

Abstract
A severe burn can trigger a hypermetabolic state which lasts for years following the injury, to the detriment of the patient. The drastic increase in metabolic demands during this phase renders it difficult to meet the body’s nutritional requirements, thus increasing muscle, bone and adipose catabolism and predisposing the patient to a host of disorders such as multi-organ dysfunction and sepsis, or even death. Despite advances in burn care over the last 50 years, due to the multifactorial nature of the hypermetabolic phenomenon it is difficult if not impossible to precisely identify and pharmacologically modulate the biological mediators contributing to this substantial metabolic derangement. Here, we discuss biomarkers and molecules which play a role in the induction and mediation of the hypercatabolic condition post-thermal injury. Furthermore, this thorough review covers the development of the factors released after burns, how they induce cellular and metabolic dysfunction, and how these factors can be targeted for therapeutic interventions to restore a more physiological metabolic phenotype after severe thermal injuries.
Keywords: Burns, Hypermetabolism, Insulin resistance, Catabolism, Trauma, Mitochondria
1. Introduction
Burns affect nearly 300 million patients annually worldwide, with an associated substantial mortality. In the USA half a million Americans per year get burned, with approximately 40,000 requiring hospitalization [1]. Thermal injuries induce systemic biomolecular changes with profound physiological alterations, such as increased muscle and bone catabolism, hepatic steatosis, higher susceptibility to infections, multiple organ dysfunction, insulin resistance and sepsis. The hypermetabolic response, a profound increase in metabolic demand reflected by an elevated resting energy expenditure (REE), is the primary contributor to aforementioned complications, and can persist for up to 3 years after a severe burn [2]. While great strides have been made in the treatment of thermal injuries, such as improved wound care, fluid and infection management and rigorous nutritional guidelines, our understanding of the hypermetabolic phenomenon which contributes drastically to patient morbidity and mortality is lagging behind. A thorough exploration of the affected tissues and the mediators of this response is required to inform the development of improved treatment options for burned patients.
The metabolic changes following burns are not dissimilar to other traumas but very different in terms of their extent and persistence; characterized primarily by an ‘ebb’ phase within 48 hours where metabolism, cardiac output and oxygen consumption are all decreased. This is typically followed by a ‘flow’ phase at around 120 hours post-burn, where these variables gradually increase and plateau [3]. There are numerous reasons to believe this adaptive response is to the patient’s benefit. For instance, the acute response allows vital organs to conserve energy, and mild to moderate hyperglycemia provides fuel for the brain and immune system after trauma. Burn injuries, however, stand out in their intensity and duration. The chronic persistence of the hypermetabolic response, which appears to be driven by catecholamines, stress hormones, and pro-inflammatory cytokines, far surpasses the ability of the patient to respond, and physiological exhaustion ensues. Augmented rates of glycolysis, lipolysis and proteolysis induce a loss of lean and total body mass which subsequently causes immune dysfunction, decreased wound healing and severe infections [4]. Left untreated, the amalgamation of these systemic injuries leads to organ dysfunction, sepsis and death.
Here, we present a comprehensive review of the downstream biomolecular changes that occur after a severe burn injury and examine recent findings to elaborate on how these may contribute to the hypermetabolic phenomenon, patient morbidity and mortality. Burn severity is normally determined via the total body surface area (TBSA) covered by the injury, with anything above 20% being considered a severe burn likely to result in the occurrence of the hypermetabolic condition. However, we recently presented new evidence that even a 10% TBSA burn can cause substantial and pathological alterations similar to a burn over 30% TBSA [5]. Current best clinical practices and areas of improvement will be discussed, with an eye towards the advancement of patient care following severe thermal injuries.
2. Cytokines and stress hormones
The immediate post-burn response encompasses a cascade of biological mediators which contribute to patient hypermetabolism. These include pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β) and interleukin 6 (IL-6) [2], [4] and [6]. A marked increase in plasma catecholamines like dopamine, epinephrine and norepinephrine can persist for years after the initial insult, contributing to muscle catabolism, insulin resistance and immunosuppression [2]. Moreover, sustained increases in the hormones cortisol and glucagon further compound the problem via enhanced breakdown of protein in muscle and the liver in addition to increasing circulating glucose and consequently, hyperglycemia (Figure 1) [2] and [7]. The complexity and redundancy of the systemic response to burns renders it difficult to identify which factors contribute most to long-term hypermetabolism and can be singled out for therapeutic interventions. Here we discuss the mediators released following thermal injury and how they may play a role in the hypermetabolic phenotype.
Figure 1.
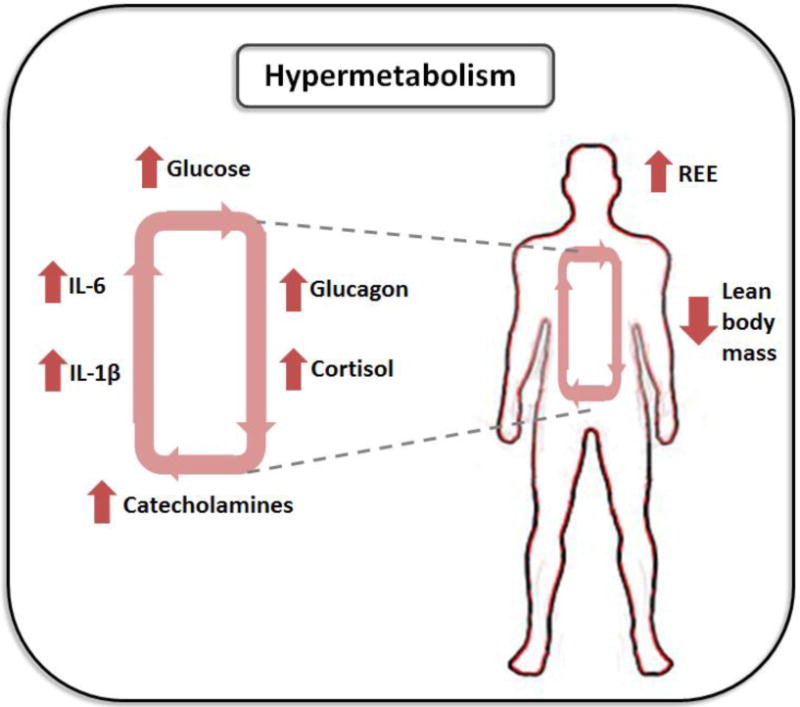
The hypermetabolic response post-burn is characterized by an increase in resting energy expenditure (REE) and loss of lean body mass driven by stress hormones and pro-inflammatory cytokines.
2.1 TNF-α
As a pro-inflammatory cytokine of the acute phase reaction, levels of TNF-α are predictably higher in burned patients when compared to healthy controls. The role of TNF-α in inflammation, which includes amplifying the cellular and mediator response, as well as inducing apoptosis of injured cells and repairing inflammatory damage, renders it essential following trauma [8]. However, this cytokine is also known to increase the formation of reactive oxygen species (ROS) and accelerate the rate of lipolysis, thus increasing adipose catabolism and the release of free fatty acids (FFAs) in patients affected by thermal injuries [9]. In differentiated human adipocytes, TNF-α activates mitogen-activated protein kinase kinase (MEK), extracellular signal-related kinase (ERK) and increases intracellular cyclic adenosine monophosphate (cAMP), subsequently initiating lipolytic pathways [10]. As circulating FFAs have been known to induce insulin resistance, a hallmark feature of the hypermetabolic response, their presence after burns is detrimental to the patient. Furthermore, these moieties accumulate in the liver and muscle, leading to cellular dysfunction [11], [12], [13] and [14].
The adverse effects of TNF-α have been demonstrated in a mouse model of non-severe burn injury (<10% TBSA) covered by scald burn [15]. Despite the lack of a hypercatabolic response in these mice, there is a clear decrease in bone volume following the injury. As TNF-α has been shown to affect bone synthesis and degradation, antibodies against this inflammatory cytokine appear to restore bone thickness and volume following burn [15]. In a 30% TBSA burn model, TNF-α induces apoptosis in gut epithelial cells at a rate three times higher than that of sham animals [16]. This cytokine has also been associated with loss of skeletal muscle and impaired wound healing post-burn [17] and [18]. Anti-TNF-α antibodies diminish the mucosal atrophy after burn by inhibiting apoptosis without affecting proliferation [16]. However, attempts to modulate inflammatory mediators in humans have been met with mixed results [6]. Fortunately, high TNF-α levels are not sustained in burn patients, rendering it likely that this mediator’s contributions to hypermetabolism are restricted to the acute phase post-burn.
2.2 IL-1β
In addition to TNF-α, circulating IL-1β has been associated with increases in REE and cachexia in critical illnesses such as cancer and AIDS. The maturation and secretion of IL-1β occurs following activation of inflammasomes, large multimeric protein complexes which recognize pathogen-associated molecular patterns [19]. One such biomolecule, the NLRP3 inflammasome, named for its Nod-like receptor domain, has been shown to detect obesity-associated endogenous damage-associated molecular patterns (DAMPs) [20]. The suggested mechanisms for the activation of NLRP3 include such factors as an increase in saturated fatty acids, mitochondrial dysfunction and augmented formation of ROS, all prominent components in the aftermath of a severe burn [19]. As mature IL-1β has been shown to interfere with insulin sensitivity via down-regulation of insulin receptor substrate-1 expression, it’s postulated that this cytokine contributes to the stress-induced diabetes and hyperglycemia which precede long-term metabolic dysfunction and hypermetabolism following thermal injury [21]. Indeed, following a severe burn, there is a 3-fold increase in leukocyte levels in the adipose tissue, indicative of inflammation [22]. The subsequent activation of NLRP3 stimulates the maturation of IL-1β which would indeed contribute to the post-burn hypermetabolic response.
2.3 IL-6
As serum IL-6 has been associated with higher REE, the sustained increase of this mediator post-burn renders it an interesting therapeutic target. However, its multifactorial role, acting as both a pro- and anti-inflammatory cytokine with hormone-like characteristics, renders it difficult to determine when and if it would be of benefit to the patient to modulate this biomolecule. Infusion of recombinant human IL-6 in healthy volunteers mimics the hypercatabolic state seen in burn patients, such as increased energy expenditure and plasma concentrations of glucose and FFAs [23]. One possible mechanism by which IL-6 can contribute to hypermetabolism is by stimulating the liver to induce an acute phase response. IL-6 signalling proceeds via binding of its membrane bound receptor which signals the JAK/STAT pathway, leading to the translocation of activated STAT proteins into the nucleus [24]. More recently, the relationship between the STAT-regulated proteins and mitochondrial bioenergetics has been outlined, and IL-6 has been implicated in the browning of white adipose tissue (WAT) [25] and [26]. Brown adipose tissue (BAT) displays higher rates of lipolysis which increases the circulation of FFAs after a thermal injury and these contribute to the hypermetabolic condition [26].
2.4 Stress hormones
2.4.1. Catecholamines
Peripheral increases of circulatory epinephrine and norepinephrine can be observed in patients with a severe burn injury. Indeed, shortly after thermal trauma, these moieties can increase up to 10 fold, raising the REE and triggering the development of hyperdynamic circulation, lipolysis, catabolism of skeletal muscle as well as the release of glucose from energy stores [4] and [27]. Mechanistically, catecholamines exert their effects by binding to α1, α2, β1 and β2 receptors which signal downstream to different classes of G protein-coupled molecules which affect intracellular Ca2+ or cAMP concentrations [28]. However, the tissue and cell specificity of this response varies greatly. It is also likely that catecholamines activate transcription factors such as CREB and c-Fos, leading to the induction of MAPK family consisting of ERK1/2, JNK1/2/3 and p38, therefore prolonging the hyperinflammatory response following a severe burn [29], [30] and [31]. Furthermore, it has been shown that, similar to IL-6, catecholamines can induce browning of inguinal WAT, thus increasing the rate of lipolysis [32]. As is the case with inflammatory cytokines, the surge in catecholamines is thought to play a crucial role in orchestrating the initial stress response, but their sustained increase imposes a significant burden on the patient. For instance, pediatric burned patients display consistently and significantly higher levels of catecholamines up to 2 years post-burn when compared to healthy volunteers [33]. In another study, urinary norepinephrine increased 10-fold post-burn and remained significantly elevated for up to 540 days [2].
2.4.2. Glucagon and cortisol
The glucagon/cortisol tandem which increases significantly following burns and other traumas exert their hypermetabolic effects via increases in lipolysis, proteolysis and hyperglycemia. Mechanistically, glucagon secreted by the pancreas elevates blood glucose by promoting gluconeogenesis and glycogenolysis in hepatocytes [34]. Signalling through G proteins increases intracellular cAMP levels, slowing glycolytic flux and releasing liver stores of glucose [35]. Cortisol, synthesized from cholesterol in the adrenal glands, increases in circulation following stress and signals intracellularly via glucocorticoid receptors [36]. This stress hormone facilitates the action of glucagon by increasing insulin resistance. By decreasing the translocation of GLUT4 to the cell membrane, cortisol decreases the peripheral use of glucose [37]. Its ability to accelerate the rate of potassium excretion also reduces bone formation and volume [38]. In physiological concentrations, this hormone also activates lipases in adipose tissue, increasing the breakdown of fat [34]. Indeed, during the post-burn hypermetabolic response, glycolytic-gluconeogenetic cycling is increased 250%, while triglyceride-fatty acid cycling is increased 450% [39]. In a pediatric study with 212 burn patients, hypercortisolemia (3 to 5 fold higher than healthy controls) persists for up to 100 days post-burn [40]. In severe burns, significantly increased cortisol levels can persist for up to 3 years [2].
