

Abstract
The mu and delta opioid receptors (MOR and DOR) are highly homologous members of the opioid family of GPCRs. There is evidence that MOR and DOR interact, however the extent to which these interactions occur in vivo and affect synaptic function is unknown. There are two stable DOR subtypes: DPDPE sensitive (DOR1) and deltorphin II sensitive (DOR2); both agonists are blocked by DOR selective antagonists. Robust motivational effects are produced by local actions of both MOR and DOR ligands in the ventral tegmental area (VTA). Here we demonstrate that a majority of both dopaminergic and non-dopaminergic VTA neurons express combinations of functional DOR1, DOR2, and/or MOR, and that within a single VTA neuron, DOR1, DOR2, and MOR agonists can differentially couple to downstream signaling pathways. As reported for the MOR agonist DAMGO, DPDPE and deltorphin II produced either a predominant K+ dependent hyperpolarization or a Cav2.1 mediated depolarization in different neurons. In some neurons DPDPE and deltorphin II produced opposite responses. Excitation, inhibition, or no effect by DAMGO did not predict the response to DPDPE or deltorphin II, arguing against a MOR-DOR interaction generating DOR subtypes. However, in a subset of VTA neurons the DOR antagonist TIPP-Ψ augmented DAMGO responses; we also observed DPDPE or deltorphin II responses augmented by the MOR selective antagonist CTAP. These findings directly support the existence of two independent, stable forms of the DOR, and show that MOR and DOR can interact in some neurons to alter downstream signaling.
Keywords: Delta opioid receptor, mu opioid receptor, Cav2.1, Ventral tegmental area
Chemical compounds studied in this article: [D-Pen2, D-Pen5]enkephalin (DPDPE) (PubChem CID:104787); [D-Ala2, Glu4]deltorphin (deltorphin II) (PubChem CID: 123795); [D-Ala2, N-Me-Phe4, Gly-ol5]-Enkephalin acetate salt (DAMGO) (PubChem CID: 5462471); D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP) (PubChem CID: 10418702); H-Tyr-Tic-psi(CH2NH)Phe-Phe-OH (TIPP-psi) (PubChem CID: 5311481)
1. Introduction
Opiate drugs and endogenous opioid peptides exert powerful behavioral actions through binding to receptors expressed on neurons in the central nervous system. These opioid receptors belong to family A of G protein-coupled receptors (GPCRs), exhibit high amino acid sequence homology in their transmembrane domains, and control neuronal activity through similar intracellular signaling pathways and ionic conductances. Of particular interest in this regard are the mu opioid receptor (MOR) and the delta opioid receptor (DOR) as they have the highest amino acid sequence homology (Chang et al., 2004), are often expressed in high density in the same brain regions (Erbs et al., 2015), and respond to similar concentrations of the endogenous opioid peptides leucine enkephalin (l-enk), methionine enkephalin (m-enk) and β-endorphin (Chang et al., 2004). Despite these molecular and cellular similarities, MOR and DOR agonists can generate different and often opposing effects on motivated behaviors (e.g. analgesia, reward, motivation) (Bals-Kubik et al., 1993; Farias et al., 2003; Hammond et al., 1998; Margolis et al., 2008a; Mitchell et al., 2014).
Complicating our understanding of the interaction between MOR and DOR is the evidence that there are two consistent functional forms of DOR: DOR1, selectively activated by the synthetic cyclic peptide [D-Pen2, D-Pen5]enkephalin (DPDPE) (Mosberg et al., 1983) and DOR2, selectively activated by the amphibian skin derived peptide [D-Ala2, Glu4]deltorphin (deltorphin II) (Erspamer et al., 1989; Kreil et al., 1989). There is also evidence for partial selectivity of antagonists: 7-benzylidenenaltrexone (BNTX) preferentially blocks DOR1 activity (Sofuoglu et al., 1993), and naltriben and 5′NTII preferentially block DOR2 actions (Jiang et al., 1991; Portoghese et al., 1991; Sofuoglu et al., 1991). However, the pharmacologic properties of these antagonists seem to be tissue dependent (Zaki et al., 1996). Further evidence for distinct actions of the two DOR forms is that in rodents both DPDPE and deltorphin II produce analgesia, however repeated exposure to either ligand does not produce cross-tolerance to the other DOR agonist (Mattia et al., 1991). Clearly, unraveling the neurobiological underpinnings of these distinct responses has the potential to improve the clinical success of DOR based therapeutics.
One proposed explanation for these DOR subtypes is receptor heterodimerization (van Rijn and Whistler, 2009). Opioid receptors typically signal through Gi/o proteins, inhibiting adenylyl cyclase, opening K+ channels to inhibit firing or closing Ca2+ channels to decrease neurotransmitter release (Williams et al., 2001). The resolution of the crystal structures of MOR and DOR supports the possibility of direct interaction: the receptors crystallized as dimers, interfacing at molecular domains that are virtually identical between MORs and DORs (Manglik et al., 2012; Provasi et al., 2015). In cultured cells MOR-DOR heterodimerization can change the intracellular signaling properties of MORor DOR ligands, conferring a preferential coupling to non-G protein mediated signaling pathways (Rozenfeld and Devi, 2007). Heterodimerization of MORs and DORs can also generate an unexpected agonist-antagonist interaction observed in heterologous expression systems, such that a DOR antagonist increases the potency and efficacy of a MOR agonist and vice-versa at G protein dependent pathways (Gomes et al., 2000, 2011), presumably by enabling the heterodimers to switch from G protein independent to G protein dependent signaling. This type of interaction at the neuronal level in vivo would complicate the interpretation of data from behavioral pharmacology experiments that use receptor selective antagonists, since it raises the possibility that an antagonist will not just block activation of the intended receptor, but may also increase the efficacy or potency of an endogenous peptide acting at a heterodimer receptor partner. Further evidence for functional MOR-DOR heterodimers is that synthetic bivalent compounds that combine MOR agonist and DOR antagonist actions show enhanced MOR analgesia and reduced MOR tolerance, dependence, and reward (Daniels et al., 2005). The atomic spacing between the MOR agonist and DOR antagonist components of the bivalent molecule is critical (must be greater than 22 Å), suggesting that the ligand’s action depends on MOR and DOR binding sites being a specific and relatively short distance from each other.
