

Abstract
Aims
The endothelium plays an important role during vascular inflammation. Previous data have demonstrated a high expression level of manganese-superoxide dismutase (MnSOD) in endothelial cells and suggested an important role of MnSOD in several cardiovascular diseases. Manganese (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) has been shown to mimic some of the effects of MnSOD and prevented the development of diabetes and obesity. However, its effect on vascular inflammation and the underlying mechanism is still unknown.
Methods and results
Leukocyte adhesion was evaluated in-vivo and in-vitro using dynamic flow chamber and intravital microscopy in mice. Expression of adhesion molecules induced by TNFα and adhesion of leukocytes to the vessel wall were inhibited by MnTBAP. The anti-inflammatory effect of MnTBAP was partly mediated by up-regulation of the BMPR-II and Smad dependent pathway. Additionally, MnTBAP decelerated the turn-over of endogenous BMPR-II.
Conclusion
Our data demonstrate that MnTBAP activates Smad signaling, preserves the turn-over of BMPR-II and elicits anti-inflammatory effects in endothelial cells, partly mediated by BMPR-II. This finding suggests a potential therapeutic impact of MnTBAP in the treatment of vascular inflammation.
Keywords: MnTBAP, Endothelial cell, Inflammation, BMPR-II
Graphical Abstract
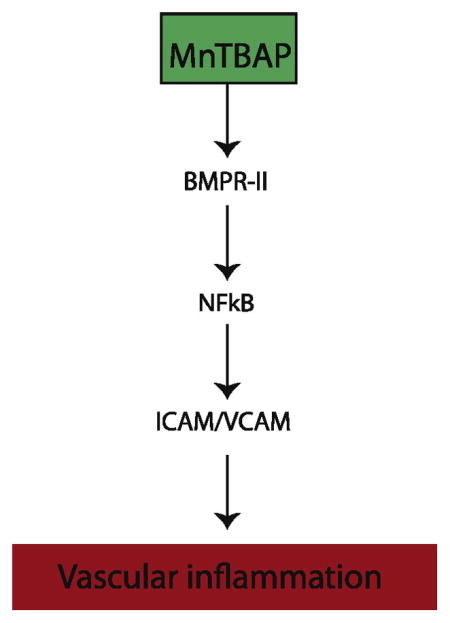
1. Introduction
Endothelial dysfunction is associated with a variety of cardiovascular diseases such as atherosclerosis and hypertension. A variety of inflammatory stimuli are able to induce endothelial dysfunction and confer a pro-adhesive endothelial phenotype resulting in increased leukocyte adhesion [1]. The manganese-superoxide dismutase (MnSOD) belongs to the superoxide dismutase (SOD) family and is the primary antioxidant defence against superoxide radicals within the mitochondria. It is highly expressed in the endothelium [2] and protects the cells against oxidative damage by converting superoxide to H2O2 and O2, and inhibiting free radical reactions [3]. MnSOD has also been found in human atherosclerotic plaques, and specifically in foam cells of the lipid-rich region [4], suggesting a potential role for MnSOD in cardiovascular diseases. MnTBAP is a manganic porphyrin complex that possesses similar effects as MnSOD in catalyzing the dismutation of superoxide radicals (O2−) [5]. We have previously demonstrated that MnTBAP prevented TNFα-induced upregulation of the intercellular adhesion molecule (ICAM) in endothelial cells [6]. This finding suggests anti-inflammatory properties for MnTBAP in endothelial cells and is further supported by works from other groups showing positive effects of MnTBAP in preventing diabetes [7] and obesity [8]. However, the underlying molecular mechanism, by which MnTBAP elicits anti-inflammatory effects in the vasculature, is still not fully understood.
The bone morphogenic protein receptor (BMPR)-II has been traditionally linked with primary pulmonary arterial hypertension (PAH) [9]. Recent studies also suggest a central role of BMPR-II in mediating vascular inflammation. While anti-inflammatory conditions, e.g. statins or stable flow have been shown to increase BMPR-II expression [10], pro-atherogenic factors downregulated BMPR-II expression in endothelial cells causing robust inflammation and atherosclerosis [11]. One mechanism, through which loss of BMPR-II caused vascular inflammation, was the increased production of reactive oxygen species (ROS).