3. Organellar contribution to hypermetabolism
3.1 Mitochondrial dysfunction
Mitochondria, the cellular powerhouses which house the electron transport chain (ETC), play a vital role in the generation of ATP via oxidative phosphorylation [41]. Additionally, this organelle orchestrates key steps in intrinsic apoptosis, heme synthesis, calcium signalling, and β-oxidation, thus rendering it indispensable to biological processes [42]. The notion that mitochondria play a causative role in hypermetabolism stems from the increase in oxygen usage during this phenomenon. As approximately 90% of systemic O2 consumption occurs in this organelle, fluctuations in mitochondrial activity may underlie the hypermetabolic phenotype [43]. Following a severe burn, it is estimated that ~50% of hypermetabolism, with a REE from 120 to 180% above normal values, is linked to ATP-consuming reactions, implying that mitochondrial bioenergetics must be increased to meet the energy demands of the patient [4] and [44]. The ability of mitochondria to generate ATP lies in their coupling efficiency, a measure of the degree to which electron transport contributes to the phosphorylation of ADP. Rather than generate energy, mitochondria which become uncoupled dissipate this potential as heat, a process known as thermogenesis [45]. This is particularly important to counter heat loss in neonates, especially in the adipose tissue.
The impact of burn injuries on mitochondria is immediate. Yasuhara et al. demonstrated in a rodent model with a 40% TBSA burn that cytochrome c leaks from the outer membrane of mitochondria from muscle tissue to the cytosol within 15 minutes, with concomitant alterations in mitochondrial membrane potential within 1 hour [46]. Moreover, it appears as though muscle bioenergetics in the area local to the burn wound are affected early on after injury, yet distal muscle function remains unaffected for days, suggesting that the extent and localization of mitochondrial dysfunction varies depending on the burn area. In mice, major bioenergetic parameters such as basal respiration, ATP synthesis and maximal respiratory capacity are significantly decreased at 3 hours post burn in the lungs and liver, coinciding with the reported hypometabolism following trauma. However, at day 10, these parameters rebound considerably and are higher than those of sham mice, perhaps indicative of a compensatory recovery and contributing to the hypermetabolic phenotype [47].
Porter et al. have done extensive work to elucidate mitochondrial function in skeletal muscle from both adult and pediatric burn patients. They’ve demonstrated that mitochondrial abnormalities accompany muscle cachexia, the wasting of tissue, and postulate that mitochondrial protein turnover is associated with increased protein damage due to hypermetabolism-induced oxidative stress [48]. Moreover, they’ve shown that mitochondrial coupling control is diminished, even at 2 years following a severe (>30% TBSA) burn, thus resulting in increased thermogenesis [49]. As such, much of the electrochemical potential is being directed to heat production, perhaps a mechanism to cope with the resultant heat loss that occurs through open wounds and a compromised skin barrier. While this may confer benefits for thermoregulation, muscle tissue mitochondria are generally well-coupled, with over >80% of the oxygen consumption tied to ATP production due to the energetic demands of these tissues, and this shift to heat production contributes to the increase in REE and the hypermetabolic, hypercatabolic condition [50]. The plasticity of mitochondria following severe burns, and the role they play in increasing systemic energy expenditure after injury, renders these organelles potential therapeutic targets for regulation after thermal trauma.
3.2 Endoplasmic reticulum stress
Hypermetabolism induced in pathological states places a significant burden on the ER and results in the accumulation of mis- and unfolded proteins within the organelle. This phenomenon, known as ER stress, is sensed by three classes of enzymes: inositol-requiring enzyme 1 (IRE1α and β), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6 α and β) [51] and [52]. Together, these proteins initiate the unfolded protein response (UPR), which works to regulate the transcription and translation of genes involved in protein folding, ER-associated protein degradation (ERAD), protein secretion and protein translocation to the ER. Further, these proteins cleave pre-mature mRNAs, inhibit protein synthesis and activate apoptosis related factors. Prolonged activation of the UPR then gives rise to cellular death which may contribute to worsened outcomes and mortality in pathophysiological conditions [52] and [53]. In burns, ER stress markers have been shown to be elevated up to 250 days post injury and occur coincident with insulin resistance, hyperglycemia and inflammation [53]. Together, these results implicate ER stress and the UPR as valuable therapeutic targets and support more mechanistic studies of this phenomenon.
While the presence of ER stress and the activation of the UPR is well established in the literature, the mechanism underlying trauma-induced ER stress is lesser known, with many authors postulating either an increase in protein synthesis demand or extracellular signalling. With regards to the latter, catecholamines may be prime mediators of trauma-induced ER stress. In animal models of cardiomyopathy, norepinephrine administration has been shown to increase ER stress markers and inhibition of ER stress with chemical chaperones has been correlated with decreased norepinephrine levels [54], [55] and [56]. Substantiating evidence for the involvement of catecholamines in ER stress is β-adrenergic receptor blockade effects; treatment with a variety of β-blockers, including both selective and non-selective blockers successfully attenuated ER stress in an array of metabolic conditions while β-agonists produced the opposite effect [57], [58], [59] and [60]. β-blockers have also been shown to modulate levels of Ca-related proteins in ER stress models and norepinephrine specifically has been shown to alter the glycosylation of its transporter (NET), further enhancing the effects of the catecholamine [54] and [59]. These effects are very similar to those of thapsigargin and tunicamycin, which are well established inducers of ER stress and which work by inhibiting N-linked glycosylation and sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA), respectively [61]. Taken alongside the fact that adrenergic receptors are g-protein coupled receptors and alter downstream Ca levels, Ca dysregulation appears to be one of the main contributors to ER stress.
Indeed, levels of proteins involved in Ca homeostasis within the cell, such as SERCA, Na-Ca exchanger (NCX1) and calnexin, along with cystolic and endoplasmic Ca levels have been reported to be modulated in both animal models of disease and in burn patients [53], [54], [59] and [62]. These changes occur coincident with ER stress marker elevations and show improvement upon β-blocker administration. Further, overexpression of these calcium-related proteins attenuates the ER stress response in cells. In models of obesity and atherosclerosis, SERCA dysregulation has been reported to be caused by lipid metabolic alterations, namely phosphotidylcholine and cholesterol [65] and [66]. It is important to note that, in burns, both high levels of catecholamines and lipid cycling have been reported (see above). Further, both have been shown to be involved in feedback cycles, enhancing their effects. Thus, part of the mechanism involved in trauma-induced ER stress appears to be related to Ca dysregulation caused by an influx of signalling molecules, including catecholamines and lipids.
Interestingly, SERCA has also been implicated in non-shivering thermogenesis. Electron micrographs of brown adipose tissue have recently demonstrated a physical link between the ER and mitochondria of BAT [67]. While it was previously known that small molecules such as Ca may be transferred between the ER and mitochondria in mitochondrial-associated membranes (MAM), this report suggests that the transfer of larger proteins such as SERCA can also occur. Evidence for the involvement of SERCA in thermogenesis is demonstrated by the presence of the SERCA1 isoform in BAT but not WAT. The SERCA1 reported in BAT also differs from that of muscle in that it has higher rates of uncoupling [68]. Further, mitochondrial Ca administration, represented in the cell as a cystolic Ca increase, enhances heat production in BAT [69]. It is suggested that the levels of Ca sufficient to uncouple SERCA and enhance heat production are similar to those that arise during adrenergic stimulation, further implicating catecholamine involvement. The rise of Ca can be attributed to the activation of a1-adrenoreceptors which increases its release from intracellular stores as well as the stimulation of B3-adrenergic receptors which further compound the effects of a1-adrenoreceptor activation [70]. Indeed, an approximate 10-fold increase in Ca, from its basal concentration of 0.05 μM to 0.2–0.7 μM raises the rate of heat production in BAT cells [69]. This is interesting, as it implies that an H+ gradient in BAT mitochondria is not a necessity for heat production, and that ATP-consuming calcium cycling by Ca2+-ATPase may also play a key role. Sarcolipin (SLN), an established regulator of SERCA which uncouples the ATPase and increases Ca leak across the membrane, has also been shown to be required for browning [71] and [72]. SLN knock outs do not show thermogenesis but regain this ability with SLN overexpression [73]. It appears that SLN works in conjunction with UCP-1 to regulate thermogenesis and browning, as the expression of one is compensated for by the other [74]. These studies suggest a link between ER stress and browning.
Overexpression of GRP78/BiP, a chemical chaperone upregulated in response to ER stress, in the ventromedial hypothalamus has been reported to decrease food intake and corresponding weight gain in animal models of obesity. These changes were accompanied by reductions in other ER stress markers such as p-IRE and peIF2α and increases in BAT darkness and temperature [75] and [76]. ATF4 and CHOP knock out mice have also been reported to induce browning while TLR4 activated browning inhibition occurred coincident with increased ER stress markers [77] and [78]. Although the mechanism is unclear, these studies suggest that the presence of ER stress markers inhibits browning of WAT and thermogenesis. Interestingly, a recent study has reported that the IRE1α-XBP1 pathway is necessary for UCP-1 transcription, despite the fact that ER stress induced activation of IRE1α-XBP1 does not induce browning [79]. Further work is necessary to define the link between ER stress and browning.
Complicating the ER stress phenomena are the myriad of other factors which have been observed to persist in pathological conditions and further, contribute to its induction. For example, in addition to lipids, cholesterol and catecholamines, the amino acid homocysteine, the pro-inflammatory cytokines TNFα, IL-6 and IL1B, the energy source glucose and free radicals including ROS and NO have all been implicated in the induction of ER stress [80]. The latter is particularly interesting in that it perpetuates a cycle; depletion of Ca from the ER and its subsequent rise in the cytosol serves as a signal for uncoupling and ROS production in the mitochondria. These ROS then have the potential to target Ca channels within the ER and affect protein folding, further contributing to ER stress. Burns are a complicated pathophysiological phenomenon in that all the aforementioned factors have been reported to be up-regulated and persistent. Thus, in hypermetabolic states, it is likely the amalgamation of several factors which work together induce and perpetuate ER stress.
4. Lipid metabolism post-burn
Lipid mobilisation is often measured by plasma free fatty acid (FFA) levels, a product of lipid oxidation. In burns, high levels of FFA have been reported in both animals and humans, suggesting high rates of lipolysis [81] and [82]. These levels appear immediately after injury and persist anywhere from 5 days to 2 months. The extent of mobilisation and the approach to pre-burn levels are likely dependent on both age and sex [83] and [84]. The potential for these elevated FFAs to persist for greater than a couple of months exists, as factors correlated with FFA levels and which contribute to the hypermetabolic response have been reported to last up to 3 years post injury [2] and [81]. Elevated and persistent levels of glycerol, a product of lipolysis, have also been reported in burns, further confirming the occurrence of high rates of lipid oxidation [81].
Lipidomic analyses have revealed greatly-altered lipid profiles in burn injuries, with enhanced negative effects in the older population [83] and [85]. Generally, levels of saturated and monounsaturated fatty acids (SFA and MUFA, respectively), such as palmitic and oleic acid, were increased, whereas levels of polyunsaturated fatty acids (PUFA), such as eicosenoic and eicosatrenoic acids, were decreased. SFAs and MUFAs have been shown to induce inflammatory responses and alter lipid-induced insulin sensitivity via toll-like receptor 4 (TLR4) pathways while PUFAs are known to produce the opposite effect, inhibiting inflammation in hypermetabolic states [86] and [87]. While initial activation of these pathways post-trauma may be a beneficial evolutionary response, prolonged lipid alterations and TLR4 activation have been linked to metabolic disorders and sepsis [87].
Increases in triglycerides have also been found in burns [81], [82], [88] and [89]. Reports of increased triglycerides seem somewhat paradoxical given the persistent lipolysis observed in burned patients. Fats are stored as tryacylglycerols (TAG), which are composed of one glycerol molecule bound to 3 fatty acids. Adipocytes are stimulated by hormones released during conditions that require the mobilisation of fats. Epinephrine, adrenocorticotropic hormone and glucagon can all activate lipolytic processes in adipocytes, producing both glycerol and FFA [34] and [90]. Thus, the increase in both triglycerides and FFA suggests that fats are increasingly broken down and re-esterified. Indeed, this futile cycling has been reported in many studies, with increases of up to 15 X, compared with controls [39], [81] and [91].