Another possibility is that the behavioral differences observed in response to DOR subtype pharmacologies is generated by functional selectivity or biased agonism. That is, structurally distinct DOR selective ligands induce different conformational changes in the same receptor that favor activation of one or another intracellular signaling pathway, thereby imposing different effects on the circuit. The first evidence that such ligand-directed alternative signaling is possible was demonstrated in studies of the β2-adrenergic receptor (Drake et al., 2008). Also, as heterodimerized receptors signal through alternative mechanisms, a heterodimer-selective ligand (Fujita et al., 2014; Gomes et al., 2013) would appear to be a biased agonist. Splice variants may also lead to different pharmacologies (Pasternak, 2001), however it is unknown if DOR splice variants are expressed in neurons and have functional consequences. While each of these possibilities for functional diversity depends upon ligand-receptor and receptor-receptor interaction within a single cell, to date, there has been no direct demonstration that the pharmacological differences between DOR1 and DOR2 ligands at the behavioral level can be explained by different molecular interactions at the single neuron level.
DOR1 activation in the ventral tegmental area (VTA) increases dopamine release in the nucleus accumbens (NAc) (Devine et al., 1993a, 1993b). Although DPDPE does not induce a conditioned place preference when infused into the VTA (Mitchell et al., 2014), animals will self-administer DPDPE directly into the VTA, suggesting that, like MOR activation, DOR1 activation in the VTA has a positive motivational effect (Devine and Wise, 1994). However, in long term alcohol drinking rats, while the MOR selective antagonist CTAP reduces alcohol consumption, the DOR selective antagonist TIPP-Ψ increases it (Margolis et al., 2008a). These complex behavioral effects of selective DOR ligands in the VTA and the evidence of MOR-DOR competitive interaction contrast with the limited number of ex vivo electrophysiologic studies investigating actions of selective DOR agonists. For instance, an early study with a small sample size (3 neurons) found that DPDPE did not elicit a postsynaptic GIRK response (Johnson and North, 1992a). While a number of studies have used the endogenous opioid peptide m-enk to characterize MOR actions in the VTA (e.g. (Ford et al., 2006; Johnson and North, 1992b)), m-enk also acts at DOR. MOR activation by the selective agonist DAMGO induces robust presynaptic inhibition of GABA release in VTA but we have detected only small DOR effects on GABA release in EtOH naive animals (Margolis et al., 2008a; Mitchell et al., 2014). Because MOR and DOR in the VTA elicit robust motivational and rewarding actions and because DOR1 and DOR2 agonists in the VTA can differ in their synaptic and behavioral actions (Margolis et al., 2008a; Mitchell et al., 2014), we investigated DOR subtype function and interactions of DOR with MORs in single neurons from throughout the VTA; we characterized the postsynaptic responses to DPDPE and deltorphin II, compared these to responses to the MOR agonist DAMGO, and probed for MOR-DOR interactions.
2. Methods
2.1. Animals
Animal care and all experimental procedures were in accordance with guidelines from the National Institutes of Health and approved in advance by the Ernest Gallo Clinic (through June 2013) and Research Center and the University of California, San Francisco Institutional Animal Care and Use Committees (after July 2013).
2.2. Slice preparation and electrophysiology
Recordings were made in control male Sprague-Dawley rats (p22 to adult). 11% of recordings were completed in rats greater than 60 days old, including experiments of all types. No differences were observed between younger and adult animals, so the data are presented together. Some data were obtained in neurons also used for previously reported experiments (Margolis et al., 2014, 2006b, 2012). Rats were deeply anesthetized with isoflurane and then decapitated. Horizontal brain slices (150 μm thick) were prepared using a vibratome (Leica Instruments). Slices were prepared in ice cold Ringer solution (in mM: 119 NaCl, 2.5 KCl, 1.3 MgSO4, 1.0 NaH2PO4, 2.5 CaCl2, 26.2 NaHCO3, and 11 glucose saturated with 95% O2–5% CO2) and allowed to recover at 33–35°C for at least 1 h. Slices were visualized under a Zeiss Axioskop or Axioskop FS 2 plus with differential interference contrast optics and infrared illumination or an Axio Examiner A1 also equipped with Dodt optics, using a Zeiss Axiocam MRm and Axiovision 4 (Zeiss) or Microlucida (MBF Biosciences, Williston, VT, USA) software. Whole cell recordings were made at 33°C using 2.5–5 MΩ pipettes containing (in mM): 123 K-gluconate, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 MgATP, 0.3 Na3GTP, and 0.1% biocytin (pH 7.2, osmolarity adjusted to 275). Liquid junction potentials were not corrected during recordings. Ih was measured by voltage clamping cells and stepping from −60 to −40, −50, −70, −80, −90, −100, −110 and −120 mV. Input resistance was monitored with hyperpolarizing pulses (0.1 Hz) throughout each experiment.
Recordings were made using Axopatch 1-D amplifiers (Axon Instruments), filtered at 2 kHz and collected at 5 kHz or filtered at 5 kHz and collected at 20 kHz using IGOR Pro (Wavemetrics, Lake Oswego, OR, USA). For all current clamp experiments I = 0. Most VTA neurons were selected in an unbiased manner from throughout the VTA by superimposing a grid on the slice, numbering each grid location, and using a random number generator to choose the grid location for recording. The closest healthy cell to the randomly generated grid location was patched.
In most cases opioid agonists were bath applied. The sequence of drug applications was varied across experiments. In a smaller set of experiments, opioid agonists were delivered via a Smart Squirt pressure ejector (Automate, Inc., Berkeley, CA, USA) for 60 s within 300 mM of the recording site. In many of these experiments agonist application sequences were repeated to test for order effects, but none were observed.
Agonists, antagonists, salts, ATP, and GTP were obtained from Sigma (St. Louis, MO, USA) or Tocris (Ballwin, MO, USA). ω-agatoxin-IVA was purchased from Alomone Labs (Jerusalem, Israel).
2.3. Cell type identification and immunocytochemistry
All cells were labeled with biocytin during whole cell recording. Slices were fixed immediately after recording in 4% formaldehyde for 2 h and then stored at 4°C in PBS. Slices were pre-blocked for 2 h at room temperature in PBS plus 0.2% BSA and 5% normal goat serum, then incubated at 4°C with a rabbit anti-TH polyclonal antibody (Antibody Registry: AB_390204, at 1:100). Slices were then washed thoroughly in PBS with 0.2% BSA before being agitated overnight at 4°C with Cy5 anti-rabbit secondary antibody (1:100) and FITC streptavidin (6.5 μL/mL). Sections were rinsed and mounted on slides using BioRad Fluoroguard Antifade Reagent mounting media and visualized under a Zeiss LSM 510 META microscope or with an Axioskop FS2 plus with an Axiocam MRm running Neurolucida software (MBF Biosciences, Williston, VT, USA). Primary antibodies were obtained from Chemicon International or Millipore (Hayward, CA, USA), secondary antibodies from Jackson ImmunoResearch Laboratories (West Grove, PA, USA), and all other reagents from SigmaMillipore (Hayward, CA, USA).
