dalton Pharma Services), and after 8 min, compounds were added with final concentrations ranging from 10 pM to 100 µM. For the Go activation assay, BRET1 signal was measured after 2 min using a Pherastar FS (BMG Labtech). For the β-arrestin recruitment assay BRET1 signal was measured after 30 min using a Mithras LB940 (Berthold Technologies). The signal was calculated as the ratio of the light emitted by mVenus (510–540 nm) over that emitted by RLuc8 (485 nm). Data were normalized to vehicle (0%) and dopamine (100%), and nonlinear regression analysis was performed using the sigmoidal dose-response function in GraphPad Prism to generate EC50 values. Data are expressed as a percentage of the maximum dopamine-stimulated response as mean ± SEM.
Supplementary Material
Acknowledgments
Support for this research was provided to MM, CAB, HY, OMB, AB, ME, TMK, VK, CZ, RV, AHN and LS by the National Institute on Drug Abuse Intramural Research Program. PD and JAJ are supported by DA022413 and MH054137. The X-ray crystallographic work was supported by NIDA through Interagency Agreement ADA 12003-005 with the Naval Research Laboratory (JRD). We acknowledge Caitlin Burzynski and Catherine Schweppe for their technical assistance with the binding assays, Ara Abramyan for assistance with analyzing the MD simulation data. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
ABBREVIATIONS USED
- DA
dopamine
- SAR
structure activity relationship
- TM
transmembrane
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- THF
tetrahydrofuran
- CDI
1,1’-carbonyldiimidazole
- NMR
nuclear magnetic resonance
- CDCl3
deuterated chloroform
- CD3OD
deuterated methanol, OBS, orthosteric binding site
- SBP
secondary binding pocket, CMA, chloroform/methanol/ammonium hydroxide
Footnotes
ASSOCIATED CONTENT
Supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
List of MD simulations, and the examples of validation analysis for PIA-GPCR. Elemental analysis results for all final compounds, HRMS data for compounds (R)- and (S)-4 and HPLC chromatogram analysis for compounds (R)- and (S)-4 and (R)- and (S)-7. X-ray structures of (R)- and (S)-7 (PDF)
Representative D3R models of in complex with (R)- and (S)-4 (PDB)
SMILES data (CSV)
Accession Codes
Atomic coordinates of (R)- and (S)-7 have been deposited with the Cambridge Crystallographic Data Centre (deposition numbers 1489644 and 1489645). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: + 44(0)-1223-336033 or deposit@ccdc.cam.ac.uk]. Authors will release the atomic coordinates and experimental data upon publication.
References
- 1.Heidbreder CA, Newman AH. Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann. N. Y. Acad. Sci. 2010;1187:4–34. doi: 10.1111/j.1749-6632.2009.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stemp G, Ashmeade T, Branch CL, Hadley MS, Hunter AJ, Johnson CN, Nash DJ, Thewlis KM, Vong AK, Austin NE, Jeffrey P, Avenell KY, Boyfield I, Hagan JJ, Middlemiss DN, Reavill C, Riley GJ, Routledge C, Wood M. Design and synthesis of trans-N-[4-[2-(6-cyano-1,2,3, 4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): A potent and selective dopamine D(3) receptor antagonist with high oral bioavailability and CNS penetration in the rat. J. Med. Chem. 2000;43:1878–1885. doi: 10.1021/jm000090i. [DOI] [PubMed] [Google Scholar]
- 3.Reavill C, Taylor SG, Wood MD, Ashmeade T, Austin NE, Avenell KY, Boyfield I, Branch CL, Cilia J, Coldwell MC, Hadley MS, Hunter AJ, Jeffrey P, Jewitt F, Johnson CN, Jones DN, Medhurst AD, Middlemiss DN, Nash DJ, Riley GJ, Routledge C, Stemp G, Thewlis KM, Trail B, Vong AK, Hagan JJ. Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J. Pharmacol. Exp. Ther. 2000;294:1154–1165. [PubMed] [Google Scholar]