Some authors have suggested that increased cycling may be caused by a decrease in plasma lipid carriers; FFA that cannot move around the body due to insufficient carriers are returned into cells and re-esterified. However, while there have been reports of decreased plasma albumin and apolipoproteins (lipid carriers) in burned patients, deficiencies in albumin expression fail to lead to lipid-related metabolic dysfunction [2], [90], [92] and [93]. Further, cycling has been observed in healthy volunteers without metabolic disorders and appears to be modulated by diet and exercise [94], [95] and [96]. This suggests that the high rates of cycling in burned patients may be mitigated with supplemental diets. Several causes for this cycling have been postulated. Stress hormones such as epinephrine and ACTH stimulate lipolysis post trauma [90]. These high levels of FFA can either be used to create energy by cells or can be re-esterified in the liver. Cells which uptake FFAs create acetyl CoA as a by-product which can be then be converted into ketone bodies and released into the serum [97]. Esterified triglycerides in the liver are released as low density lipoproteins (LDL) in the plasma. Low levels of ketone bodies and high levels of LDL and triglycerides in the plasma suggest that the majority of FFA are re-esterified [98]. Other reasons for high triglyceride levels may include stimulation of fatty acid synthesis by high plasma glucose levels, promotion of LDL production due to low plasma LDL and decreased removal of plasma triglycerides due to low plasma HDL [92].
In healthy individuals and in the acute phase of burn trauma, this cycling may serve a beneficial purpose; thermogenesis and substrate sensitivity, which are intimately linked, are both stimulated by cycling [99]. The non-equilibrium state induced by substrate cycling allows enzymes to become more sensitive to substrates and ultimately, become more adaptable to metabolic changes – which is a hallmark feature of burns. It remains unclear whether the benefits of substrate sensitivity and metabolic adaptability outweigh the energy expenditures caused by these elevations in metabolic rate. It is important to note that elevated levels of FFA and triglycerides have been linked to poor outcomes in burn patients [81] and [87]. As previously mentioned, this cycling is regulated by catecholamines, corticosteroids and cytokines. FFAs also participate in negative feedback loops whereby FFAs such as oleic, palmitic and linoleic acids inhibit further lipolysis and growth hormones, which have been shown to induce lipolysis [100] and [101]. Surprisingly in burns, both palmitic and oleic acids have been reported to be increased while linoleic acid has been reported to be decreased [83] and [87].
4.1 Adipose metabolism and cytokines
In other metabolic diseases, such as diabetes, the lipolytic influx of FFAs into the serum occurs coincident with cytokine release and accumulation of inflammatory markers. While in diabetes, the adipose tissue secretes cytokines into the serum, cell and clinical work suggest a modulatory role of FFAs on cytokine levels; addition of fatty acids to cultured cells is reported to increase levels of IL-6, IL-8, IL-18, IL-β, pro-matrix metalloproteinase-1 (MMP-1) and pro-MMP3 while decreasing levels of IL-10, IL-12, interferon gamma (IFNγ) and CCL2 [102], [103], [104], [105] and [106]. Further, insulin-induced inhibition of FFA release reduces levels of IL-18, but not of IL-6 or TNFα [107]. The saturation of the administered FFA along with the dose appear to affect the subsequent regulation of inflammatory markers, as differences both between and within studies have been reported. MMP1 and MMP2 are matrix metalloproteinases which are a class of proteins involved in cytokine and chemokine processing and leukocyte migration [108]. IL-6, 8 and 18 have been reported to have pro-inflammatory roles in immunity, while IL-10 has been reported to have anti-inflammatory ones [109]. Thus, modulation of these cytokines in the acute phase after a burn serves as a pro-survival mechanism. It is important to note that persistent activation of these pro-inflammatory responses lead to a myriad of other disorders, further complicating the recovery phase of a burn.
Mechanistically, the rise of cytokines and chemokines may be induced by the activation of protein kinase C (PKC) and nuclear factor-kappa beta (NF-κB), both which have been shown to be up-regulated in response to FFA [110]. PKC has been suggested as an activator of NF-κB, which is involved in the transcription of chemokines and cytokines, among other molecules involved in the immune and inflammatory responses [111]. This activation may occur in conjunction with activation NLRP3 inflammasome [112] and [113]. Activation of this inflammasome has also been linked to insulin resistance in diabetes [114]. In burns, NLRP3 inflammasome mRNA has been reported to be upregulated and inflammasome activity correlated with the percent of total body surface area burned and correlated with leukocyte infiltration in tissue [113]. Specifically, high levels of M2 macrophages have been reported in the adipose tissue. Macrophage polarization has been shown to be induced by palmitate, which also inhibits HLA-DR expression in differentiated cells. Interestingly, palmitate affects differentiated and undifferentiated cells differently, inducing a pro-inflammatory response in differentiating cells and an anti-inflammatory one in differentiated ones. Other lipid mediators, including prostaglandins, leukotrienes and resolvins are derived from polyunsaturated fats using cyclooxygenase and lipoxygenase enzymes. These molecules have diverse effects, ranging from neutrophil chemotaxis and vascular dilation to inhibition of mitogen-induced lymphocyte proliferation [115].
Interestingly, cytokines have also been implicated in the induction of lipolysis, suggesting a feedback cycle wherein cytokines stimulate the breakdown of triglycerides, which then initiate processes involved in the development of lipid mediators and inflammatory markers. These inflammatory markers and FFA can then stimulate greater lipolysis and systemically affect other organ systems. For example, the cytokines IL-6 and IL-4 have been reported to recruit macrophages to WAT and initiate the process of browning. BAT has a characteristic brown colour caused by high levels of mitochondria (Figure 2). Despite these high levels, the electron transport chains within are inefficient with regards to ATP production; high rates of uncoupling, namely caused by uncoupling protein-1 (UCP-1) dissipates much of the energy as heat [45]. Although the mechanism with which UCP-1 transports H+ across the inner mitochondrial membrane (IMM) remains unclear, several hypotheses involving fatty acids have been put forth; that of it being an H+ or –OH uniporter with allosteric long chain fatty acids (LCFA) as activators and that of it facilitating transport of H+ bound to LCFA across the membrane. Current evidence suggests that UCP-1 is activated by LCFA which are bound to the protein and move within the IMM, transporting H+ [116]. However, a recent study by Chouchani et al. suggests fatty acids are not required for uncoupling and instead, proposes reactive oxygen species (ROS) as the prime mediators of this phenomenon [117]. In burns, both increased ROS production and FFA are prominent and persistent.
Figure 2.
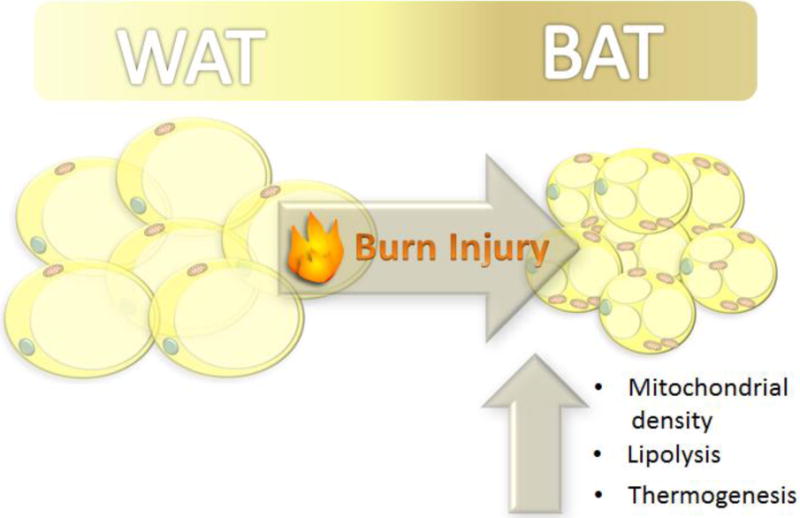
Following a severe burn, white adipose tissue (WAT) adopts the characteristics of brown adipose tissue (BAT).
The phenotypic conversion of WAT to BAT has also been observed in both animal models and human burn patients [32], [118] and [119]. This switch has been shown to be accompanied by increased UCP-1 expression, mitochondrial density and leak respiratory capacity. While this conversion may be hailed as beneficial with regards to weight loss and heat production, the reasoning behind its appearance in pathological conditions such as burns remains unclear – does BAT serve a protective role in the acute phase of burn injury or is it simply a by-product of other hypermetabolic changes? Complicating the answer to this question is the uncertainty of the mechanism underlying browning. Browning appears to be an amalgamation of several events that occur in response to stressors including catecholamine release, transcription regulation and ROS production.
As previously mentioned, circulating levels of epinephrine, norepinephrine and other stress hormones are elevated immediately post burn and contribute to lipolysis and an influx of FFA into the serum. This catecholamine release may serve as one of the main initiators of browning as pharmacological β-adrenergic receptor blockade has been reported to attenuate browning in adipocytes [32] and [119]. Further, β-adrenergic agonists appear to activate browning markers and increase resting energy expenditure, in vivo [120] and [121]. Downstream, the cytokines IL-4 and IL-13 have been linked to browning using gene knock outs; IL-4, IL-13−/− mice failed to demonstrate up-regulations in thermogenesis markers such as UCP-1, Ppargc1, Acox1 and Asc11, as was seen in wild type mice. These mice further demonstrated hypothermia in response to cold stress. This response appeared to be modulated by IL-4Rα activity, whereby IL-4RαL/L reduced BAT markers [121] and [122]. Complicating this picture is the involvement of alternatively activated macrophages; pharmacological inhibition of these macrophages with clodronate reduced cold-induced browning. Macrophages are prime producers of IL-4, IL-13 and further, cause the phosphorylation of protein kinase A (PKA) and perilipin, which in turn stimulate lipolysis. However, palmitate, a product of β-oxidation, has been reported to polarize macrophages, leading to a ‘chicken and egg’ dilemma.
It is important to note that clodronate has been reported to mediate anti-inflammatory effects by reducing levels of IL-1β, IL-6 and TNFα [123]. IL-6 has been reported to mediate the expression of IL-4R and augment the polarization of macrophages, suggesting that the aforementioned reduction in browning caused by clodronate may in fact have been a result of reduced IL-6 signalling [124]. Indeed, IL-6 levels are significantly elevated in burn patients and gene knock-outs of this cytokine prevent WAT browning, a phenomenon rescued with WT administration of IL-6 [32], [118] and [125]. Other molecular modulators of browning such as peroxisome proliferator-activated receptors (PPAR-y) have also been proposed; expression of BAT markers such as increased UCP-1 production have been reported with ginsenoside Rb1, lactate, thymol and erythropoietin activity [126], [127], [128] and [129]. The up-regulation of browning markers caused by these factors has been shown to be correlated with the expression PPAR, a group of transcription factors involved in the regulation of cellular differentiation and metabolic genes. In hypermetabolic conditions such as burns, multiple metabolic adaptations involving lipids occur. Unfortunately, many of the products of these changes are also involved in the initial driving steps, rendering the development of a mechanistic approach to decipher lipodomic changes difficult. The resultant web of changes drives the hypermetabolic state and leads to the persistence of factors which ultimately contributes to patient morbidity and mortality.
5. Glucose metabolism and insulin resistance post-burn
Rapid-onset hyperinsulinemia is a well-established metabolic change that has been reported to occur following burn injury [2], [130] and [131]. As with diabetes, these changes occur coincident with high levels of glucose and infer resistance to the hormone [2], [130], [131] and [132]. Trauma-induced insulin resistance (IR), however, is different in that a myriad of factors can contribute to hyperglycemia and therefore, the onset and persistence of IR. In particular, much of the initial hyperglycemia that occurs post trauma is a part of the fight or flight stress response, whereby plasma glucose is increased via stimulation of glucose secretion, gluconeogenesis and glycogenolysis [130] and [133]. Some of the earliest work performed on the dynamics of glucose and insulin resistance following thermal injury illustrated the complexity of these fluctuations, the scope of which may be beyond this review [134], [135] and [136]. Burns have been reported to increase levels of the glucose transporter, GLUT1, in cells that primarily utilize glycolysis as a main energy source, suggesting that the initial response is geared towards the uptake of glucose [137]. However, soon thereafter, cells lose sensitivity towards insulin, as demonstrated by the reduction of GLUT4 transporter expression observed in adipocytes and reduced clearance of glucose despite raised levels of insulin [138].
Initially, elevated levels of glucose serve as an efficient energy source and have been reported to increase cell proliferation, foster wound healing and mediate a switch from oxidative phosphorylation to glycolysis [139], [140] and [141]. However, these effects appear to be tissue specific, with much of the literature suggesting apoptotic effects with increasing levels of glucose [142] and [143]. Clinically, hyperglycemia has been linked to increased incidences of sepsis, longer hospital stays and higher mortality – outcomes which are reversed with continuous infusions of insulin [144], [145], [146] and [147]. Interestingly, while burn size, age and sex are all important variables with regards to hyperglycemia and insulin resistance, wound healing does not appear to affect insulin resistance as abnormal plasma insulin, fasting glucose and insulin sensitivity have been reported in patients at discharge, when wounds were 95% healed [148]. Persistent levels of hyperglycemia lead to insulin resistance, but in pathophysiological conditions, the presence of these changes are not always synchronized; in burns, hyperglycemia has been reported to be elevated for 6 months, before gradually decreasing to physiological levels, while insulin has been observed to be elevated for 3 years, suggesting the influence of many other factors in its persistence [2] and [129]. It has been postulated that persistent insulin resistance is mediated by hyperglycemia induced by other hypermetabolic changes such as catecholamines, inflammatory markers, ER stress and FFA.