In neurons where immunocytochemistry was inconclusive, Ih(−) neurons were classified as non-dopaminergic (Margolis et al., 2006b). While false negatives are possible using immunocytochemical techniques, we previously demonstrated that the experimental methods for both recording and cytochemistry used here produce reliable TH results (Margolis et al., 2010). Further, our percentage of TH(+) neurons out of the total population of cyto-chemically identified neurons when Ih(−) neurons are included in the total (51%: 87/170 neurons) is remarkably similar to the 55% TH(+) we determined in tissue from rats where no recordings were done. In the latter anatomical study we performed a systematic, unbiased analysis of horizontal sections cytochemically processed for TH and NeuN from throughout the VTA, similar to the extent of the recordings reported here (Margolis et al., 2006b).
2.4. Single cell RT-PCR
Individual VTA neurons were recorded in whole cell configuration for at least 3 min. At the termination of recording, the cytoplasm of the neuron was aspirated into the recording pipette, the pipette retracted from the slice, and the pipette contents were ejected into an RNase free centrifuge tube pre-chilled to −20°C. Samples were stored at −80°C until they were processed. cDNA was synthesized from single cell VTA neurons using the Message-BOOSTER cDNA Synthesis from Cell Lysates Kit (MBCL90310, Epicentre, Madison, WI, USA) according to the manufacturer’s protocol. Real-time PCR was performed using the Power SYBR Green qPCR Master Mix (Applied Biosystems, Foster City, CA, USA). The PCR template source was 4 μL of 10-times diluted first-strand cDNA. Amplification was performed with an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA). After an initial denaturation step at 95°C for 10 min, amplification was performed using 45 cycles of denaturation (95°C for 15 s), annealing (55°C for 30 s), and extension (72°C for 30 s). We amplified GAPDH, a housekeeping gene, as a control. The data were analyzed using the sequence detection system software (version 2.2.1, Applied Biosystems, Foster City, CA, USA) as described in Data Analysis.
The forward (F) and reverse (R) primers are as follows:
GAPDH-F TCAAGAAGGTGGTGAAGCAG
GAPDH-R AGGTGGAAGAATGGGAGTTG
OPRD1-F TGCTCGTCATGTTTGGAATC
OPRD1-R CCAAGGCCAGATTGAAGATG
OPRM1-F TCGGTCTGCCTGTAATGTTC
OPRM1-R CAGATTTTGAGCAGGTTCTCC
TH-F AGGGGTACAAAACCCTCCTC
TH-R CGCACAAAATACTCCAGGTG
2.5. nCounter Single Cell Gene Expression Assay (Nanostring Technologies®)
Individual VTA neurons were acquired as described above and stored at −80°C until processed. Samples were transferred into sterilized PCR tubes containing 5 μL of iScript RT-qPCR Sample Preparation Reagent (Biorad, Hercules, CA, USA) and then stored again at −80°C. Gene expression was then determined using a digital approach by nCounter Single Cell Gene Expression Assay. All the steps including cDNA conversion of whole single cell using SuperScript VILO MasterMix (Thermo Fisher Scientific, Waltham, MA, USA) followed by multiplexed target enrichment (MTE) with 22 amplification cycles using MTE primers and hybridization were performed by Nanostring Technologies® Inc. (Seattle, WA) (http://www.nanostring.com/products/single_cell). mRNA expression was measured in 5 μL whole-cell lysate (single cell) with a custom nCounter reporter probe set including the housekeeping gene GAPDH (NM_017008.4) as a control, OPRD1 (NM_012617.1), OPRM1 (NM_001038601.2) and TH (NM_012740.3). The raw data produced by nCounter Analyzer were exported as a Reporter Code Count (RCC) file containing the raw counts for each gene in a sample. Data Analysis was then performed according to manufacturer’s nCounter Expression Data Analysis Guide as described in Data Analysis.
The forward (F) and reverse (R) MTE primers are as follows:
GAPDH-F GAAGGTGGTGAAGCAGGC
GAPDH-R GCCATGTAGGCCATGAGG
OPRD1-F CCATCACCGCGCTCTACT
OPRD1-R GGTACTTGGCGCTCTGGA
OPRM1-F CAGGCAGGGGTCCATAGAT
OPRM1-R CTTTGGAGCCCGATAGCA
TH-F CAGGGCCTTTCCCAAAGT
TH-R TCCTTTATTGAGAGAATAATCAGGG
2.6. Data Analysis
Ih magnitude was measured as the difference between the initial response following the capacitative charge to a voltage step from −60 to −120 mV and the final current during the same 200 ms step. Neurons were considered Ih(−) if the slope of the I–V curve for hyperpolarizing steps from −60 to −90, −100, −110, and −120 mV was 0.
Results are presented as mean ± S.E.M. Drug effects observed during bath application were statistically evaluated in each neuron by binning data into 30 s data points and comparing the last 4 min of baseline (8 data points) to the last 4 min of drug application, i.e., when, on average, the drug effect had stabilized, using Student’s unpaired t-test (Excel, Microsoft, Redmond, WA, USA). In neurons that were firing spontaneously, firing rate was analyzed. In neurons that were quiescent, membrane potential was analyzed. In experiments where drug was applied multiple times, this analysis was applied to each individual drug application, where baseline data was the 4 min preceding each individual drug application. Effects of antagonists or blockers were assessed with Student’s paired t tests comparing the response to the first agonist application in control aCSF to the second agonist application in the presence of the blocker. When the same agonist was applied twice there was no significant difference between the first and second applications (e.g. see Fig. 4A). In cells where agonists were applied via pressure ejection, baseline was measured as the mean membrane potential over a 2 min baseline period just prior to drug application, and the drug measurement was the mean membrane potential over the last 30 s of drug application. Statistical comparisons between groups of neurons were made using one way ANOVAs on interval data and Fisher’s exact tests or Chi-Square Tests on nominal data (Excel, Microsoft, Redmond, WA, USA or vassarstats.net). Where possible, differences between group means were also compared using a permutation test, the advantage of which is that it is an assumption-free method of comparison (custom written script available for download at https://osf.io/mx7pc/). p < 0.05 was required for significance in all analyses.
Fig. 4. Both DOR subtypes hyperpolarize VTA neurons via K+ channel activation and excite VTA neurons through activation of Cav2.1.