Based on our previous finding that MnTBAP has ROS-scavenging properties and prevented the expression of ICAM within an inflammatory milieu, we hypothesized that MnTBAP would mediate part of its anti-inflammatory effects through regulation of BMPR-II. In the present study, we provide evidence that MnTBAP upregulates BMPR-II expression and elicits anti-inflammatory effects in endothelial cells. Additionally, we demonstrate for the first time, that MnTBAP decelerates the turn-over of endogenous BMPR-II and partially restores BMPR-II expression in endothelial cells, in which protein synthesis of BMPR-II was blocked by cycloheximide.
2. Materials and methods
2.1. Animals
4–8-week-old male C57/BL6J mice (Charles River Laboratories) were used for the study. Animals were maintained in the animal facility at the University Hospital Freiburg. The Standing Committee on Animals at University Freiburg approved all protocols pertaining to experimentation with animals. The animals were maintained in a 22 °C room with a 12-hour light/dark cycle and received drinking water ad libitum. C57/BL6 mice were fed regular chow and treated subcutaneously with either NaCl or with MnTBAP (Calbiochem 475,870, USA).
2.2. Cell cultures
All experiments were performed according to the principles outlined in the Declaration of Helsinki for the use of human tissue and were approved by the ethics review board of the University Hospital Freiburg. Human umbilical vein endothelial cells (HUVECs) were freshly isolated from human umbilical veins of newborns and cultured as previously described [12].
2.3. Dynamic adhesion assay
HUVECs were plated in a 35-mm dish, cultured to a confluence of 90%, and treated with MnTBAP and/or TNFα as indicated. PBMCs from healthy donors were isolated by biocoll separation and stained with CFDA (10 μM). PBMCs were activated with PMA (200 ng/ml) and allowed to adhere for 10 min to the endothelial surface. In some cases, PBMCs were pre-treated with MnTBAP (50 μM) before activated by PMA. In these settings, PBMCs were then incubated with HUVECs that were only activated with TNFα, but no additional MnTBAP. For the dynamic adhesion assay of neutrophils, neutrophils were isolated with heavy and light Histopaque® (Histopaque-1077 and Histopaque-1119; Sigma-Aldrich) as previously described [13]. The GlykoTech flow chamber was assembled with the dish at the bottom of the resulting parallel flow chamber. Chambers and tubes were filled with PBS without serum before the experiment. Shear stress was applied with a syringe pump (Harvard apparatus PHD 2000) with flow rates of 1.0 dyne/cm2 for a total of 10 min/well. Adherent cells were counted under a microscope by blinded investigators. All experiments were performed at least 3 times or as indicated.
2.4. Intravital microscopy of mesenteric venules
Mice were pre-treated with MnTBAP (5 mg/kg body weight, i.p.) for 3 days and murine TNFα (10 ng/g body weight, i.p.) 4 h before surgery. Animals were anaesthetized with ketamine (100 m/kg) and xylazin (2 mg/kg). Leukocytes were fluorescently labeled in-vivo by retroorbital injection of 60 μl Rhodamine 6G (10 μg/ml, Sigma-Aldrich). A loop of ileal mesentery was exteriorized through a midline incision in the abdominal wall and placed on a plastic chamber to observe the peri-intestinal microcirculation by intravital microscopy. Leukocyte interactions with the endothelial vessel wall were recorded for 1 min at least in 3 veins/mouse. Leukocytes stationary for >30 s were defined as adherent and were counted as cells that adhere to 100 μm vessel length. The average diameter of examined venules was normalized to 100 μm. Leukocyte adhesion was quantified by two independent investigators in a blinded way.
2.5. Transendothelial migration assay
HUVECs were seeded at 1.5 × 10 [5] cells in gelatine coated transwell dishes with 5 μm pores (Corning Costar; Pittston, PA, USA) and allowed to grow for 3 days. HUVEC monolayers were pre-treated with MnTBAP and/or TNFα as indicated. PBMCs were isolated as previously described and labeled with Calcein AM. 1 × 10 [5] of labeled PMBCs were added to the HUVEC monolayer. The cell culture inserts were then placed into 24 well plates filled with 600 μl of cell culture medium and MCP-1 (50 ng/ml) as chemoattractant and incubated for 2 h. Transmigrated PBMCs were collected from the lower compartment and counted.