5.1 Catecholamines and glucose control
Epinephrine and its derivative, norepinephrine are two stress-related hormones which are systemically released and which activate pathways involved in glucose metabolism and alertness. During periods of stress, glucose is mobilized and glycogenesis inhibited in an effort to make energy readily available [89]. Acute administration of epinephrine reduces the insulin sensitivity of most tissues and while a definitive mechanism has not yet been determined, it has recently been suggested to increase PDK mRNA via a pathway independent of p38 [149]. As mentioned, in burns, epinephrine and norepinephrine have been reported to be elevated up to 540 days post injury, implicating their involvement in the perpetuation of hyperglycemia and consequently insulin resistance [2]. Interestingly, studies examining the effect of chronic epinephrine using animal models of obesity have reported protective effects, with epinephrine enhancing insulin sensitivity and increasing glucose uptake in some tissues [150] and [151]. Cortisol, a longer lasting stress hormone which is released alongside epinephrine and norepinephrine during stress, has also been shown to increase glucose production and impair glucose uptake in response to insulin [152]. Periods of chronic stress (or multiple acute stressors spaced closely together) result in the persistent elevation of cortisol which participates in a feedback loop that further increases levels of cortisol [153]. In such states, cortisol can give rise to persistent hyperglycemia which can lead to subsequent insulin resistance.
5.2 Inflammatory markers and insulin resistance
The presence of inflammatory cytokines such as TNFα, IL-1 and IL-6 has been heavily reported in the literature to occur coincident with insulin resistance seen in diabetes [154]. These findings strongly implicate a role for these markers in the development of insulin resistance. TNFα, an inflammatory cytokine that recruits activated macrophages, was the first inflammatory cytokine that was reported to alter glucose uptake in response to insulin [155]. Involvement of its receptors, P55 and P75, and tyrosine phosphorylation of IRS-1 have been suggested to contribute to TNFα-mediated insulin sensitivity [156]. The IL-1 family is an interesting group of cytokines as both the inflammatory IL-1α and β and their antagonist, IL-1 receptor antagonist (RA) have been shown to be present in adipocytes [157]. IL-1β has been shown to decrease IRS-1 expression and consequently levels of tyrosine phosphorylation, protein kinase B activation and AS160 phosphorylation [158]. These effects also decrease GLUT4 translocation to the plasma membrane, directly linking this class of cytokines to insulin resistance. IL-6 has also been implicated in the development of insulin resistance by its elevation during obesity and insulin resistant states which parallel its reduction during weight loss [159]. However, the effects of this cytokine are debated as IL-6 deficient (−/−) mice display obesity, insulin resistance and hepatic inflammation [160] and [161]. Recently, exercise was shown to increase glucagon-like peptide-1 (GLP-1) and stimulate insulin secretion via IL-6 [162]. This resulted in better glycemic control and suggested a mediating role for IL-6, whereby the cytokine modulates insulin levels in response to plasma glucose. In burns, this endocrine loop may initially work to reduce glucose levels by stimulating insulin production (thereby serving a protective role). Unfortunately, the persistence of hyperglycemia, as caused by the amalgamation of several hypermetabolic changes, leads to chronic elevations in insulin.
5.3 ER stress as an inducer of insulin resistance
When the protein folding burden on the endoplasmic reticulum (ER) is too high, ER stress occurs. Several pathways that are part of the unfolded protein response (UPR) are then activated to reduce this burden. Inositol requiring enzyme-1α (IRE1α)-dependant activation of c-JUN N-terminal kinase (JNK), a downstream molecule of the UPR, leads to serine phosphorylation of IRS-1 and a subsequent reduction in insulin sensitivity (Figure 3) [163]. ER stress is a prominent feature of burns and JNK activation along with serine phosphorylation of IRS-1 has been observed in both patients and animal models of the disease [53] and [131]. Interestingly, JNK has also been reported to be activated by TNFα and IL-6, suggesting multiple mechanisms by which it can induce insulin resistance [164]. In burns, an insulinemic flux is initially triggered by rapid elevations in glucose caused by the catecholamine stress response. This response is later sustained by other components of the hypermetabolic response such as ER stress and inflammation, which produce more lasting changes.
Figure 3.
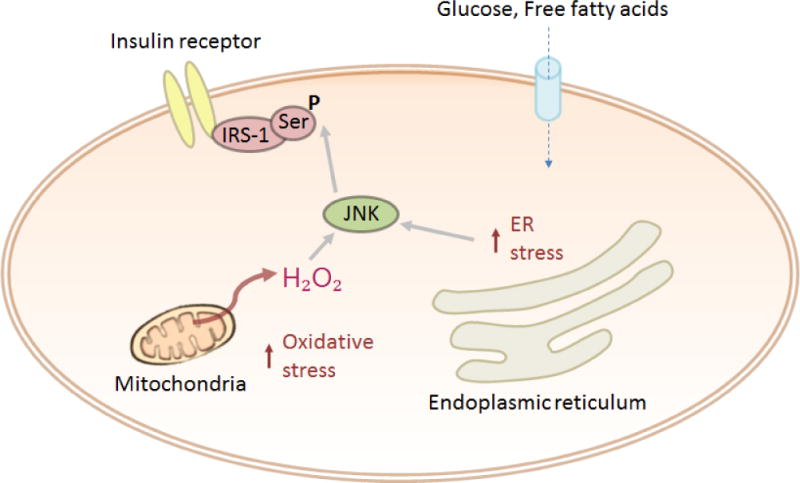
Factors which induce ER stress subsequently increase the cellular resistance to insulin signalling via JNK.
5.4 FFA and insulin resistance
The induction of insulin release following lipolytic flux has been reported in the literature since the late 60’s [165], where oleate administration raised insulin levels and consequently, lowered those of glucose. Further studies of this effect – particularly in pathophysiological conditions – have revealed a complicated web of interactions that have blurred the lines between cause and effect. Recent mechanistic studies have proposed the involvement of g-protein coupled receptor 40 (GPR40), coenzymes, g-protein coupled receptor 120 (GPR120) and ultimately, insulin itself in the progression of insulin resistance. GPR40 has been shown to be a receptor for FFA on pancreatic β-cells that amplifies cellular response to glucose by activating cyclic AMP (cAMP) and protein lipase C (PLC) that together, mobilise calcium and induce insulin secretion [166]. Interestingly, persistent calcium mobilisation, as caused by elevated levels of FFA (a common feature of burn patients), may be linked to ER stress, which itself plays a role in insulin resistance. β-cell responsiveness has also been reported to be dependant on long chain coenzyme A (LC-CoA), an enzyme which facilitates fatty acid breakdown and malonyl CoA, an inhibitor of the enzyme that regulates LC-CoA [167]. CoA thioesters have also been shown to inhibit insulin degrading enzyme, thus contributing to elevated levels of insulin [168]. Lastly, GPR120, another FFA receptor, has been reported to activate glucagon-like peptide 1 (GLP-1) and induce insulin secretion [169]. Insulin itself has been proposed to induce insulin resistance (alongside FFA), based upon the observance that GPR40 KO mice that lacked hyperinsulinemia also did not develop insulin resistance [166] and [170]. Inflammation, ER stress, FFA and insulin have all been implicated in the phosphorylation and alteration of IRS-1, leading to insulin resistance.
6. Treatment options to alleviate the hypermetabolic response
6.1 Insulin
The modulation of blood glucose levels following severe burns is paramount in reducing the morbidity and mortality stemming from stress-induced hyperglycemia. Insulin therapy, wherein glucose levels are kept at around 130 mg/dL, significantly decreases infection instances, sepsis, sepsis-associated organ failure and mortality in critically ill patients [171]. Indeed, a lack of early glycemic control is associated with higher mortality. Insulin treatment mediates peripheral glucose uptake into the skeletal muscle and adipose tissue, while also suppressing hepatic gluconeogenesis. Beyond its ability to restore euglycemia, insulin has also been shown to increase DNA replication and fatty acid synthesis, as well as inhibiting the production of pro-inflammatory mediators such as IL-5 and IL-6 and reducing the donor site healing time in burn patients [172], [173], [174] and [175]. One means by which insulin opposes the hypercatabolic response post-burn is by shifting the balance between protein breakdown and synthesis, particularly in muscle tissue. Despite adequate nutritional intake, burn patients have higher rates of protein catabolism which can be ameliorated dramatically via insulin treatment, improving protein synthesis rates by nearly 400%. This likely occurs by the stimulating the inward transport of amino acids, as phenylalanine uptake was increased by 535% in patients following insulin infusion [176] and [177]. Insulin has also been shown to improve mitochondrial function by improving coupling efficiency [178]. Despite the benefits, aggressive insulin therapy has been associated with episodes of hypoglycemia which can worsen patient outcomes and therefore, the close monitoring of glucose levels is crucial [171]. Combinatorial treatment, such as the use of insulin with exogenous glucagon-like peptide-1 (GLP-1), can reduce the amount of insulin required to achieve normoglycemia. Fenofibrate, a lipolysis agonist, reduces insulin resistance and when used in tandem with insulin, can result in fewer hypoglycemic outcomes [179].
6.2 Metformin
Metformin’s high safety profile, exhibiting minimal risk of hypoglycemia, renders it a strong alternative to insulin for achieving euglycemia in patients with severe burns. Via the activation of AMP-activated protein kinase (AMPK), metformin supresses hepatic gluconeogenesis and is known to increase insulin sensitivity and thus, peripheral glucose uptake. By inhibiting complex I of the electron transport chain, metformin may also lower the mitochondrial hyperactivity seen following a severe burn [180]. Recently we have demonstrated that metformin decreases glucose in burn patients equally as effectively as insulin with the additional benefit of a reduction in hypoglycemic episodes [181]. Furthermore, metformin significantly decreased the circulation of saturated fatty acids and down-regulated the concentrations of serum IL-1β and monocyte chemoattractant protein-1, thus making it likely that this agent has anti-inflammatory effects [181]. Interestingly, other studies have shown that metformin does not improve the rate of protein breakdown, and that the anti-catabolic benefits of this intervention are mainly attributable to increased protein synthesis, thus improving net muscle protein balance and preventing tissue wasting [182]. The use of metformin has been associated with a rare condition known as lactic acidosis, particularly in patients with some degree of renal dysfunction, and caution should be taken. However, the ease of oral administration and economic benefits of metformin makes it an attractive therapeutic option for the long-term control of insulin resistance and tissue catabolism [181].
6.3 Propranolol
As outlined above, catecholamines have a major contribution to the hypermetabolic response, with sustained increases that can persist for months following the injury. Propranolol is a non-selective β-blocker used to attenuate the hyperactive sympathetic system seen in burn patients. Inhibition of β-adrenergic signalling following thermal injuries has been shown to decrease the cardiac workload, supraphysiological thermogenesis, peripheral lipolysis and ultimately, REE [177]. This β-blocker has also been shown to reduce ER stress, subsequently improving insulin sensitivity [183]. Work by Herndon et al. has demonstrated in severely burned (>40% TBSA) children that propranolol treatment increases the net muscle-protein balance by 82% over base-line values, while it decreases by 27% in the control group. These changes in skeletal muscle protein dynamics are associated with a decrease in REE as well as the preservation of lean body mass, thus counteracting patient hypermetabolism [182]. Moreover, Barrow et al. showed in severely burned children that propranolol is capable of preventing hepatomegaly through a reduction in hepatic free fatty acid uptake. Indeed, in 80% of children not receiving propranolol, liver sizes increased by 100% or more as measured by ultrasonic scanning. Over the same time period, 86% of children who received the β-blocker showed no changes or a decrease in liver size. This was accompanied by a down-regulation of 11 genes related to lipid metabolism [184]. A recent systemic review on the use of propranolol confirms the decreased REE, improved lean mass and insulin resistance attributed to this compound, however, further trials on the adult population are warranted [185].
6.4 Recombinant human growth hormone
Disruptions in the growth hormone (GH)/insulin-like growth factor-1 (IGF-1)/IGF binding protein-3 (IGFBP-3) hormone axis following a severe burn are known to persist for years. This is likely due to the overexpression of proinflammatory cytokines such as TNF-α and IL-1β which have been shown to inhibit this system [177]. In children over this timespan, physical growth and development are blunted, as IGF-1 and IFGBP-3 remain significantly diminished. The daily intramuscular injection of the anabolic agent recombinant human growth hormone (rHGH) has been shown to improve weight gain, height velocities, lean body mass, nitrogen and potassium balance and cardiac function in burn patients. Administration of rHGH seemingly confers these benefits via IGF-1, as this biomolecule increases in the serum by 100% in patients receiving treatments versus healthy individuals [182]. Additionally, growth hormone has been shown to induce short-term anti-apoptotic effects via mitochondrially-associated mi-RNAs [186]. Children receiving rHGH following a severe burn show significant increases in lean body mass and improvement in skeletal muscle protein kinetics, even at a year after treatments are stopped [187]. While rHGH appears to attenuate the hypermetabolic response in children, administration of this agent in adults has been linked to hyperglycemia and hypermetabolism. Indeed, in a prospective, multicenter, double-blind, randomized, placebo-controlled trial with 285 critically ill non-burned patients, it was shown that high doses of rHGH are associated with a 40% increase in morbidity and mortality [188]. Therefore, age may be a predictive variable of outcome for rHGH interventions, suggesting that the target patient demographic must be considered before initiating treatment.