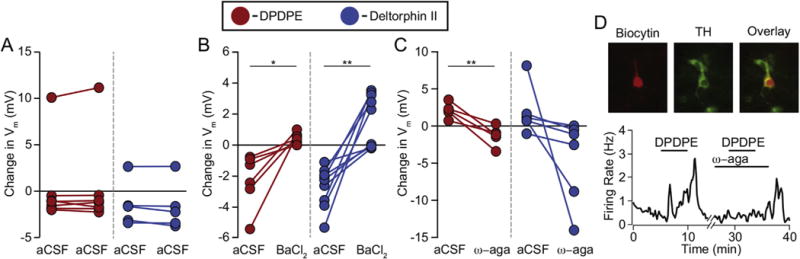
(A) In control experiments, VTA neurons respond similarly to the first and second application of the same DOR agonist, regardless of the magnitude or direction of the response. (1 μM DPDPE, n = 7; 1 μM deltorphin II, n = 5) (B) In neurons responding to DPDPE or deltorphin II with a hyperpolarization, the K+ channel blocker BaCl2 (100 μM) prevented a hyperpolarization in response to a second agonist application (DPDPE n = 6; deltorphin II n = 9). (C) In neurons where the initial response to DPDPE (n = 5) or deltorphin II (n = 5) was a depolarization, this response was blocked by the Cav2.1 blocker ω-agatoxin-IVA (100 nM). In an additional neuron that first responded to deltorphin II with a hyperpolarization, the response was larger in the presence of ω-agatoxin-IVA. Paired t-tests, *p < 0.05; **p < 0.01 (D) Example VTA recording of a spontaneously firing neuron where bath application of DPDPE (1 mM) increased the firing rate of the cell, and this increase was prevented by 100 nM of ω-agatoxin-IVA. Inset: this cell was filled with biocytin (red) during recording and cytochemically identified as TH(+) (green). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The RT-PCR data were analyzed using the sequence detection system software (version 2.2.1, Applied Biosystems, Foster City, CA, USA). The software generates the baseline subtracted amplification plot of normalized reporter values (ΔRn) versus cycle number. The amplification threshold was set at 6–7 of the ΔRn linear dynamic range (50–60% of maximum ΔRn). The fractional cycle at which the intersection of amplification threshold and the plot occurs is defined as the threshold cycle (Ct-value) for the plot. Samples that gave a Ct-value within 45 cycles were considered to be positive for the mRNA expression. The samples for which Ct-values were not observed within 45 cycles (i.e., undetected) were considered to be negative for the mRNA expression as we described previously (Margolis et al., 2014). Data Analysis of nCounter Single Cell Gene Expression Assay was performed according to manufacturer’s nCounter Expression Data Analysis Guide. Briefly, raw counts were normalized by 6 positive controls, and then background (average of 8 negative controls) was subtracted. Finally, housekeeping gene normalization was performed. Samples that gave normalized counts over 0 were considered to be positive for the mRNA expression. The samples for which counts were not detected were considered to be negative for mRNA expression.
3. Results
In this study we investigated whether we could detect a consistent relationship between DOR subtype actions in VTA neurons, and whether either DOR1 or DOR2 actions could be related to a postsynaptic MOR agonist action in the same neuron. Because of the variability of the response to DOR1 and DOR2 agonists from neuron to neuron, we systematically tested a large number of neurons from throughout the VTA (511 neurons in total) for DOR agonist responses.
3.1. DOR1 subtype agonist postsynaptic actions in VTA neurons
We tested for responses to the DOR1 subtype agonist DPDPE (1 μM bath application or 10 μM localized pressure ejection) in 362 VTA neurons. DPDPE inhibited 193 and excited 74 VTA neurons (Fig. 1). Among neurons identified as dopaminergic (with TH immunocytochemistry) 38% (28/73) were inhibited (Fig. 1A) and 19% (14/73) were excited by DPDPE (Fig. 1B). Population results are summarized in Table 1. DPDPE effects were completely blocked by the DOR selective antagonist TIPP-Ψ (Fig. 1F and G; comparison of second DPDPE response in aCSF (104 ± 10% of first DPDPE response; n = 8) v. in TIPP-Ψ (1 ± 8%; n = 9): unpaired two tailed Student’s t-test p = 0.0000009, two tailed permutation test p = 0.00001). TIPP-Ψ alone did not cause a change in membrane potential (baseline −53 ± 2, TIPP-Ψ −53 ± 2, n = 19; Supplementary Fig. 1).
Fig. 1. The DOR1 agonist DPDPE inhibits and excites different subsets of VTA neurons.
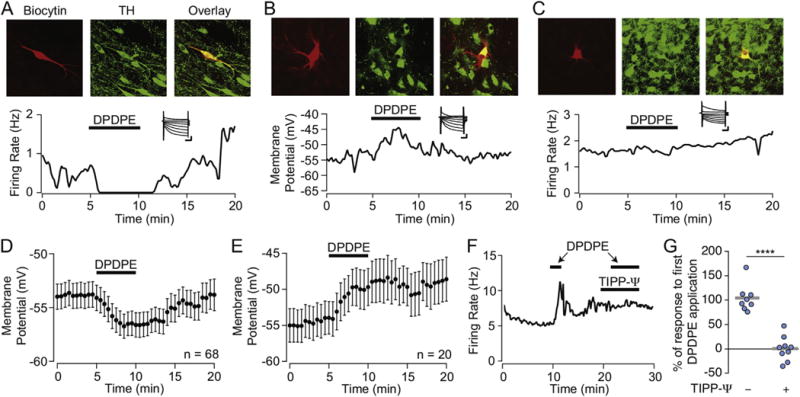
Example recordings of identified VTA dopamine neurons filled with biocytin (red) and cytochemically labeled for TH (green) that were (A) inhibited by, (B) depolarized by, or (C) insensitive to bath application of DPDPE (1 μM). Insets, all 3 example neurons were Ih(+). Scale bars 100 ms and 200 pA. Summary data showing the time course of the DPDPE induced hyperpolarizations (D) and depolarizations (E) in quiescent VTA neurons. Across populations, more reversal was evident during DPDPE washout following hyperpolarizations compared to depolarizations. Example recording (F) showing that the DOR selective antagonist TIPP-Ψ (100 nM) completely blocked the response to DPDPE. In control neurons, a second application of DPDPE induces a response of the same magnitude as the first application (G, left, n = 8), however when the second application is completed in the presence of TIPP-Ψ (100 nM) DPDPE responses are blocked (G, right, n = 9) Circles show individual neurons, grey bars indicate means. ****p ≤ 0.0001. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 1. DPDPE effects by VTA cell type measured in current clamp.
Firing rate was analyzed in neurons firing spontaneously, and membrane potential was analyzed in quiescent neurons. Changes from baseline were determined statistically as described in the Methods.
| Cell type | Inhibited | No change | Excited |
|---|---|---|---|
| All cells | 49% (193/396) | 33% (129/396) | 19% (74/396) |
| TH(+)* | 38% (28/73) | 42% (31/73) | 19% (14/73) |
| Ih(+)TH(−) | 63% (22/35) | 23% (8/35) | 14% (5/35) |
| Ih(−) | 50% (17/34) | 24% (8/34) | 26% (9/34) |
Distribution of response types was statistically different between dopamine (TH(+)) neurons and non-dopamine (Ih(+)TH(−) and Ih(−)) neurons, 2 × 3 Freeman-Halton extension of the Fisher Exact Test, p = 0.03.