2.6. Permeability assay
Permeability across HUVEC monolayers was measured using fluorescein isothiocyanate (FITC)-dextran (molecular mass: 40 kDa; Sigma-Alrdrich). Briefly, HUVECs were plated at a density of 1.5 × 10 [5] cells into gelatin-coated Tranwell units (6.5 mm diameter; 0.4 μm pore size polycarbonate filter; Corning Costar; Pittston, PA, USA) and allowed to grow to confluence monolayers for 3 days. HUVECs were then treated with MnTBAP (50 μM) or/and TNFα (100 ng/ml) as indicated. FITC-dextran (1 mg/ml) was added into the upper chamber. Samples were collected at different time points from the lower compartment and fluorescence was measured using a fluorescent microplate reader.
2.7. Western blot analysis
For Western blotting, cells were lysed using RIPA buffer. Equal amounts of protein (30–50 μg) were loaded and separated by 7.5% SDS-PAGE. Membranes were incubated overnight at 4 °C with the appropriate primary antibody followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG (Dako). Blots were visualized by an ECL system (Pierce). ICAM-1, VCAM-1, PECAM-1and phospho Smad 1/5 antibodies were purchased from Cell Signaling, BMPR-Ia antibody was from Abcam, and BMPR-II antibody from BD Biosciences.
2.8. Quantitative real-time PCR
Total RNA was extracted using Aurum RNA Mini Kit (Bio-Rad). Quantitative real-time PCR was performed using MyiQ lightcycler software (Bio-Rad). The following primers were used: MCP-1 (Forward: 5′-ACT GAA GCT CGT ACT CTC-3′; Reverse: 5′-CTT GGG TTG TGG AGT GAG-3′), RANTES (Forward: 5′-ACA GGT ACC ATG AAG GTC TC – 3′; Reverse: 5′-TCC TAG CTC ATC TCC AAA GA –3′), BMPR-II (Forward: 5′-CAA ATC TGT GAG CCC AAC AGT CAA-3′; Reverse: 5′-GAG GAA GAA TAA TCT GGA TAA GGA CCA AT-3′), hRP (Forward: 5′-GCA CCA CGT CCA ATG ACA T-3′; Reverse: 5′-GTG CGG CTG CTT CCA TAA-3′). hRP was used as an endogenous control and quantification was calculated using the ΔΔCT method.
2.9. Measurement of MCP-1, RANTES and PF4
Mice were pre-treated with MnTBAP (5 mg/kg body weight, i.p.) for 3 days and murine TNFα (10 ng/g body weight, i.p.) 4 h before blood sampling. Serum concentration of mouse MCP-1, RANTES, and PF4 were measured using ELISA kits (RayBio Mouse ELISA Kit) according to the manufacturer’s protocol.
2.10. siRNA transfection
BMPR-II siRNA was purchased from Life Technologies. Scrambled negative control Alexa Fluor was purchased from Qiagen, Hilden, Germany. For transfection, Lipofectamine RNAiMAx was used according to the manufacturer’s protocol (Life Technologies). Transfection efficiency was confirmed by Western blot.
2.11. Transfection and luciferase assay
NIH3T3 cells were plated in a 96 well plate, cultured to 70% confluence, and either transfected with 50 ng NF-κB plasmid or the corresponding control plasmid (all GeneCopoeia™/tebu-bio, Le Perray-en-Yvelines Cedex, France), both containing the firefly luciferase gene from Photinus pyralis. For determination of transfection efficacy, a plasmid containing the renilla gene from Renilla reniformis was used. 24 h after transfection, Dual-Glow-luciferase assay (GeneCopoeia™) was performed according to manufacturer’s instructions using a Glomax, 96 microplate luminometer from Promega Madison, WI, USA and obtained firefly activity was normalized to renilla activity. Data are shown as mean ± SD of at least 3 independent experiments.
2.12. Statistical analysis
All experiments were performed in triplicate or as indicated. Animal numbers are stated with the different experimental results. Results are given as mean ± SEM. Statistical analysis was performed using GraphPad Prism 4.0, USA and comparisons were calculated by Student’s t-test or Mann-Whitney U test. A p value <0.05 was considered significant.