6.5 Oxandrolone
While it is known that testosterone is an anabolic agent which increases lean body mass, the potential hepatotoxicity and masculinizing effects of this compound limits its therapeutic value. Oxandrolone is a synthetic analog to testosterone which is safe to use in males or females due to the lack of hepatotoxicity, ease of clearance by the kidneys and because it possesses of only 5% of the virilizing androgenic effects of testosterone [182]. In severely burned adults with an average TBSA of 44%, oxandrolone treatment resulted in a significantly lower mortality rate [189]. Jeschke et al. showed that 0.1 mg/kg of oxandrolone given every 12 h resulted in shortened length of hospital stay, maintenance of lean body mass and improved hepatic protein synthesis [190]. Indeed, oxandrolone likely combats the hypermetabolic response in muscle via a decrease in protein breakdown in adults, while increasing protein synthesis in children, thus implicating age as a key determinant in this agent’s effects [182]. Versus unburned controls, oxandrolone significantly increased lean body mass and bone mineral content at 6, 9, and 12 months following the injury. When combined with exercise in burned children, oxandrolone’s beneficial effects are further amplified, leading to improved rehabilitation [191].
7. Conclusions and future directions
Following a severe burn, the ubiquitous and essential hypermetabolic response required to survive, are undone by the severity and length of this physiological response to trauma. Months to years of persistent hypercatabolism, increased lipolysis, supraphysiological thermogenesis and hyperactivity of the sympathetic system and inflammatory response entail the wasting of patient tissues and organs, predisposing them to a host of disorders and death. Decades of advances in burn care have greatly transformed our understanding and treatment of the hypermetabolic response, increasing the rate of survival, particularly for pediatric and adult burns (Figure 4). Despite these efforts, we are only capable of managing this phenomenon, and lack the ability and knowledge to completely eliminate hypermetabolism. Furthermore, the morbidity and mortality in the LD50 burn size of elderly patients (>65 years) has not improved throughout this time period, indicating a dire need for better treatment options in this demographic [192]. More studies are required to determine biomarkers of the hypermetabolic condition that can be used for rapid assessment and intervention in burn patients. Combinatorial treatments which modulate glucose levels and inflammatory mediators in addition to anabolic agents may further subdue the hypermetabolic response, leading to improved outcomes for severe thermal injuries.
Figure 4.
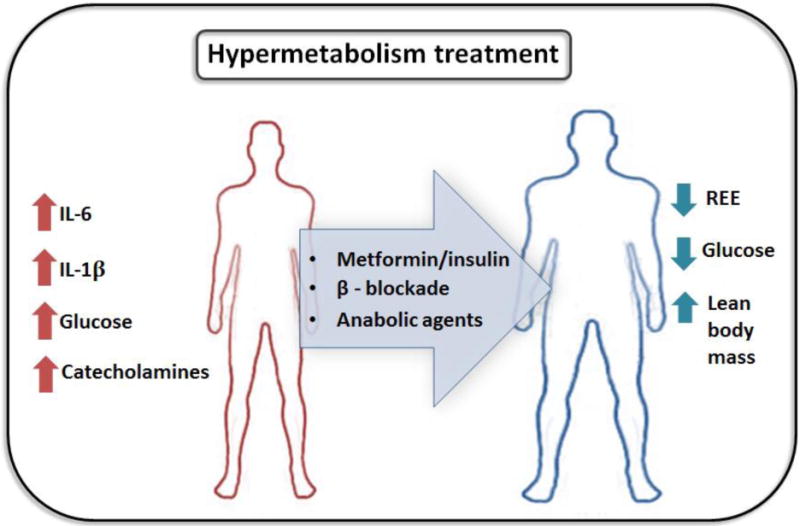
Glucose control, beta-blockade and anabolic agents can be used to counter the systemic derangements initiated by the hypermetabolic response.
Highlights.
Severe burns trigger chronic hypermetabolism.
Hypermetabolism increases incidences of infection and multi-organ dysfunction.
Cytokines, catecholamines and hormones induce the hypermetabolic state.
ER stress, abnormal mitochondria and insulin resistance post-burn are interconnected.
Beta blockade and glucose control agents can limit hypermetabolism post-thermal injury.
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research (#123336), the Canada Foundation for Innovation Leader’s Opportunity Fund (#25407) and National Institutes of Health (2R01GM087285-05A1).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Health Organization. The injury chartbook: a graphical overview of the global burden of injuries. Geneva: 2002. [Google Scholar]
- 2.Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, Herndon DN. Long-term persistance of the pathophysiologic response to severe burn injury. PloS one. 2011;6(7):e21245. doi: 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolfe RR. Review: acute versus chronic response to burn injury. Circulatory shock. 1980;8(1):105–115. [PubMed] [Google Scholar]
- 4.Jeschke MG, Chinkes DL, Finnerty CC, Kulp G, Suman OE, Norbury WB, Herndon DN. The pathophysiologic response to severe burn injury. Annals of surgery. 2008;248(3):387. doi: 10.1097/SLA.0b013e3181856241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeschke MG, Mlcak RP, Finnerty CC, Norbury WB, Gauglitz GG, Kulp GA, Herndon DN. Burn size determines the inflammatory and hypermetabolic response. Critical care. 2007;11(4):1. doi: 10.1186/cc6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wieser V, Moschen AR, Tilg H. Inflammation, cytokines and insulin resistance: a clinical perspective. Archivum immunologiae et therapiae experimentalis. 2013;61(2):119–125. doi: 10.1007/s00005-012-0210-1. [DOI] [PubMed] [Google Scholar]
- 7.Simmons PS, Miles JM, Gerich JE, Haymond MW. Increased proteolysis. An effect of increases in plasma cortisol within the physiologic range. Journal of Clinical Investigation. 1984;73(2):412. doi: 10.1172/JCI111227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochemical pharmacology. 2003;66(8):1403–1408. doi: 10.1016/s0006-2952(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 9.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 10.Rydén M, Dicker A, van Harmelen V, Hauner H, Brunnberg M, Perbeck L, Arner P. Mapping of early signaling events in tumor necrosis factor-α-mediated lipolysis in human fat cells. Journal of Biological Chemistry. 2002;277(2):1085–1091. doi: 10.1074/jbc.M109498200. [DOI] [PubMed] [Google Scholar]
- 11.Barrow RE, Hawkins HK, Aarsland A, Cox R, Rosenblatt J, Barrow LN, Herndon DN. Identification of factors contributing to hepatomegaly in severely burned children. Shock. 2005;24(6):523–528. doi: 10.1097/01.shk.0000187981.78901.ee. [DOI] [PubMed] [Google Scholar]
- 12.Machado MV, Ferreira DM, Castro RE, Silvestre AR, Evangelista T, Coutinho J, Cortez-Pinto H. Liver and muscle in morbid obesity: the interplay of fatty liver and insulin resistance. PloS one. 2012;7(2):e31738. doi: 10.1371/journal.pone.0031738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao CL, Zhu C, Zhao YP, Chen XH, Ji CB, Zhang CM, Guo XR. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Molecular and cellular endocrinology. 2010;320(1):25–33. doi: 10.1016/j.mce.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 14.Van Herpen NA, Schrauwen-Hinderling VB. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiology & behavior. 2008;94(2):231–241. doi: 10.1016/j.physbeh.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 15.O’Halloran E, Kular J, Xu J, Wood F, Fear M. Non-severe burn injury leads to depletion of bone volume that can be ameliorated by inhibiting TNF-α. Burns. 2015;41(3):558–564. doi: 10.1016/j.burns.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Spies M, Chappell VL, Dasu MR, Herndon DN, Thompson JC, Wolf SE. Role of TNF-α in gut mucosal changes after severe burn. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2002;283(3):G703–G708. doi: 10.1152/ajpgi.00149.2001. [DOI] [PubMed] [Google Scholar]
- 17.Reid MB, Li YP. Tumor necrosis factor-α and muscle wasting: a cellular perspective. Respiratory research. 2001;2(5):1. doi: 10.1186/rr67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashcroft GS, Jeong MJ, Ashworth JJ, Hardman M, Jin W, Moutsopoulos N, Song XY. Tumor necrosis factor-alpha (TNF-α) is a therapeutic target for impaired cutaneous wound healing. Wound Repair and Regeneration. 2012;20(1):38–49. doi: 10.1111/j.1524-475X.2011.00748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nature Reviews Immunology. 2013;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nature Reviews Immunology. 2010;10(12):826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature medicine. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stanojcic M, Chen P, Harrison RA, Wang V, Antonyshyn J, Zúñiga-Pflücker JC, Jeschke MG. Leukocyte infiltration and activation of the NLRP3 inflammasome in white adipose tissue following thermal injury. Critical care medicine. 2014;42(6):1357. doi: 10.1097/CCM.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolsk E, Mygind H, Grøndahl TS, Pedersen BK, van Hall G. IL-6 selectively stimulates fat metabolism in human skeletal muscle. American Journal of Physiology-Endocrinology and Metabolism. 2010;299(5):E832–E840. doi: 10.1152/ajpendo.00328.2010. [DOI] [PubMed] [Google Scholar]
- 24.Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochemical journal. 1998;334(2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier JA, Larner AC. In Seminars in immunology. 1. Vol. 26. Academic Press; 2014. Feb, Toward a new STATe: the role of STATs in mitochondrial function; pp. 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdullahi A, Jeschke MG. White Adipose Tissue Browning: A Double-edged Sword. Trends in Endocrinology & Metabolism. 2016;27(8):542–552. doi: 10.1016/j.tem.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herndon DN, Hart DW, Wolf SE, Chinkes DL, Wolfe RR. Reversal of catabolism by beta-blockade after severe burns. New England Journal of Medicine. 2001;345(17):1223–1229. doi: 10.1056/NEJMoa010342. [DOI] [PubMed] [Google Scholar]
- 28.Taylor MRG. Pharmacogenetics of the human beta-adrenergic receptors. The pharmacogenomics journal. 2007;7(1):29–37. doi: 10.1038/sj.tpj.6500393. [DOI] [PubMed] [Google Scholar]
- 29.Obata T, Brown GE, Yaffe MB. MAP kinase pathways activated by stress: the p38 MAPK pathway. Critical care medicine. 2000;28(4):N67–N77. doi: 10.1097/00003246-200004001-00008. [DOI] [PubMed] [Google Scholar]
- 30.LAZOU A, SUGDEN PH, CLERK A. Activation of mitogen-activated protein kinases (p38-MAPKs, SAPKs/JNKs and ERKs) by the G-protein-coupled receptor agonist phenylephrine in the perfused rat heart. Biochemical Journal. 1998;332(2):459–465. doi: 10.1042/bj3320459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meitzen J, Luoma JI, Stern CM, Mermelstein PG. β1-Adrenergic receptors activate two distinct signaling pathways in striatal neurons. Journal of neurochemistry. 2011;116(6):984–995. doi: 10.1111/j.1471-4159.2010.07137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patsouris D, Qi P, Abdullahi A, Stanojcic M, Chen P, Parousis A, Jeschke MG. Burn induces browning of the subcutaneous white adipose tissue in mice and humans. Cell reports. 2015;13(8):1538–1544. doi: 10.1016/j.celrep.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kulp GA, Herndon DN, Lee JO, Suman OE, Jeschke MG. Extent and magnitude of catecholamine surge in pediatric burned patients. Shock (Augusta, Ga) 2010;33(4):369. doi: 10.1097/SHK.0b013e3181b92340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nature Reviews Molecular Cell Biology. 2015;16(11):678–689. doi: 10.1038/nrm4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proceedings of the National Academy of Sciences. 2001;98(24):13710–13715. doi: 10.1073/pnas.231370798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. Journal of Allergy and Clinical Immunology. 2013;132(5):1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dimitriadis G, Leighton B, Parry-Billings M, Sasson S, Young M, Krause U, Newsholme EA. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochemical Journal. 1997;321(3):707–712. doi: 10.1042/bj3210707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmer BF. Regulation of potassium homeostasis. Clinical Journal of the American Society of Nephrology. 2015;10(6):1050–1060. doi: 10.2215/CJN.08580813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolfe RR, Herndon DN, Jahoor F, Miyoshi H, Wolfe M. Effect of severe burn injury on substrate cycling by glucose and fatty acids. New England Journal of Medicine. 1987;317(7):403–408. doi: 10.1056/NEJM198708133170702. [DOI] [PubMed] [Google Scholar]
- 40.Norbury WB, Herndon DN, Branski LK, Chinkes DL, Jeschke MG. Urinary cortisol and catecholamine excretion after burn injury in children. The Journal of Clinical Endocrinology & Metabolism. 2008;93(4):1270–1275. doi: 10.1210/jc.2006-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2009;1787(11):1324–1333. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 42.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nature reviews Drug discovery. 2010;9(6):447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 43.Herst PM, Tan AS, Scarlett DJG, Berridge MV. Cell surface oxygen consumption by mitochondrial gene knockout cells. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2004;1656(2):79–87. doi: 10.1016/j.bbabio.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Hart DW, Wolf SE, Mlcak R, Chinkes DL, Ramzy PI, Obeng MK, Herndon DN. Persistence of muscle catabolism after severe burn. Surgery. 2000;128(2):312–319. doi: 10.1067/msy.2000.108059. [DOI] [PubMed] [Google Scholar]