3.2. DOR2 agonist postsynaptic actions in VTA neurons
We tested for responses to the DOR2 subtype agonist deltorphin II (1 μM bath application or 10 μM localized pressure ejection) in 264 VTA neurons. Deltorphin II inhibited 113 VTA neurons and excited 55 neurons (Fig. 2). Among TH(+) VTA neurons, 33% (13/39) were inhibited and only 10% (4/39) were excited by deltorphin II. This is statistically less frequent than deltorphin II induced excitations in non-dopamine neurons (Table 2). Population results are summarized in Table 2. Like DPDPE, deltorphin II effects were completely blocked by TIPP-Ψ (Fig. 2C and D; comparison of second deltorphin II response in aCSF (113 ± 7% of first deltorphin II response; n = 5) v. in TIPP-Ψ (4 ± 5%; n = 9): unpaired two tailed Student’s t-test p = 0.00000002, two tailed permutation test p = 0). Distributions of the magnitudes of DPDPE and deltorphin II effects across all neurons and by cell type are displayed in Fig. 3 for comparison.
Fig. 2. The DOR2 agonist deltorphin II inhibits some and excites other VTA neurons.
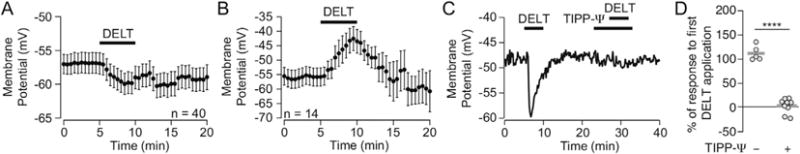
Summary data showing the time course of deltorphin II induced hyperpolarizations (A) and depolarizations (B) in quiescent VTA neurons. Across all neurons, deltorphin II induced depolarizations tended to be followed by more rapid and complete reversal during washout than hyperpolarizations. Example recording (C) showing that 100 nM of the DOR selective antagonist TIPP-Ψ completely blocked the response to deltorphin II in VTA neurons. (D) Summary data showing that in control neurons reapplication of deltorphin II causes a second response of equal magnitude to the first response (right), however when the second deltorphin II application is performed in the presence of TIPP-Ψ (100 nM), the deltorphin II response is eliminated (right). Circles show individual neurons, grey bars indicate means. ****p ≤ 0.0001.
Table 2. Deltorphin II effects by VTA cell type measured in current clamp.
Firing rate was analyzed in neurons firing spontaneously, and membrane potential was analyzed in quiescent neurons. Changes from baseline were determined statistically as described in the Methods.
| Cell type | Inhibited | No change | Excited |
|---|---|---|---|
| All cells | 43% (113/264) | 36% (96/264) | 21% (55/264) |
| TH(+)* | 33% (13/39) | 56% (22/39) | 10% (4/39) |
| Ih(+)TH(−) | 33% (9/27) | 33% (9/27) | 33% (9/27) |
| Ih(−) | 54% (13/24) | 21% (5/24) | 25% (6/24) |
Distribution of response types was statistically different between dopamine (TH(+)) neurons and non-dopamine (Ih(+)TH(−) and Ih(−)) neurons, 2 × 3 Freeman-Halton extension of the Fisher Exact Test, p = 0.01.
Fig. 3. Distributions of DOR1 and DOR2 effects in different subpopulations of VTA neurons.
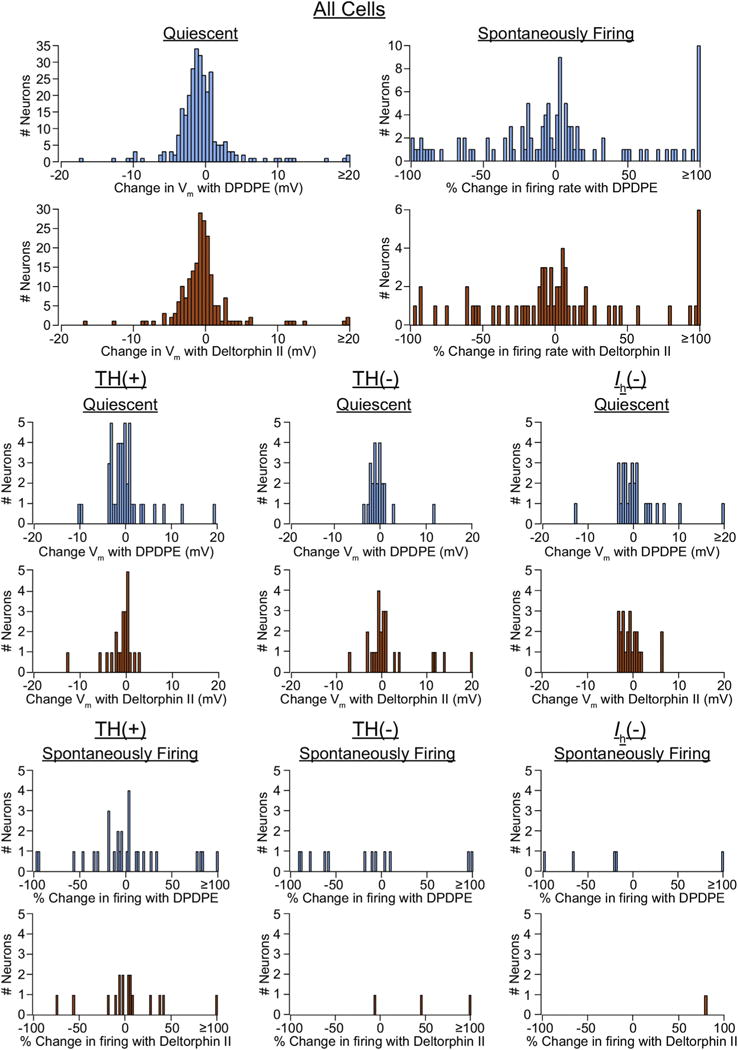
Neither DPDPE (blue) nor deltorphin II (orange) effects in VTA neurons appear to sort with TH content or Ih expression. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. DOR1 and DOR2 agonists postsynaptically inhibit VTA neurons via K+ channel activation
Canonically, direct somatodendritic inhibition by opioid receptor activation is mediated by opening G protein coupled inwardly rectifying K+ channels (GIRKs) (Williams et al., 2001). To test if the DOR agonist induced inhibitions in VTA neurons are mediated by K+ channels, we tested if hyperpolarizations were blocked by the K+ channel blocker BaCl2. In control experiments, applying either DPDPE or deltorphin II twice to the same neuron yielded responses of similar magnitude (Figs. 1G, 2D and 4A). However, in neurons where either agonist caused a hyperpolarization with the first application, in the presence of 100 μM BaCl2 the same agonist no longer caused a hyperpolarization (Fig. 4B; baseline DPDPE effect −2.3 ± 0.7 mV, DPDPE response in the presence of BaCl2 0.4 ± 0.1 mV, n = 8; paired two tailed Student’s t-test p = 0.01; two tailed permutation test p = 0; baseline deltorphin II effect −2.7 ± 0.4 mV, deltorphin II response in the presence of BaCl2 1.6 ± 0.6 mV, n = 9; paired two tailed Student’s t-test p = 0.001; two tailed permutation test p = 0.000001). In fact, in many cases, neurons initially hyperpolarized by an agonist responded with depolarizations in the presence of BaCl2. This effect was more pronounced in a subset of deltorphin II responding neurons. This is similar to what we observed with the MOR selective agonist DAMGO in VTA neurons (Margolis et al., 2014). As expected, bath application of BaCl2 alone caused most VTA neurons to depolarize (baseline −65 ± 3 mV, BaCl2 −53 ± 3 mV, n = 12; Supplementary Fig. 2).