3. Results
3.1. MnTBAP prevents TNF-induced expression of adhesion molecules
Upon exposure to inflammatory stimuli, endothelial cells (EC) express adhesion molecules, allowing monocytes to adhere to ECs. To explore whether MnTBAP affects the expression of adhesion molecules, HUVECs pre-treated with MnTBAP were challenged to TNFα and expression of ICAM and VCAM were detected. Stimulation with TNFα resulted in a 57.9-fold increase of ICAM mRNA and 138.3-fold upregulation of VCAM mRNA, while treatment with MnTBAP did not affect ICAM or VCAM mRNA (Fig. 1A, B). Concurrently, stimulation with TNFα increased ICAM and VCAM protein expression by 11.6-fold and 14.6-fold, respectively (Fig. 1C–E). Pre-treatment with MnTBAP prevented upregulation of ICAM and VCAM mRNA and protein by TNFα (Fig. 1A–E). Interestingly, MnTBAP not only inhibited the expression of adhesion molecules, but also reduced secretion and deposition of the chemokines MCP-1, RANTES, and PF-4 in-vitro and in-vivo (Supplemental Fig. 1A–D), underlining an anti-inflammatory effect of MnTBAP in the vasculature.
Fig. 1.

MnTBAP prevents TNFα-induced upregulation of adhesion molecules. TNFα upregulates mRNA expression of the adhesion molecules ICAM (A) and VCAM (B), which is partly inhibited by MnTBAP. A similar effect is also demonstrated on the protein level (C–E). Data are represented as mean ± SEM and are representative for at least 3 independent experiments. n.s., not significant.
3.2. TNFa-induced leukocyte adhesion is prevented by MnTBAP
To evaluate whether MnTBAP reduced leukocyte adhesion to ECs under inflammatory conditions, dynamic adhesion assay was performed. PBMCs were allowed to adhere to control or TNFα-activated ECs. While adhesion of leukocytes to TNFα-activated ECs was upregulated, this effect was reduced by 66% in the presence of MnTBAP (Fig. 2A, B).
Fig. 2.
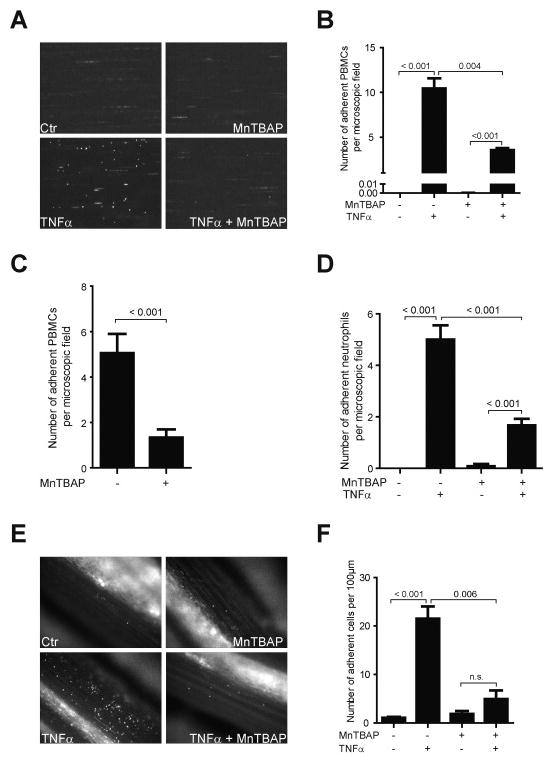
TNFα-induced leukocyte adhesion is prevented by MnTBAP. Adhesion of leukocytes to activated endothelial cells is impaired by MnTBAP in the dynamic flow chamber (A). Total number of adherent cells per microscopic field is shown in the bar graph (B). Pre-treatment of PBMCs with MnTBAP prevented their adhesion to the endothelial monolayer in the dynamic flow chamber (C). Adhesion of neutrophils to activated endothelial cells is reduced by MnTBAP in the dynamic flow chamber (D), n = 2. Leukocyte adhesion is reduced in a mouse model of systemic inflammation after treatment with MnTBAP (E). Total number of adherent cells are shown in the bar graphs (F), n = 5. Data are represented as mean ± SEM and are representative for 3 independent experiments or as indicated.
Conversely, MnTBAP also reduced adhesion of PBMCs to the endothelium, when PBMCs were pre-treated with MnTBAP, but not the ECs (Fig. 2C). Additionally, MnTBAP also prevented the adhesion of neutrophils to ECs in the dynamic adhesion assay (Fig. 2D), suggesting an anti-inflammatory effect of MnTBAP. To further characterize the effect of MnTBAP on leukocyte adhesion in-vivo, intravital microscopy of the mesenteric arteries was performed. Pre-treatment of MnTBAP clearly reduced leukocyte adhesion to the vessel wall (Fig. 2E, F).