- 45.Matthias A, Ohlson KB, Fredriksson JM, Jacobsson A, Nedergaard J, Cannon B. Thermogenic responses in brown fat cells are fully Ucp1-dependent UCP2 or UCP3 do not substitute for UCP1 in adrenergically or fatty acid-induced thermogenesis. Journal of Biological Chemistry. 2000;275(33):25073–25081. doi: 10.1074/jbc.M000547200. [DOI] [PubMed] [Google Scholar]
- 46.Yasuhara S, Perez ME, Kanakubo E, Yasuhara Y, Shin YS, Kaneki M, Martyn JJ. Skeletal muscle apoptosis after burns is associated with activation of proapoptotic signals. American Journal of Physiology-Endocrinology And Metabolism. 2000;279(5):E1114–E1121. doi: 10.1152/ajpendo.2000.279.5.E1114. [DOI] [PubMed] [Google Scholar]
- 47.Szczesny B, Brunyánszki A, Ahmad A, Oláh G, Porter C, Toliver-Kinsky T, Szabo C. Time-Dependent and Organ-Specific Changes in Mitochondrial Function, Mitochondrial DNA Integrity, Oxidative Stress and Mononuclear Cell Infiltration in a Mouse Model of Burn Injury. PloS one. 2015;10(12):e0143730. doi: 10.1371/journal.pone.0143730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porter C, Herndon DN, Sidossis LS, Børsheim E. The impact of severe burns on skeletal muscle mitochondrial function. Burns. 2013;39(6):1039–1047. doi: 10.1016/j.burns.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porter C, Herndon DN, Børsheim E, Bhattarai N, Chao T, Reidy PT, Sidossis LS. Long-term skeletal muscle mitochondrial dysfunction is associated with hypermetabolism in severely burned children. Journal of Burn Care & Research. 2016;37(1):53–63. doi: 10.1097/BCR.0000000000000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiological reviews. 1997;77(3):731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 51.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nature reviews Drug discovery. 2013;12(9):703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 52.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO reports. 2006;7(9):880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jeschke MG, Finnerty CC, Herndon DN, Song J, Boehning D, Tompkins RG, Gauglitz GG. Severe injury is associated with insulin resistance, endoplasmic reticulum stress response, and unfolded protein response. Annals of surgery. 2012;255(2):370. doi: 10.1097/SLA.0b013e31823e76e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mao W, Iwai C, Qin F, Liang CS. Norepinephrine induces endoplasmic reticulum stress and downregulation of norepinephrine transporter density in PC12 cells via oxidative stress. American Journal of Physiology-Heart and Circulatory Physiology. 2005;288(5):H2381–H2389. doi: 10.1152/ajpheart.00904.2004. [DOI] [PubMed] [Google Scholar]
- 55.Wei SG, Yu Y, Weiss RM, Felder RB. Endoplasmic reticulum stress increases brain MAPK signaling, inflammation and renin-angiotensin system activity and sympathetic nerve activity in heart failure. American Journal of Physiology-Heart and Circulatory Physiology. 2016;311(4):H871–H880. doi: 10.1152/ajpheart.00362.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei SG, Yu Y, Weiss RM, Felder RB. Inhibition of brain mitogen-activated protein kinase signaling reduces central endoplasmic reticulum stress and inflammation and sympathetic nerve activity in heart failure rats. Hypertension. 2016;67(1):229–236. doi: 10.1161/HYPERTENSIONAHA.115.06329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brooks NC, Song J, Boehning D, Kraft R, Finnerty CC, Herndon DN, Jeschke MG. Propranolol improves impaired hepatic phosphatidylinositol 3-kinase/akt signaling after burn injury. Molecular Medicine. 2012;18(4):707. doi: 10.2119/molmed.2011.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ni L, Zhou C, Duan Q, Lv J, Fu X, Xia Y, Wang DW. β-AR blockers suppresses ER stress in cardiac hypertrophy and heart failure. PLoS One. 2011;6(11):e27294. doi: 10.1371/journal.pone.0027294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.George I, Sabbah HN, Xu K, Wang N, Wang J. β-Adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in a coronary embolization model of heart failure in canines. Cardiovascular research. 2011;91(3):447–455. doi: 10.1093/cvr/cvr106. [DOI] [PubMed] [Google Scholar]
- 60.Haas MJ, Kurban W, Shah H, Onstead-Haas L, Mooradian AD. Beta Blockers Suppress Dextrose-Induced Endoplasmic Reticulum Stress, Oxidative Stress, and Apoptosis in Human Coronary Artery Endothelial Cells. American journal of therapeutics. 2015 doi: 10.1097/MJT.0000000000000200. [DOI] [PubMed] [Google Scholar]
- 61.Foufelle F, Fromenty B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacology research & perspectives. 2016;4(1) doi: 10.1002/prp2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song J, Finnerty CC, Herndon DN, Boehning D, Jeschke MG. Severe burn-induced endoplasmic reticulum stress and hepatic damage in mice. Mol Med. 2009;15(9–10):316–320. doi: 10.2119/molmed.2009.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park SW, Zhou Y, Lee J, Lee J, Ozcan U. Sarco (endo) plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proceedings of the National Academy of Sciences. 2010;107(45):19320–19325. doi: 10.1073/pnas.1012044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeschke MG, Gauglitz GG, Song J, Kulp GA, Finnerty CC, Cox RA, Boehning D. Calcium and ER stress mediate hepatic apoptosis after burn injury. Journal of cellular and molecular medicine. 2009;13(8b):1857–1865. doi: 10.1111/j.1582-4934.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li Y, Ge M, Ciani L, Kuriakose G, Westover EJ, Dura M, Tabas I. Enrichment of Endoplasmic Reticulum with Cholesterol Inhibits Sarcoplasmic-Endoplasmic Reticulum Calcium ATPase-2b Activity in Parallel with Increased Order of Membrane Lipids Implications For Depletion Of Endoplasmic Reticulum Calcium Stores And Apoptosis In Cholesterol-Loaded Macrophages. Journal of Biological Chemistry. 2004;279(35):37030–37039. doi: 10.1074/jbc.M405195200. [DOI] [PubMed] [Google Scholar]
- 67.De Meis L, Ketzer LA, Da Costa RM, De Andrade IR, Benchimol M. Fusion of the endoplasmic reticulum and mitochondrial outer membrane in rats brown adipose tissue: activation of thermogenesis by Ca 2+ PloS one. 2010;5(3):e9439. doi: 10.1371/journal.pone.0009439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Meis L. Brown adipose tissue Ca2+-ATPase uncoupled ATP hydrolysis and thermogenic activity. Journal of Biological Chemistry. 2003;278(43):41856–41861. doi: 10.1074/jbc.M308280200. [DOI] [PubMed] [Google Scholar]
- 69.de Meis L, Arruda AP, da Costa RM, Benchimol M. Identification of a Ca2+-ATPase in Brown Adipose Tissue Mitochondria Regulation Of Thermogenesis By Atp And Ca2+ Journal of Biological Chemistry. 2006;281(24):16384–16390. doi: 10.1074/jbc.M600678200. [DOI] [PubMed] [Google Scholar]
- 70.Leaver EV, Pappone PA. β-adrenergic potentiation of endoplasmic reticulum Ca2+ release in brown fat cells. American Journal of Physiology. 2002;282(5):1016–1024. doi: 10.1152/ajpcell.00204.2001. [DOI] [PubMed] [Google Scholar]
- 71.Smith WS, Broadbridge R, East JM. Sarcolipin uncouples hydrolysis of ATP from accumulation of Ca2+ by the Ca2+-ATPase of skeletal-muscle sarcoplasmic reticulum. Biochemical Journal. 2002;361(2):277–286. doi: 10.1042/0264-6021:3610277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mall S, Broadbridge R, Harrison SL, Gore MG, Lee AG, East JM. The presence of sarcolipin results in increased heat production by Ca2+-ATPase. Journal of Biological Chemistry. 2006;281(48):36597–36602. doi: 10.1074/jbc.M606869200. [DOI] [PubMed] [Google Scholar]
- 73.Bal NC, Maurya SK, Sopariwala DH, Sahoo SK, Gupta SC, Shaikh SA, Tupling AR. Sarcolipin is a newly identified regulator of muscle-based thermogenesis in mammals. Nature Medicine. 2012;18(10):1575–1579. doi: 10.1038/nm.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rowland LA, Bal NC, Kozak LP, Periasamy M. Uncoupling protein 1 and sarcolipin are required to maintain optimal thermogenesis, and loss of both systems compromises survival of mice under cold stress. Journal of Biological Chemistry. 2015;290(19):12282–12289. doi: 10.1074/jbc.M115.637603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Contreras C, González-García I, Seoane-Collazo P, Martínez-Sánchez N, Liñares-Pose L, Rial-Pensado E, Nogueiras R. Reduction of Hypothalamic ER Stress Activates Browning of White Fat and Ameliorates Obesity. Diabetes. 2016:db151547. doi: 10.2337/db15-1547. [DOI] [PubMed] [Google Scholar]
- 76.Contreras C, González-García I, Martínez-Sánchez N, Seoane-Collazo P, Jacas J, Morgan DA, Nogueiras R. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell reports. 2014;9(1):366–377. doi: 10.1016/j.celrep.2014.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F. ATF4 regulates lipid metabolism and thermogenesis. Cell research. 2010;20(2):174–184. doi: 10.1038/cr.2010.4. [DOI] [PubMed] [Google Scholar]
- 78.Okla M, Wang W, Kang I, Pashaj A, Carr T, Chung S. Activation of Toll-like receptor 4 (TLR4) attenuates adaptive thermogenesis via endoplasmic reticulum stress. Journal of Biological Chemistry. 2015;290(44):26476–26490. doi: 10.1074/jbc.M115.677724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Asada R, Kanemoto S, Matsuhisa K, Hino K, Cui M, Cui X, Imaizumi K. IRE1α-XBP1 is a novel branch in the transcriptional regulation of Ucp1 in brown adipocytes. Scientific reports. 2015;5 doi: 10.1038/srep16580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int J Clin Exp Med. 2010;3(1):33–40. [PMC free article] [PubMed] [Google Scholar]
- 81.Kraft R, Herndon DN, Finnerty CC, Hiyama Y, Jeschke MG. Association of postburn fatty acids and triglycerides with clinical outcome in severely burned children. The Journal of Clinical Endocrinology & Metabolism. 2012;98(1):314–321. doi: 10.1210/jc.2012-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolfe RR, Herndon DN, Peters EJ, Jahoor FAROOK, Desai MH, Holland OB. Regulation of lipolysis in severely burned children. Annals of surgery. 1987;206(2):214. doi: 10.1097/00000658-198708000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeschke MG, Patsouris D, Stanojcic M, Abdullahi A, Rehou S, Pinto R, Amini-Nik S. Pathophysiologic response to burns in the elderly. EBioMedicine. 2015;2(10):1536–1548. doi: 10.1016/j.ebiom.2015.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jeschke MG, Przkora R, Suman OE, Finnerty CC, Mlcak RP, Pereira CT, Herndon DN. Sex differences in the long-term outcome after a severe thermal injury. Shock. 2007;27(5):461–465. doi: 10.1097/01.shk.0000238071.74524.9a. [DOI] [PubMed] [Google Scholar]
- 85.Cetinkale O, Yazici Z. Early postburn fatty acid profile in burn patients. Burns. 1997;23(5):392–399. doi: 10.1016/s0305-4179(97)89764-1. [DOI] [PubMed] [Google Scholar]
- 86.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid–induced insulin resistance. The Journal of clinical investigation. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singer P, Shapiro H, Theilla M, Anbar R, Singer J, Cohen J. Anti-inflammatory properties of omega-3 fatty acids in critical illness: novel mechanisms and an integrative perspective. Intensive care medicine. 2008;34(9):1580–1592. doi: 10.1007/s00134-008-1142-4. [DOI] [PubMed] [Google Scholar]
- 88.Galster AD, Bier DM, Cryer PE, Monafo WW. Plasma palmitate turnover in subjects with thermal injury. Journal of Trauma and Acute Care Surgery. 1984;24(11):938–945. doi: 10.1097/00005373-198411000-00003. [DOI] [PubMed] [Google Scholar]
- 89.Vega GL, Baxter CR. Induction of hypertriglyceridemia in rabbits by thermal injury: I. Time course of elevated plasma triglyceride levels. Journal of Burn Care & Research. 1988;9(3):266–270. doi: 10.1097/00004630-198805000-00006. [DOI] [PubMed] [Google Scholar]
- 90.Vaughan M, Steinberg D. Effect of hormones on lipolysis and esterification of free fatty acids during incubation of adipose tissue in vitro. Journal of lipid research. 1963;4(2):193–199. [PubMed] [Google Scholar]
- 91.Wolfe RR, Klein S, Herndon DN, Jahoor F. Substrate cycling in thermogenesis and amplification of net substrate flux in human volunteers and burned patients. Journal of Trauma and Acute Care Surgery. 1990;30:6–9. doi: 10.1097/00005373-199012001-00004. [DOI] [PubMed] [Google Scholar]
- 92.Coombes EJ, Shakespeare PG, Batstone GF. Lipoprotein changes after burn injury in man. Journal of Trauma and Acute Care Surgery. 1980;20(11):971–975. doi: 10.1097/00005373-198011000-00012. [DOI] [PubMed] [Google Scholar]