3.4. DOR1 and DOR2 can directly excite VTA neurons by activating a Cav2.1 channel
We previously reported that DAMGO directly depolarizes a subset of VTA neurons by opening a Ca2+ channel, specifically Cav2.1 (Margolis et al., 2014). We therefore tested if the same mechanism was responsible for the depolarizations and increases in firing rate observed with the DOR agonists. Excitations were blocked by the selective Cav2.1 blocker ω–agatoxin IVA (Fig 4C; baseline DPDPE effect 2.1 ± 0.4 mV, DPDPE response in the presence of ω–agatoxin IVA −1.3 ± 0.6 mV, n = 7; paired two tailed Student’s t-test p = 0.006; two tailed permutation test p = 0.002; baseline deltorphin II effect 1.9 ± 1.3 mV, deltorphin response in the presence of ω–agatoxin IVA −4.5 ± 2.3 mV, n = 6; paired two tailed Student’s t-test p = 0.1; two tailed permutation test p = 0.004). Two of these DPDPE excited cells and one deltorphin II excited cell were firing spontaneously during the experiment; in all 3 cases the DOR agonist elicited both an increase in firing rate and a depolarization; in these neurons the increase in firing rate was also blocked by ω–agatoxin IVA (example in Fig 4D; baseline agonist induced change in firing rate 0.8 ± 0.3 Hz, change in firing rate in ω–agatoxin IVA −0.02 ± 0.03 Hz; paired two tailed Student’s t-test p = 0.1; two tailed permutation test p = 0). In a subset of cells, an inhibition was observed when the excitation was blocked, again most markedly among deltorphin II responses. Bath application of ω–agatoxin IVA did not by itself induce a change in the membrane potential (baseline 53 ± 4, ω–agatoxin IVA −53 ± 4, n = 9; Supplementary Fig 2). Together, these data indicate that the DOR mediated excitations observed here require Cav2.1 activity.
3.5. Comparison of DOR1 and DOR2 actions in single VTA neurons
The biological underpinnings of DOR subtype responses are as yet undetermined. In heterologous DOR expression systems DPDPE and deltorphin II generally evoke similar responses, yet these agonists have different behavioral effects and do not produce cross tolerance (Zaki et al., 1996). These observations indicate that some neural elements respond differently to DOR1 and DOR2 selective agonists or that DOR subtype selective agonists differentially activate signaling pathways in the same cells, i.e., functional selectivity. To examine these two possibilities, in some of the neurons described above we investigated whether these DOR subtype selective agonists produce similar physiological effects. 211 neurons were tested with both DPDPE and deltorphin II, in no particular order. Interestingly, many neurons showed differential sensitivities to DPDPE and deltorphin II. For instance, 41 out of 74 neurons that did not respond to DPDPE were either inhibited or excited by deltorphin II (Table 3). There was even a subset of neurons (23) that were excited by one DOR agonist and inhibited by the other (Table 3 and Fig 5A–C). Interestingly, 178 of 211 neurons tested (84%) responded to at least one DOR agonist, indicating that the vast majority of VTA neurons express functional somatodendritic DORs. There was a significant relationship between the types of responses (Table 3), likely driven by the fact that for each agonist independently there are more than twice as many inhibitions as there are excitations (Tables 1 and 2). Importantly, more than half of the neurons that were insensitive to DPDPE responded to deltorphin II, and vice versa; no order effect was observed. Therefore, failure to observe a response when only a DOR1 or only a DOR2 type ligand is applied does not indicate absence of a functional DOR in that neuron. Additionally, there is no clear topographical organization of DOR1 or DOR2 responses in different regions of the VTA (Fig. 6).
Table 3. Responsiveness to DPDPE and deltorphin II are not correlated.
All measurements included here were made in current clamp, table entries indicate number of statistically determined types of observations for each combination of effect or lack thereof.
| DPDPE inhibited | DPDPE no change | DPDPE excited | |
|---|---|---|---|
| Deltorphin II inhibited | 56 | 24 | 12 |
| Deltorphin II no change | 31 | 33 | 12 |
| Deltorphin II excited | 11 | 17 | 15 |
3 × 3 χ2 Test, χ2 = 20.58, p = 0.0004.
Fig. 5. Individual VTA neural responses to DOR1, DOR2, or MOR do not predict responses to the other agonists.
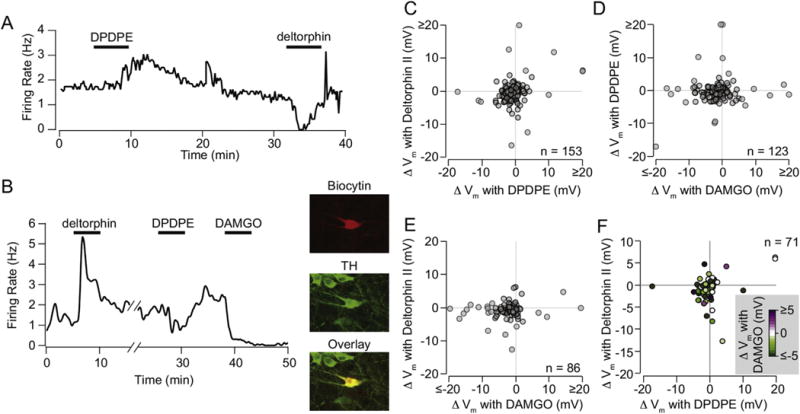
(A) Example VTA neuron that was excited by DPDPE (1 μM) and inhibited by deltorphin II (1 μM). (B) Example recorded VTA neuron tested with all three ligands. This neuron was filled with biocytin during recording (red), and cytochemically identified as TH(+) (green). (C) Among quiescent neurons tested for responses to DPDPE and deltorphin II, subsets of neurons responded to just one or the other agonist. Further, among neurons tested for responses to DPDPE and DAMGO (D) or deltorphin II and DAMGO (E), no clear relationship between responses to the two subtype agonists were observed. (F) Among quiescent neurons tested for responses to all three agonists, no relationship between responses was observed. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 6. Different DPDPE and deltorphin II responses were distributed throughout the VTA.