3.3. MnTBAP modulates cell permeability and transendothelial migration
While adhesion of leukocytes to the endothelium is the initiating step in cellular infiltration of the vessel wall, cell permeability and transmigration through the endothelium also play critical roles during vascular inflammation. To assess whether MnTBAP modulates cell permeability, expression of the platelet endothelial cell adhesion molecule (PECAM)-1 was determined. PECAM-1 protein was expressed abundantly in HUVECs and was markedly down-regulated following treatment with TNFα (30 ng/ml). Pre-treatment with MnTBAP (50 nM) abrogated this down-regulation partly (Supplemental Fig. 2A, B). Because PECAM-1 has been shown to be critical for cell integrity and transmigration of leukocytes, permeability assay and transendothelial migration assay were further performed. Treatment with TNFα increased permeability of the endothelial monolayer, reflected by an increased passage of FITC-dextran. Cell permeability was decreased after pre-treatment with MnTBAP (Supplemental Fig. 2C), suggesting a protective effect of MnTBAP on EC integrity. In line with this finding, pre-treatment of the HUVEC monolayer with MnTBAP also reduced transendothelial migration of PBMCs towards MCP-1 (Supplemental Fig. 2D).
3.4. MnTBAP increases expression of BMPR-II in endothelial cells
Previous reports have suggested a central role of the BMPR-II in inhibiting leukocyte adhesion. Treatment of HUVECs with MnTBAP increased BMPR-II mRNA by 1.3-fold at 2 h, but did not affect protein expression of BMPR-II during a time course of 16 h (Fig. 3A–C). Phosphorylation of Smad 1/5 was increased upon treatment with MnTBAP, suggesting modulation of the BMP signaling pathway by MnTBAP (Supplemental Fig. 3A). Interestingly, BMPR-II expression was inhibited by TNFα, and MnTBAP was able to restore the inhibition of BMPR-II (Fig. 3D, E). This finding suggests a central role of BMPR-II in mediating the anti-inflammatory effect of MnTBAP.
Fig. 3.

MnTBAP modulates expression of BMPR-II in HUVECs. Incubation with MnTBAP increases mRNA expression of BMPR-II (A), but does not show effects on protein expression of BMPR-II (B, C). TNFα inhibits BMPR-II protein expression, which is partially restored by MnTBAP (D, E). Data are represented as mean ± SEM and are representative for at least 3 independent experiments. n.s., not significant.
To further evaluate how MnTBAP affects the dynamics of BMPR-II expression, protein synthesis of BMPR-II was blocked with cycloheximide. HUVECs were treated with 20 μg/ml cycloheximide, and BMPR-II protein expression was assessed. Loss of BMPR-II protein was observed in a time-dependent manner (Fig. 4A–B). Surprisingly, pre-treatment with MnTBAP for 16 h prior to addition of cycloheximide preserved BMPR-II protein expression, despite the addition of cycloheximide (Fig. 4C–D). This effect was most impressively seen after 2 h incubation with cycloheximide, when MnTBAP increased the reduced expression of BMPR-II by 2-fold. These data suggest that MnTBAP modulates protein degradation of BMPR-II.
Fig. 4.

MnTBAP restores inhibitory effects of cyclohexamide on BMPR-II protein expression. Treatment with cyclohexamide (CHX) inhibits protein synthesis of BMPR-II (A, B). Pre-treatment with MnTBAP preserves loss of BMPR-II protein following blockade of protein synthesis by CHX (C, D). Data are represented as mean ± SEM and are representative for at least 3 independent experiments. n.s., not significant.
3.5. Anti-inflammatory effect on MnTBAP is mediated by BMPR-II dependent regulation of NFκB
To further evaluate the anti-inflammatory effect of MnTBAP and BMPR-II, NFκB activation was evaluated. Treatment with TNFα increased the NFκB promoter activity by 3.8-fold. This effect was reduced by 40.9% after pre-treatment with MnTBAP (Fig. 5A). However, when BMPR-II was silenced using siRNA, the anti-inflammatory effect of MnTBAP was partly abolished. In particular, the counter-regulation of TNFα-induced activation of the NFκB promoter by MnTBAP was reduced in BMPR-II depleted cells compared to control siRNA transfected cells (Fig. 5B). Although the difference between the siBMRP-II transfected and control siRNA transfected groups was statistically not significant, the partial loss of MnTBAP’s anti-inflammatory effect in the presence of TNFα was reproducible in several experiments, suggesting that the anti-inflammatory effect of MnTBAP is – at least partly – dependent on the BMPR-II signaling pathway.