- 4.Vorel SR, Ashby CR, Jr, Paul M, Liu X, Hayes R, Hagan JJ, Middlemiss DN, Stemp G, Gardner EL. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. J. Neurosci. 2002;22:9595–9603. doi: 10.1523/JNEUROSCI.22-21-09595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pilla M, Perachon S, Sautel F, Garrido F, Mann A, Wermuth CG, Schwartz JC, Everitt BJ, Sokoloff P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature. 1999;400:371–375. doi: 10.1038/22560. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Ladona FJ, Cox BF. BP 897, a selective dopamine D3 receptor ligand with therapeutic potential for the treatment of cocaine-addiction. CNS Drug Rev. 2003;9:141–158. doi: 10.1111/j.1527-3458.2003.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keck TM, Burzynski C, Shi L, Newman AH. Beyond small-molecule SAR: using the dopamine D3 receptor crystal structure to guide drug design. Adv. Pharmacol. 2014;69:267–300. doi: 10.1016/B978-0-12-420118-7.00007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keck TM, John WS, Czoty PW, Nader MA, Newman AH. Identifying medication targets for psychostimulant addiction: unraveling the dopamine D3 receptor hypothesis. J. Med. Chem. 2015;58:5361–5380. doi: 10.1021/jm501512b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreira FA, Dalley JW. Dopamine receptor partial agonists and addiction. Eur. J. Pharmacol. 2015;752:112–115. doi: 10.1016/j.ejphar.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Appel NM, Li SH, Holmes TH, Acri JB. Dopamine D3 receptor antagonist (GSK598809) potentiates the hypertensive effects of cocaine in conscious, freely-moving dogs. J. Pharmacol. Exp. Ther. 2015;354:484–492. doi: 10.1124/jpet.115.224121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newman AH, Beuming T, Banala AK, Donthamsetti P, Pongetti K, LaBounty A, Levy B, Cao J, Michino M, Luedtke RR, Javitch JA, Shi L. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012;55:6689–6699. doi: 10.1021/jm300482h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michino M, Donthamsetti P, Beuming T, Banala A, Duan L, Roux T, Han Y, Trinquet E, Newman AH, Javitch JA, Shi L. A single glycine in extracellular loop 1 is the critical determinant for pharmacological specificity of dopamine D2 and D3 receptors. Mol. Pharmacol. 2013;84:854–864. doi: 10.1124/mol.113.087833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ballesteros JA, Weinstein H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 19.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE. Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18684–18689. doi: 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swaminath G, Xiang Y, Lee TW, Steenhuis J, Parnot C, Kobilka BK. Sequential binding of agonists to the beta2 adrenoceptor. Kinetic evidence for intermediate conformational states. J. Biol. Chem. 2004;279:686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- 22.Newman AH, Cao J, Bennett CJ, Robarge MJ, Freeman RA, Luedtke RR. N-(4-[4-(2,3-dichlorophenyl)piperazin-1-yl]butyl, butenyl and butynyl)arylcarboxamides as novel dopamine D(3) receptor antagonists. Bioorg. Med. Chem. Lett. 2003;13:2179–2183. doi: 10.1016/s0960-894x(03)00389-5. [DOI] [PubMed] [Google Scholar]
- 23.Banala AK, Levy BA, Khatri SS, Furman CA, Roof RA, Mishra Y, Griffin SA, Sibley DR, Luedtke RR, Newman AH. N-(3-fluoro-4-(4-(2-methoxy or 2,3-dichlorophenyl)piperazine-1-yl)butyl)arylcarboxamides as selective dopamine D3 receptor ligands: critical role of the carboxamide linker for D3 receptor selectivity. J. Med. Chem. 2011;54:3581–3594. doi: 10.1021/jm200288r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman AH, Grundt P, Cyriac G, Deschamps JR, Taylor M, Kumar R, Ho D, Luedtke RR. N-(4-(4-(2,3-dichloro- or 2-methoxyphenyl)piperazin-1-yl)butyl)heterobiarylcarboxamides with functionalized linking chains as high affinity and enantioselective D3 receptor antagonists. J. Med. Chem. 2009;52:2559–2570. doi: 10.1021/jm900095y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michino M, Free RB, Doyle TB, Sibley DR, Shi L. Structural basis for Na(+)-sensitivity in dopamine D2 and D3 receptors. Chem. Commun. (Cambridge, U. K.) 2015;51:8618–8621. doi: 10.1039/c5cc02204e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stolzenberg S, Michino M, LeVine MV, Weinstein H, Shi L. Computational approaches to detect allosteric pathways in transmembrane molecular machines. Biochim. Biophys. Acta. 2016;1858:1652–1662. doi: 10.1016/j.bbamem.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weichert D, Banerjee A, Hiller C, Kling RC, Hubner H, Gmeiner P. Molecular determinants of biased agonism at the dopamine D(2) receptor. J. Med. Chem. 2015;58:2703–2717. doi: 10.1021/jm501889t. [DOI] [PubMed] [Google Scholar]