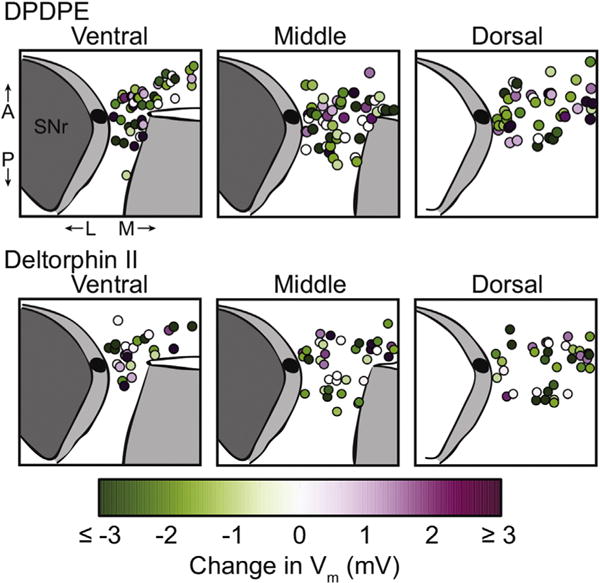
Recordings were made in horizontal midbrain slices and distributed throughout the VTA. Changes from baseline membrane potential are color coded in quiescent neurons here. There is no observed relationship between DOR response and recording location. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.6. Is there a relationship between MOR response and DOR subtype?
One proposal for the difference between DOR1 and DOR2 is that the DOR1 subtype is generated by heterodimerization of MORs with DORs (van Rijn and Whistler, 2009). Heterodimerization between DOR and MOR can differentially affect downstream signaling pathways (Fujita et al., 2015; Rozenfeld and Devi, 2007). For example, since we observed both excitations and inhibitions with application of the MOR selective agonist DAMGO (Margolis et al., 2014), it could be that the excitations represent ligand action on a heterodimer and the inhibitions on the homodimer or vice versa. Alternatively, it could be the case that either DPDPE or deltorphin II predominately target either the homodimer or the heterodimer. To address this possibility, in a subset of the neurons we tested MOR responses in addition to DPDPE or deltorphin II or both. We found no relationship between responses to DAMGO and to either DPDPE or deltorphin II (Fig. 5B,D–F). These data demonstrate that MOR and DOR are often expressed in the same VTA neuron, a prerequisite for heterodimerization. However, co-expression with MOR does not confer a consistent pattern of downstream signaling following application of either a DOR1 or a DOR2 agonist. Furthermore, DOR1 and DOR2 agonists can produce the same or different effects in the same cell and the pattern is not predicted by the response (or lack thereof) to DAMGO.
Although these agonist responses do not support the idea that a DOR subtype is accounted for by MOR-DOR heterodimers in VTA neurons, the fact that both receptors are expressed in most VTA neurons raises the possibility that MOR-DOR heterodimerization occurs in these neurons. Previous studies characterizing MOR-DOR in heterologous expression systems show that pretreatment with a MOR antagonist increases the potency and efficacy of a DOR agonist (and vice versa) for G protein signaling pathways (Gomes et al., 2000, 2004; Rozenfeld and Devi, 2007). Here we tested for a similar MOR-DOR interaction for signaling through postsynaptic G protein mediated K+ channel activation. In 6 out of 11 neurons, application of the DOR antagonist TIPP-Ψ increased the magnitude of the hyperpolarization induced by a saturating dose of the MOR selective agonist DAMGO (Fig. 7A–C). Likewise, in a subset of neurons tested, the MOR selective antagonist CTAP increased the magnitude of DPDPE effects (4/6 neurons (3 hyperpolarizations and one depolarization augmented by CTAP); Fig. 7D and E). For deltorphin II, we observed either augmented hyperpolarizations (2/6 neurons) or a switch from hyperpolarization to depolarization (3/6 neurons) (Fig. 7F and G, Supplementary Fig. 3). Two possibilities are that the antagonist reverses ongoing endogenous opioid signaling in the slice, or acts as an inverse agonist, turning off constitutive activity of the receptors. These possibilities are unlikely since TIPP-Ψ application had no effect by itself (Supplementary Fig. 1), and there was no effect on membrane potential induced by CTAP application alone (baseline −51 ± 2, CTAP −50 ± 2, n = 9; Supplementary Fig. 1). It is interesting to note that in these examples, while the baseline membrane potential before the second DOR agonist application is more hyperpolarized than the first, thereby closer to the K+ reversal potential and decreasing the K+ driving force, the second agonist response is still larger than the first. The fact that CTAP can augment responses to either DPDPE or deltorphin II suggests that either DOR subtype can interact with MOR. These observations also support the conclusions that either subtype is capable of forming a MOR-DOR heterodimer, and that the difference in signaling between DOR1 and DOR2 is not simply explained by heterodimerization.
Fig. 7. MOR and DOR interact in a subset of VTA neurons.
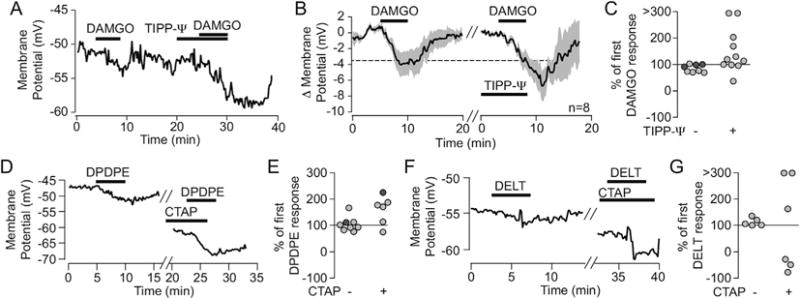
(A) Example VTA neuron recording in which a small DAMGO-induced hyperpolarization (saturating dose of 500 nM) is substantially larger when DAMGO was re-applied in the presence of the DOR selective antagonist, TIPP-Ψ (100 nM). (B) Time course average across 8 neurons in which this experiment was performed. (C) The population of DAMGO responses is shifted to larger than baseline DAMGO responses when the second application is in the presence of TIPP-Ψ. (D) Example recording in which the MOR selective antagonist CTAP (500 nM) augmented the hyperpolarization response to DPDPE (1 μM). (E) Summary across cells showing that DPDPE applied in the presence of CTAP tends to have an augmented response compared to the first response to DPDPE alone. (F) Example recording in which CTAP augmented a hyperpolarization response to deltorphin II (1 μM). (G) Summary across cells shows that CTAP markedly changes a cell’s response to deltorphin II, either increasing the magnitude of the response or even switching the effect from hyperpolarizing to depolarizing (indicated by negative values here). Light grey circles are cells where the first response was a hyperpolarization; dark grey circles are cells where the first response was a depolarization.