- 93.Roopenian DC, Low BE, Christianson GJ, Proetzel G, Sproule TJ, Wiles MV. InMAbs. 2. Vol. 7. Taylor & Francis; 2015. Mar, Albumin-deficient mouse models for studying metabolism of human albumin and pharmacokinetics of albumin-based drugs; pp. 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hammond VA, Johnston DG. Substrate cycling between triglyceride and fatty acid in human adipocytes. Metabolism. 1987;36(4):308–313. doi: 10.1016/0026-0495(87)90199-5. [DOI] [PubMed] [Google Scholar]
- 95.Klein S, Wolfe RR. Whole-body lipolysis and triglyceride-fatty acid cycling in cachectic patients with esophageal cancer. Journal of Clinical Investigation. 1990;86(5):1403. doi: 10.1172/JCI114854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Romijn JA, Klein S, Coyle EF, Sidossis LS, Wolfe RR. Strenuous endurance training increases lipolysis and triglyceride-fatty acid cycling at rest. Journal of Applied Physiology. 1993;75(1):108–113. doi: 10.1152/jappl.1993.75.1.108. [DOI] [PubMed] [Google Scholar]
- 97.McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annual review of biochemistry. 1980;49(1):395–420. doi: 10.1146/annurev.bi.49.070180.002143. [DOI] [PubMed] [Google Scholar]
- 98.Harris RL, Frenkel RA, Cottam GL, Baxter CR. Lipid mobilization and metabolism after thermal trauma. Journal of Trauma and Acute Care Surgery. 1982;22(3):194–198. doi: 10.1097/00005373-198203000-00004. [DOI] [PubMed] [Google Scholar]
- 99.Newsholme EA, Crabtree B. Substrate cycles in metabolic regulation and in heat generation. In Biochemical Society Symposium. 1975 Dec;(41):61–109. [PubMed] [Google Scholar]
- 100.Burns TW, Langley PE, Terry BE, Robinson GA. The role of free fatty acids in the regulation of lipolysis by human adipose tissue cells. Metabolism. 1978;27(12):1755–1762. doi: 10.1016/0026-0495(78)90261-5. [DOI] [PubMed] [Google Scholar]
- 101.Buhl M, Gjedsted J, Granfeldt A, Larsen PØ, Schmitz O, Tønnesen E, Møller N. Metabolic effects of free fatty acids during endotoxaemia in a porcine model–free fatty acid inhibition of growth hormone secretion as a potential catabolic feedback mechanism. Hormone and metabolic research. 2010;42(05):348–352. doi: 10.1055/s-0030-1248297. [DOI] [PubMed] [Google Scholar]
- 102.Loaiza A, Carretta MD, Taubert A, Hermosilla C, Hidalgo MA, Burgos RA. Differential intracellular calcium influx, nitric oxide production, ICAM-1 and IL8 expression in primary bovine endothelial cells exposed to nonesterified fatty acids. BMC veterinary research. 2016;12(1):1. doi: 10.1186/s12917-016-0654-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Simon MC, Bilan S, Nowotny B, Dickhaus T, Burkart V, Schloot NC. Fatty acids modulate cytokine and chemokine secretion of stimulated human whole blood cultures in diabetes. Clinical & Experimental Immunology. 2013;172(3):383–393. doi: 10.1111/cei.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Frommer KW, Schäffler A, Rehart S, Lehr A, Müller-Ladner U, Neumann E. Free fatty acids: potential proinflammatory mediators in rheumatic diseases. Annals of the rheumatic diseases. 2015;74(1):303–310. doi: 10.1136/annrheumdis-2013-203755. [DOI] [PubMed] [Google Scholar]
- 105.Shikama Y, Ishimaru N, Kudo Y, Bando Y, Aki N, Hayashi Y, Funaki M. Effects of free fatty acids on human salivary gland epithelial cells. Journal of dental research. 2013;92(6):540–546. doi: 10.1177/0022034513487378. [DOI] [PubMed] [Google Scholar]
- 106.Xiu F, Diao L, Qi P, Catapano M, Jeschke MG. Palmitate differentially regulates the polarization of differentiating and differentiated macrophages. Immunology. 2016;147(1):82–96. doi: 10.1111/imm.12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lindegaard B, Ditlevsen S, Plomgaard P, Mittendorfer B, Pedersen BK. Acute reduction of lipolysis reduces adiponectin and IL-18: evidence from an intervention study with acipimox and insulin. Diabetologia. 2013;56(9):2034–2043. doi: 10.1007/s00125-013-2964-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Van Lint P, Libert C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. Journal of leukocyte biology. 2007;82(6):1375–1381. doi: 10.1189/jlb.0607338. [DOI] [PubMed] [Google Scholar]
- 109.Dinarello CA. Historical insights into cytokines. European journal of immunology. 2007;37(S1):S34–S45. doi: 10.1002/eji.200737772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li H, Li H, Bao Y, Zhang X, Yu Y. Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-κB pathway in rat aorta. International journal of cardiology. 2011;152(2):218–224. doi: 10.1016/j.ijcard.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 111.Barnes PJ, Karin M. Nuclear factor-κB—a pivotal transcription factor in chronic inflammatory diseases. New England Journal of Medicine. 1997;336(15):1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 112.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MTH, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nature immunology. 2011;12(5):408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stanojcic M, Chen P, Harrison RA, Wang V, Antonyshyn J, Zúñiga-Pflücker JC, Jeschke MG. Leukocyte infiltration and activation of the NLRP3 inflammasome in white adipose tissue following thermal injury. Critical care medicine. 2014;42(6):1357. doi: 10.1097/CCM.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature medicine. 2011;17(2):179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kang JX, Weylandt KH. In Lipids in Health and Disease. Springer; Netherlands: 2008. Modulation of inflammatory cytokines by omega-3 fatty acids; pp. 133–143. [DOI] [PubMed] [Google Scholar]
- 116.Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell. 2012;151(2):400–413. doi: 10.1016/j.cell.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J, Robinson AJ. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016 doi: 10.1038/nature17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Petruzzelli M, Schweiger M, Schreiber R, Campos-Olivas R, Tsoli M, Allen J, Zechner R. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell metabolism. 2014;20(3):433–447. doi: 10.1016/j.cmet.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 119.Sidossis LS, Porter C, Saraf MK, Børsheim E, Radhakrishnan RS, Chao T, Hawkins HK. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell metabolism. 2015;22(2):219–227. doi: 10.1016/j.cmet.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cypess AM, Weiner LS, Roberts-Toler C, Elía EF, Kessler SH, Kahn PA, Kolodny GM. Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell metabolism. 2015;21(1):33–38. doi: 10.1016/j.cmet.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nguyen KD, Qiu Y, Cui X, Goh YS, Mwangi J, David T, Chawla A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature. 2011;480(7375):104–108. doi: 10.1038/nature10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, Chawla A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell. 2014;157(6):1292–1308. doi: 10.1016/j.cell.2014.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pennanen N, Lapinjoki S, Urtti A, Mönkkönen J. Effect of liposomal and free bisphosphonates on the IL-1β, IL-6 and TNFα secretion from RAW 264 cells in vitro. Pharmaceutical research. 1995;12(6):916–922. doi: 10.1023/a:1016281608773. [DOI] [PubMed] [Google Scholar]
- 124.Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Estevez E. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nature immunology. 2014;15(5):423–430. doi: 10.1038/ni.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Abdullahi A, Chen P, Stanojcic M, Sadri AR, Coburn N, Jeschke MG. IL-6 Signal from the Bone Marrow is Required for the Browning of White Adipose Tissue Post Burn Injury. Shock. 2016 doi: 10.1097/SHK.0000000000000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mu Q, Fang X, Li X, Zhao D, Mo F, Jiang G, Liu JL. Ginsenoside Rb1 promotes browning through regulation of PPARγ in 3T3-L1 adipocytes. Biochemical and biophysical research communications. 2015;466(3):530–535. doi: 10.1016/j.bbrc.2015.09.064. [DOI] [PubMed] [Google Scholar]
- 127.Carrière A, Jeanson Y, Berger-Müller S, André M, Chenouard V, Arnaud E, Villageois P. Browning of white adipose cells by intermediate metabolites: an adaptive mechanism to alleviate redox pressure. Diabetes. 2014;63(10):3253–3265. doi: 10.2337/db13-1885. [DOI] [PubMed] [Google Scholar]
- 128.Choi JH, Kim SW, Yu R, Yun JW. Monoterpene phenolic compound thymol promotes browning of 3T3-L1 adipocytes. European Journal of Nutrition. 2016:1–13. doi: 10.1007/s00394-016-1273-2. [DOI] [PubMed] [Google Scholar]
- 129.Wang L, Teng R, Di L, Rogers H, Wu H, Kopp JB, Noguchi CT. PPARα and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes. 2013;62(12):4122–4131. doi: 10.2337/db13-0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gauglitz GG, Herndon DN, Kulp GA, Meyer WJ, III, Jeschke MG. Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. The Journal of Clinical Endocrinology & Metabolism. 2009;94(5):1656–1664. doi: 10.1210/jc.2008-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gauglitz GG, Halder S, Boehning DF, Kulp GA, Herndon DN, Barral JM, Jeschke MG. Post-burn hepatic insulin resistance is associated with ER stress. Shock (Augusta, Ga.) 2010;33(3) doi: 10.1097/SHK.0b013e3181b2f439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wolfe RR, Miller HI. Cardiovascular and metabolic responses during burn shock in the guinea pig. American Journal of Physiology–Legacy Content. 1976;231(3):892–897. doi: 10.1152/ajplegacy.1976.231.3.892. [DOI] [PubMed] [Google Scholar]
- 133.Marik PE, Bellomo R. Stress hyperglycemia: an essential survival response! Critical care. 2013;17(2):1. doi: 10.1186/cc12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wilmore DW, Mason AD, Pruitt BA. Alterations in glucose kinetics following thermal injury. Surgical Forum. 1975;26:81–83. [PubMed] [Google Scholar]
- 135.Wilmore DW, Mason AD, Pruitt BA. Insulin response to glucose in hypermetabolic burn patients. Annals of Surgery. 1976;183(3):314–320. doi: 10.1097/00000658-197603000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bessey PQ, Wilmore DW. Beta-adrenergic regulation of glucose disposal: a reciprocal relationship with insulin release. Journal of Surgical Research. 1983;34(5):404–414. doi: 10.1016/0022-4804(83)90089-6. [DOI] [PubMed] [Google Scholar]
- 137.Gamelli RL, Liu H, He LK, Hofmann CA. Augmentations of glucose uptake and glucose transporter-1 in macrophages following thermal injury and sepsis in mice. Journal of leukocyte biology. 1996;59(5):639–647. doi: 10.1002/jlb.59.5.639. [DOI] [PubMed] [Google Scholar]
- 138.Waller AP, Kohler K, Burns TA, Mudge MC, Belknap JK, Lacombe VA. Naturally occurring compensated insulin resistance selectively alters glucose transporters in visceral and subcutaneous adipose tissues without change in AS160 activation. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2011;1812(9):1098–1103. doi: 10.1016/j.bbadis.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lopez R, Arumugam A, Joseph R, Monga K, Boopalan T, Agullo P, Lakshmanaswamy R. Hyperglycemia enhances the proliferation of non-tumorigenic and malignant mammary epithelial cells through increased leptin/IGF1R signaling and activation of AKT/mTOR. PloS one. 2013;8(11):e79708. doi: 10.1371/journal.pone.0079708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu G, Takashi E, Kudo M, Ishiwata T, Naito Z. Contradictory effects of short-and long-term hyperglycemias on ischemic injury of myocardium via intracellular signaling pathway. Experimental and molecular pathology. 2004;76(1):57–65. doi: 10.1016/j.yexmp.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 141.Moruzzi N, Del Sole M, Fato R, Gerdes JM, Berggren PO, Bergamini C, Brismar K. Short and prolonged exposure to hyperglycaemia in human fibroblasts and endothelial cells: metabolic and osmotic effects. The international journal of biochemistry & cell biology. 2014;53:66–76. doi: 10.1016/j.biocel.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 142.Hou Q, Lei M, Hu K, Wang M. The effects of high glucose levels on reactive oxygen species-induced apoptosis and involved signaling in human vascular endothelial cells. Cardiovascular toxicology. 2015;15(2):140–146. doi: 10.1007/s12012-014-9276-9. [DOI] [PubMed] [Google Scholar]
- 143.Liu D, Zhang H, Gu W, Zhang M. Effects of exposure to high glucose on primary cultured hippocampal neurons: involvement of intracellular ROS accumulation. Neurological Sciences. 2014;35(6):831–837. doi: 10.1007/s10072-013-1605-4. [DOI] [PubMed] [Google Scholar]