Fig. 5.

MnTBAP elicits anti-inflammatory effects through NFκB. Incubation with MnTBAP prevented activation of the NFκB promoter (A). The preventive effect of MnTBAP on TNFα-induced activation of the NFκB promoter was partly abrogated in BMPR-II depleted cells (B). Data are represented as mean ± SEM and are representative for at least 3 independent experiments. n.s., not significant.
4. Discussion
The mitochondrial manganese SOD (MnSOD) belongs to the SOD superfamily. The importance of MnSOD in cell biology is evident in MnSOD knock-out mice, where administration of the MnSOD mimetic MnTBAP rescued part of their phenotypes and prolonged their survival. Recent studies have also demonstrated a potential therapeutic relevance of MnTBAP in preventing diabetes and obesity [7,8]. While most of the protective effects of MnTBAP are attributed to its anti-oxidative property, the present work demonstrates that MnTBAP also directly interacts with other signaling pathways of inflammation. Here, we show that MnTBAP mediates anti-inflammatory effects through upregulation of BMPR-II and inhibition of the NFκB signaling. We, and others have previously demonstrated that MnTBAP prevented the increase of ICAM in endothelial cells upon inflammatory stimuli [6,14]. Using flow chamber and intravital microscopy, we now demonstrate that treatment with MnTBAP decreased the expression of adhesion molecules in endothelial cells and reduced adhesion of leukocytes to the endothelium. MnTBAP also protected against TNFα-induced increase in cell permeability and transendothelial migration, contributing to the preservation of endothelial cell integrity. Moreover, MnTBAP inhibited activation of the NFκB promoter by TNFα, suggesting an anti-inflammatory effect of MnTBAP.
The BMPR-II receptor has been mainly associated with primary PAH. But recent findings suggest that BMPR-II also plays an important role in vascular inflammation. Indeed, loss of BMPR-II in endothelial cells caused robust inflammation and atherosclerosis [11], while statins or stable flow, which has anti-inflammatory properties, increased BMPR-II expression in endothelial cells [10]. In line with this finding, our data demonstrate that TNFα reduced BMPR-II expression, which could be partly rescued by MnTBAP. To further evaluate whether the effect of MnTBAP on the TNFα-NFκB pathway is medicated by BMPR-II, we conducted BMPR-II loss of function experiments. In BMPR-II depleted cells, the preventive effect of MnTBAP on TNFα-induced activation of the NFκB promoter was reduced compared to control. However, this effect was not statistically significant. A possible explanation for this result could be that MnTBAP elicits its anti-inflammatory effects through multiple signaling pathways, which may compensate the depletion of BMPR-II signaling. Indeed, previous data have shown that MnTBAP reduced ROS generation and elicited anti-oxidative effects in a concentration-dependent manner [6,15]. This anti-oxidative effect may have been sufficient enough to counter-regulate a further increase of the NFκB promoter activity in siBMPR-II depleted cells independently of BMPR-II. Another possibility could be that MnTBAP modulates the turn-over of BMPR-II, in which case the deceleration of BMPR-II protein degradation could compensate for the depleted BMPR-II. This is supported by our data, demonstrating for the first time that MnTBAP inhibits BMPR-II turn-over following protein synthesis blockade. Previously, Dunmore et al. have demonstrated a similar phenomenon in pulmonary artery endothelial cells for chloroquine. Chloroquine also restored BMPR-II expression after BMPR-II mRNA was knocked down [16]. Because BMPR-II is targeted for ubiquitination and degradation via the lysosome [17], it was suggested that chloroquine preserves BMPR-II protein expression through inhibition of its lysosomal degradation [18]. Whether this is also a potential mechanism for MnTBAP, needs to be further investigated in future studies.