- 29.Kota K, Kuzhikandathil EV, Afrasiabi M, Lacy B, Kontoyianni M, Crider AM, Song D. Identification of key residues involved in the activation and signaling properties of dopamine D3 receptor. Pharmacol. Res. 2015;99:174–184. doi: 10.1016/j.phrs.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 30.Cheng J, Sun X, Li W, Liu G, Tu Y, Tang Y. Molecular switches of the kappa opioid receptor triggered by 6’-GNTI and 5’-GNTI. Sci. Rep. 2016;6:18913. doi: 10.1038/srep18913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rives ML, Rossillo M, Liu-Chen LY, Javitch JA. 6’-Guanidinonaltrindole (6’-GNTI) is a G protein-biased kappa-opioid receptor agonist that inhibits arrestin recruitment. J. Biol. Chem. 2012;287:27050–27054. doi: 10.1074/jbc.C112.387332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. “Molecular switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry. 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joseph-McCarthy D, Campbell AJ, Kern G, Moustakas D. Fragment-based lead discovery and design. J. Chem. Inf. Model. 2014;54:693–704. doi: 10.1021/ci400731w. [DOI] [PubMed] [Google Scholar]
- 34.Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 36.John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 38.Mackerell AD, Jr, Feig M, Brooks CL., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 39.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, Mackerell AD., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang L, Roux B. Automated force field parameterization for non-polarizable and polarizable atomic models based on target data. J. Chem. Theory Comput. 2013;9:3543–3556. doi: 10.1021/ct4003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stolzenberg S, Quick M, Zhao C, Gotfryd K, Khelashvili G, Gether U, Loland CJ, Javitch JA, Noskov S, Weinstein H, Shi L. Mechanism of the association between Na+ binding and conformations at the intracellular gate in neurotransmitter:sodium symporters. J. Biol. Chem. 2015;290:13992–14003. doi: 10.1074/jbc.M114.625343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao C, Stolzenberg S, Gracia L, Weinstein H, Noskov S, Shi L. Ion-controlled conformational dynamics in the outward-open transition from an occluded state of LeuT. Biophys. J. 2012;103:878–888. doi: 10.1016/j.bpj.2012.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernandez CX, Schwantes CR, Wang LP, Lane TJ, Pande VS. MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 2015;109:1528–1532. doi: 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robarge MJ, Husbands SM, Kieltyka A, Brodbeck R, Thurkauf A, Newman AH. Design and synthesis of [(2,3-dichlorophenyl)piperazin-1-yl]alkylfluorenylcarboxamides as novel ligands selective for the dopamine D3 receptor subtype. J. Med. Chem. 2001;44:3175–3186. doi: 10.1021/jm010146o. [DOI] [PubMed] [Google Scholar]
- 47.Keck TM, Banala AK, Slack RD, Burzynski C, Bonifazi A, Okunola-Bakare OM, Moore M, Deschamps JR, Rais R, Slusher BS, Newman AH. Using click chemistry toward novel 1,2,3-triazole-linked dopamine D3 receptor ligands. Bioorg. Med. Chem. 2015;23:4000–4012. doi: 10.1016/j.bmc.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grundt P, Prevatt KM, Cao J, Taylor M, Floresca CZ, Choi JK, Jenkins BG, Luedtke RR, Newman AH. Heterocyclic analogues of N-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butyl)arylcarboxamides with functionalized linking chains as novel dopamine D3 receptor ligands: potential substance abuse therapeutic agents. J. Med. Chem. 2007;50:4135–4146. doi: 10.1021/jm0704200. [DOI] [PubMed] [Google Scholar]
- 49.Kumar V, Rahbek-Clemmensen T, Billesbolle CB, Jorgensen TN, Gether U, Newman AH. Novel and high affinity fluorescent ligands for the serotonin transporter based on (s)-citalopram. ACS Med. Chem. Lett. 2014;5:696–699. doi: 10.1021/ml5000806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parsons S, Flack HD, Wagner T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2013;69:249–259. doi: 10.1107/S2052519213010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J, Levant B, Jiang C, Keck TM, Newman AH, Wang S. Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D(3) receptor antagonists. J. Med. Chem. 2014;57:4962–4968. doi: 10.1021/jm401798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 53.Donthamsetti P, Quejada JR, Javitch JA, Gurevich VV, Lambert NA. Using Bioluminescence Resonance Energy Transfer (BRET) to characterize agonist-induced arrestin recruitment to modified and unmodified G protein-coupled receptors. Curr. Protoc. Pharmacol. 2015;70:2.14.1–2.14.14. doi: 10.1002/0471141755.ph0214s70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.