- 144.Furnary AP, Wu Y, Bookin SO. Effect of hyperglycemia and continuous intravenous insulin infusions on outcomes of cardiac surgical procedures: the Portland Diabetic Project. Endocrine Practice. 2004;10(Supplement 2):21–33. doi: 10.4158/EP.10.S2.21. [DOI] [PubMed] [Google Scholar]
- 145.Furnary AP, Zerr KJ, Grunkemeier GL, Starr A. Continuous intravenous insulin infusion reduces the incidence of deep sternal wound infection in diabetic patients after cardiac surgical procedures. The Annals of thoracic surgery. 1999;67(2):352–360. doi: 10.1016/s0003-4975(99)00014-4. [DOI] [PubMed] [Google Scholar]
- 146.Gore DC, Chinkes D, Heggers J, Herndon DN, Wolf SE, Desai M. Association of hyperglycemia with increased mortality after severe burn injury. Journal of Trauma-Injury Infection and Critical Care. 2001;51(3):540–544. doi: 10.1097/00005373-200109000-00021. [DOI] [PubMed] [Google Scholar]
- 147.Holm C, Hörbrand F, Mayr M, von Donnersmarck GH, Mühlbauer W. Acute hyperglycaemia following thermal injury: friend or foe? Resuscitation. 2004;60(1):71–77. doi: 10.1016/j.resuscitation.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 148.Fram RY, Cree MG, Wolfe RR, Barr D, Herndon DN. Impaired glucose tolerance in pediatric burn patients at discharge from the acute hospital stay. Journal of burn care & research: official publication of the American Burn Association. 2010;31(5) doi: 10.1097/BCR.0b013e3181eebe63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wan Z, Frier BC, Williams DB, Wright DC. Epinephrine induces PDK4 mRNA expression in adipose tissue from obese, insulin resistant rats. Obesity. 2012;20(2):453–456. doi: 10.1038/oby.2011.252. [DOI] [PubMed] [Google Scholar]
- 150.Ziegler MG, Milic M, Sun P, Tang CM, Elayan H, Bao X, O’Connor DT. Endogenous epinephrine protects against obesity induced insulin resistance. Autonomic Neuroscience. 2011;162(1):32–34. doi: 10.1016/j.autneu.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jensen J, Ruzzin J, Jebens E, Brennesvik EO, Knardahl S. Improved insulin-stimulated glucose uptake and glycogen synthase activation in rat skeletal muscles after adrenaline infusion: role of glycogen content and PKB phosphorylation. Acta physiologica scandinavica. 2005;184(2):121–130. doi: 10.1111/j.1365-201X.2005.01437.x. [DOI] [PubMed] [Google Scholar]
- 152.Rizza RA, Mandarino LJ, Gerich JE. Cortisol-Induced Insulin Resistance in Man: Impaired Suppression of Glucose Production and Stimulation of Glucose Utilization due to a Postreceptor Defect of Insulin Action*. The Journal of Clinical Endocrinology & Metabolism. 1982;54(1):131–138. doi: 10.1210/jcem-54-1-131. [DOI] [PubMed] [Google Scholar]
- 153.Palmieri TL, Levine S, Schonfeld-Warden N, O’Mara MS, Greenhalgh DG. Hypothalamic–pituitary–adrenal axis response to sustained stress after major burn injury in children. Journal of burn care & research. 2006;27(5):742–748. doi: 10.1097/01.BCR.0000238098.43888.07. [DOI] [PubMed] [Google Scholar]
- 154.Wieser V, Moschen AR, Tilg H. Inflammation, cytokines and insulin resistance: a clinical perspective. Archivum immunologiae et therapiae experimentalis. 2013;61(2):119–125. doi: 10.1007/s00005-012-0210-1. [DOI] [PubMed] [Google Scholar]
- 155.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose Expression of Tumor Necrosis Factor-: Direct Role in Obesity-Linked Insulin Resistance. Science-New York Then Washington. 1993;259:87–87. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 156.Hotamisligil GS. Mechanisms of TNF-α-induced insulin resistance. Experimental and clinical endocrinology & diabetes. 1999;107(02):119–125. doi: 10.1055/s-0029-1212086. [DOI] [PubMed] [Google Scholar]
- 157.Juge-Aubry CE, Somm E, Giusti V, Pernin A, Chicheportiche R, Verdumo C, Meier CA. Adipose Tissue Is a Major Source of Interleukin-1 Receptor Antagonist Upregulation in Obesity and Inflammation. Diabetes. 2003;52(5):1104–1110. doi: 10.2337/diabetes.52.5.1104. [DOI] [PubMed] [Google Scholar]
- 158.Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1β-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. 2007;148(1):241–251. doi: 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.De Filippo G, Rendina D, Moccia F, Rocco V, Campanozzi A. Interleukin-6, soluble interleukin-6 receptor/interleukin-6 complex and insulin resistance in obese children and adolescents. Journal of endocrinological investigation. 2015;38(3):339–343. doi: 10.1007/s40618-014-0176-4. [DOI] [PubMed] [Google Scholar]
- 160.Wallenius V, Wallenius K, Ahrén B, Rudling M, Carlsten H, Dickson SL, Jansson JO. Interleukin-6-deficient mice develop mature-onset obesity. Nature medicine. 2002;8(1):75–79. doi: 10.1038/nm0102-75. [DOI] [PubMed] [Google Scholar]
- 161.Matthews VB, Allen TL, Risis S, Chan MHS, Henstridge DC, Watson N, Jowett JB. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia. 2010;53(11):2431–2441. doi: 10.1007/s00125-010-1865-y. [DOI] [PubMed] [Google Scholar]
- 162.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Hansen AMK. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nature medicine. 2011;17(11):1481–1489. doi: 10.1038/nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Özcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Özdelen E, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 164.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. Journal of Biological Chemistry. 1996;271(40):24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 165.Greenough W, Crespin S, Steinberg D. Hypoglycaemia and hyperinsulinaemia in response to raised free-fatty-acid levels. The Lancet. 1967;290(7530):1334–1336. doi: 10.1016/s0140-6736(67)90917-8. [DOI] [PubMed] [Google Scholar]
- 166.Steneberg P, Rubins N, Bartoov-Shifman R, Walker MD, Edlund H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell metabolism. 2005;1(4):245–258. doi: 10.1016/j.cmet.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 167.Ruderman NB, Dean D. Malonyl CoA, long chain fatty acyl CoA and insulin resistance in skeletal muscle. Journal of basic and clinical physiology and pharmacology. 1998;9(2–4):295–308. doi: 10.1515/jbcpp.1998.9.2-4.295. [DOI] [PubMed] [Google Scholar]
- 168.Hamel FG, Upward JL, Bennett RG. In vitro inhibition of insulin-degrading enzyme by long-chain fatty acids and their coenzyme A thioesters. Endocrinology. 2003;144(6):2404–2408. doi: 10.1210/en.2002-0007. [DOI] [PubMed] [Google Scholar]
- 169.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nature medicine. 2005;11(1):90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 170.Ye J. Role of insulin in the pathogenesis of free fatty acid-induced insulin resistance in skeletal muscle. Endocrine, Metabolic & Immune Disorders-Drug Targets (Formerly Current Drug Targets-Immune, Endocrine & Metabolic Disorders) 2007;7(1):65–74. doi: 10.2174/187153007780059423. [DOI] [PubMed] [Google Scholar]
- 171.Jeschke MG. Clinical review: Glucose control in severely burned patients-current best practice. Critical Care. 2013;17(4):1. doi: 10.1186/cc12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Bouillon R. Intensive insulin therapy in the medical ICU. New England Journal of Medicine. 2006;354(5):449–461. doi: 10.1056/NEJMoa052521. [DOI] [PubMed] [Google Scholar]
- 173.Van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Bouillon R. Intensive insulin therapy in critically ill patients. New England journal of medicine. 2001;345(19):1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- 174.Van den Berghe G, Wouters PJ, Bouillon R, Weekers F, Verwaest C, Schetz M, Lauwers P. Outcome benefit of intensive insulin therapy in the critically ill: insulin dose versus glycemic control. Critical care medicine. 2003;31(2):359–366. doi: 10.1097/01.CCM.0000045568.12881.10. [DOI] [PubMed] [Google Scholar]
- 175.de Oliveira Martins J, Meyer-Pflug AR, Alba-Loureiro TC, Melbostad H, da Cruz JWMC, Coimbra R, Sannomiya P. Modulation of lipopolysaccharide-induced acute lung inflammation: role of insulin. Shock. 2006;25(3):260–266. doi: 10.1097/01.shk.0000194042.18699.b4. [DOI] [PubMed] [Google Scholar]
- 176.Stanojcic M, Finnerty CC, Jeschke MG. Anabolic and anticatabolic agents in critical care. Current opinion in critical care. 2016 doi: 10.1097/MCC.0000000000000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gauglitz GG, Williams FN, Herndon DN, Jeschke MG. BURNS: WHERE ARE WE STANDING WITH PROPRANOLOL, OXANDROLONE, rhGH, AND THE NEW INCRETIN ANALOGUES? Current opinion in clinical nutrition and metabolic care. 2011;14(2):176. doi: 10.1097/MCO.0b013e3283428df1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Nisr RB, Affourtit C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2014;1837(2):270–276. doi: 10.1016/j.bbabio.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Williams FN, Herndon DN, Jeschke MG. The hypermetabolic response to burn injury and interventions to modify this response. Clinics in plastic surgery. 2009;36(4):583–596. doi: 10.1016/j.cps.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Viollet B, Guigas B, Garcia NS, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clinical science. 2012;122(6):253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jeschke MG, Abdullahi A, Burnett M, Rehou S, Stanojcic M. Glucose control in severely burned patients using metformin: an interim safety and efficacy analysis of a phase II randomized controlled trial. Annals of Surgery. 2016;264(3):518–527. doi: 10.1097/SLA.0000000000001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Diaz EC, Herndon DN, Porter C, Sidossis LS, Suman OE, Børsheim E. Effects of pharmacological interventions on muscle protein synthesis and breakdown in recovery from burns. Burns. 2015;41(4):649–657. doi: 10.1016/j.burns.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Brooks NC, Song J, Boehning D, Kraft R, Finnerty CC, Herndon DN, Jeschke MG. Propranolol improves impaired hepatic phosphatidylinositol 3-kinase/akt signaling after burn injury. Molecular Medicine. 2012;18(4):707. doi: 10.2119/molmed.2011.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Barrow RE, Wolfe RR, Dasu MR, Barrow LN, Herndon DN. The use of beta-adrenergic blockade in preventing trauma-induced hepatomegaly. Annals of surgery. 2006;243(1):115–120. doi: 10.1097/01.sla.0000193834.07413.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Flores O, Stockton K, Roberts JA, Muller MJ, Paratz JD. The efficacy and safety of adrenergic blockade after burn injury: A systematic review and meta-analysis. Journal of Trauma and Acute Care Surgery. 2016;80(1):146–155. doi: 10.1097/TA.0000000000000887. [DOI] [PubMed] [Google Scholar]
- 186.Keane J, Tajouri L, Gray B. The effect of growth hormone administration on the regulation of mitochondrial apoptosis in-vivo. International journal of molecular sciences. 2015;16(6):12753–12772. doi: 10.3390/ijms160612753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Przkora R, Herndon DN, Suman OE, Jeschke MG, Meyer WJ, Chinkes DL, Barrow RE. Beneficial effects of extended growth hormone treatment after hospital discharge in pediatric burn patients. Annals of surgery. 2006;243(6):796–803. doi: 10.1097/01.sla.0000219676.69331.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Takala J, Ruokonen E, Webster NR, Nielsen MS, Zandstra DF, Vundelinckx G, Hinds CJ. Increased mortality associated with growth hormone treatment in critically ill adults. New England Journal of Medicine. 1999;341(11):785–792. doi: 10.1056/NEJM199909093411102. [DOI] [PubMed] [Google Scholar]
- 189.Bakhtyar N, Sivayoganathan T, Jeschke MG. Therapeutic Approaches to Combatting Hypermetabolism in Severe Burn Injuries. Journal of Intensive and Critical Care 2015 [Google Scholar]
- 190.Jeschke MG, Finnerty CC, Suman OE, Kulp G, Mlcak RP, Herndon DN. The effect of oxandrolone on the endocrinologic, inflammatory, and hypermetabolic responses during the acute phase postburn. Annals of surgery. 2007;246(3):351–362. doi: 10.1097/SLA.0b013e318146980e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wolf SE, Edelman LS, Kemalyan N, Donison L, Cross J, Underwood M, Lawless M. Effects of oxandrolone on outcome measures in the severely burned: a multicenter prospective randomized double-blind trial. Journal of burn care & research. 2006;27(2):131–139. doi: 10.1097/01.BCR.0000202620.55751.4F. [DOI] [PubMed] [Google Scholar]
- 192.Pham TN, Kramer CB, Wang J, Rivara FP, Heimbach DM, Gibran NS, Klein MB. Epidemiology and outcomes of older adults with burn injury: an analysis of the National Burn Repository. Journal of burn care & research: official publication of the American Burn Association. 2009;30(1):30. doi: 10.1097/BCR.0b013e3181921efc. [DOI] [PMC free article] [PubMed] [Google Scholar]