In summary, we demonstrate that MnTBAP decreases the expression of adhesion molecules within an inflammatory milieu and subsequently prevents adhesion of leucocytes to the endothelium, and their transmigration through the vessel wall. Additionally, MnTBAP prevents loss of BMPR-II following blockade of new protein synthesis and elicits anti-inflammatory effects in endothelial cells, partly mediated by BMPR-II. Further studies will be necessary to dissect the underlying mechanism, through which MnTBAP regulates BMPR-II turnover.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Funding sources
This work is supported by grants from the German Heart Foundation (F/33/12) to Q. Zhou.
We thank Dr. Ute Woelfle, University Hospital Freiburg, Germany for providing the NFκB plasmid and the renilla control plasmid.
Footnotes
Disclosures
None.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.vph.2016.07.001.
References
- 1.Berk BC, Abe JI, Min W, Surapisitchat J, Yan C. Endothelial atheroprotective and anti-inflammatory mechanisms. Ann N Y Acad Sci. 2001;947:93–109. doi: 10.1111/j.1749-6632.2001.tb03932.x. (discussion 109–111) [DOI] [PubMed] [Google Scholar]
- 2.Abid MR, Tsai JC, Spokes KC, Deshpande SS, Irani K, Aird WC. Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J. 2001;15:2548–2550. doi: 10.1096/fj.01-0338fje. [DOI] [PubMed] [Google Scholar]
- 3.Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 4.Perrotta I, Perrotta E, Sesti S, Cassese M, Mazzulla S. MnSOD expression in human atherosclerotic plaques: an immunohistochemical and ultrastructural study. Cardiovasc Pathol. 2013;22:428–437. doi: 10.1016/j.carpath.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 5.Faulkner KM, Liochev SI, Fridovich I. Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. J Biol Chem. 1994;269:23471–23476. [PubMed] [Google Scholar]
- 6.Zhou Q, Gensch C, Keller C, Schmitt H, Esser J, Moser M, et al. MnTBAP stimulates angiogenic functions in endothelial cells through mitofusin-1. Vasc Pharmacol. 2015;72:163–171. doi: 10.1016/j.vph.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 8.Pires KM, Ilkun O, Valente M, Boudina S. Obesity. Silver Spring; 2014. Treatment with a SOD Mimetic Reduces Visceral Adiposity, Adipocyte Death, and Adipose Tissue Inflammation in High Fat-Fed Mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu H, Sung A, Zhao G, Shi L, Qiu D, Nishimura T, et al. Simvastatin enhances bone morphogenetic protein receptor type II expression. Biochem Biophys Res Commun. 2006;339:59–64. doi: 10.1016/j.bbrc.2005.10.187. [DOI] [PubMed] [Google Scholar]
- 11.Kim CW, Song H, Kumar S, Nam D, Kwon HS, Chang KH, et al. Anti-inflammatory and antiatherogenic role of BMP receptor II in endothelial cells. Arterioscler Thromb Vasc Biol. 2013:1350–1359. doi: 10.1161/ATVBAHA.112.300287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinke J, Wehofsits L, Zhou Q, Zoeller C, Baar KM, Helbing T, et al. BMPER is an endothelial cell regulator and controls bone morphogenetic protein-4-dependent angiogenesis. Circ Res. 2008;103:804–812. doi: 10.1161/CIRCRESAHA.108.178434. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y, Kucik DF, Szalai AJ, Edberg JC. Human neutrophil flow chamber adhesion assay. J Vis Exp. 2014 doi: 10.3791/51410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quagliaro L, Piconi L, Assaloni R, Da Ros R, Maier A, Zuodar G, et al. Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: the distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis. 2005;183:259–267. doi: 10.1016/j.atherosclerosis.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 15.Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys. 1997;347:256–262. doi: 10.1006/abbi.1997.0341. [DOI] [PubMed] [Google Scholar]
- 16.Dunmore BJ, Drake KM, Upton PD, Toshner MR, Aldred MA, Morrell NW. The lysosomal inhibitor, chloroquine, increases cell surface BMPR-II levels and restores BMP9 signalling in endothelial cells harbouring BMPR-II mutations. Hum Mol Genet. 2013;22:3667–3679. doi: 10.1093/hmg/ddt216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durrington HJ, Upton PD, Hoer S, Boname J, Dunmore BJ, Yang J, et al. Identification of a lysosomal pathway regulating degradation of the bone morphogenetic protein receptor type II. J Biol Chem. 2010;285:37641–37649. doi: 10.1074/jbc.M110.132415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, et al. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res. 2013;112:1159–1170. doi: 10.1161/CIRCRESAHA.111.300483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.