3.7. OPRD1 mRNA expression is ubiquitous in VTA neurons
Finally, to molecularly confirm the expression of DOR we utilized two different techniques to probe for OPRD1 mRNA in individual VTA neurons. We performed conventional RT-PCR in 21 cells, and nCounter analysis in 4 additional cells. Representative amplification curves and all Ct values for single cell RT-PCR, and all nCounter data are presented in Supplementary Fig. 4 and Supplementary Tables 1–3. As the outcomes are similar, the results from the two methods are combined here. OPRD1 mRNA was detected in all 25 neurons. Sixteen of the analyzed neurons also expressed TH mRNA (64%), a roughly similar proportion to the physiology presented above and the overall proportion of dopamine neurons in the VTA (Margolis et al., 2006b; Swanson, 1982). Further, 19 of the 25 neurons analyzed also expressed OPRM1, indicating that many VTA neurons express mRNA for both of these receptors, consistent with the electrophysiological observations reported above.
4. Discussion
4.1. MOR, DOR1 and/or DOR2 produce stable postsynaptic excitations and inhibitions in many VTA neurons
In this study we tested a large and spatially extensive population of neurons randomly sampled from throughout the VTA for postsynaptic responses to MOR and DOR selective agonists. A large proportion of VTA neurons show either the canonical postsynaptic K+ channel mediated inhibition or a Cav2.1 dependent excitation to DOR1, DOR2, or MOR agonists. Importantly, applying the same agonist sequentially in the same neuron resulted in effects of the same sign and similar magnitude, and interleaving agonist applications did not cause a change in subsequent responses, consistent with the idea that DOR1 and DOR2 responses are stable in individual acutely recorded neurons. Because these direct DPDPE and deltorphin II cellular effects are both blocked by bath application of the highly selective DOR antagonist TIPP-Ψ, but not the MOR selective antagonist CTAP, both DOR1 and DOR2 agonists produce their effects largely, if not exclusively, through direct activation of DOR in the VTA.
While opioid receptor effects are generally presumed to be inhibitory, here we report that a subset of neurons responded to DOR activation with an increase in firing rate or depolarization. This excitatory effect requires Cav2.1. We previously demonstrated that MOR activation in some VTA neurons, including dopamine neurons, is excitatory, an effect that requires Cav2.1 and G protein activity in the recorded neuron, and is independent of synaptic input (Margolis et al., 2014). MOR induced activation of Cav2.1 has also been reported in the cerebellum (Iegorova et al., 2010), and somatodendritic Cav2.1 expression (Ishibashi et al., 1995) and currents (Philippart et al., 2016) have been reported in substantia nigra and VTA neurons. Together, these data indicate that activation of Cav2.1 is a common mechanism for both MORs and DORs in VTA neurons.
We have demonstrated that DOR is expressed in most VTA neurons: over 80% of VTA neurons responded to DOR activation (Table 3). This is true for both confirmed dopamine neurons as well as non-dopamine neurons, and is consistent with our single cell detection of OPRD1 mRNA. To the best of our knowledge, this is the first comparison of DPDPE and deltorphin II actions within individual neurons. The importance of studying agonists for both DOR subtypes is evident from our findings. One third showed no response to DPDPE (Table 1) and over 1/3 showed no response to deltorphin II (Table 2). However, when both agonists were tested in the same neuron, more than half of the VTA neurons that showed no response to one of the DOR agonist classes did respond when tested with the other class of agonist. Furthermore, in these neurons, only half had similar responses (or lack of response) to both DOR1 and DOR2 agonists (104/211 neurons). Additionally, only a subset of small molecule agonists and antagonists that have been proposed to be DOR subtype specific show selectivity in this system (example data in Supplementary Figs. 5–7). Consequently, conclusions drawn from experiments using a single DOR agonist, especially if negative, must be considered tentative.
Prior electrophysiological studies have reported no postsynaptic actions for DOR agonists in the VTA in opioid naïve animals. Johnson and North (1992a) reported that effects of enkephalin on VTA neurons were blocked by a MOR selective antagonist (CTOP) but not a DOR selective antagonist (naltrindole), and that DPDPE did not affect the 3 VTA neurons tested. Given that 129 out of 396 of our neurons were insensitive to DPDPE, there is a reasonable probability that randomly selecting neurons will yield 3 that are insensitive to DPDPE, as in the Johnson and North study. Further, many of the responses reported in the present study, while statistically significant, are relatively small and therefore may have been overlooked. Small effects may be due to low receptor expression or location interior to the plasma membrane. An alternative possibility is that DOR effects are more profound on distal dendrites, which may be difficult to detect in somatic whole cell recordings given the long dendrites and low input resistances of these neurons. In view of the growing appreciation that VTA dopamine neurons are a pharmacologically heterogeneous population (Ford et al., 2006; Korotkova et al., 2003, 2006; Margolis et al., 2006a, 2008b, 2012b, 2014), it is also possible that different populations of VTA neurons were studied here compared to Johnson and North (1992a); while our recordings were made from throughout the VTA (Fig. 6), in many studies recordings are limited to the very lateral aspects of the VTA where indirect identification of putative dopamine neurons may be more reliable (e.g. Wanat et al., 2008; Williams et al., 2014; Zhang et al., 2010). The fact that our DPDPE effects were observed at a low micromolar concentration and were blocked by the highly DOR selective antagonist TIPP-Ψ (that exhibits over 10,000 fold selectivity for DOR over MOR in cell culture (Schiller et al., 1993)), confirms that our observations are due to activation of the DOR. Our results are also consistent with in vivo behavioral and microdialysis studies demonstrating functional DOR in the VTA of naïve rats (Devine et al., 1993a, 1993b; Devine and Wise, 1994; Khaimova et al., 2004; Ragnauth et al., 1997).
4.2. DOR subtypes are not a form of biased agonism
How can DPDPE and deltorphin II stably activate different signaling pathways within the same neuron? The data presented here are not consistent with the concept that DOR subtype pharmacology is a form of biased agonism. First, if DOR subtypes represented signaling bias, all DOR-expressing neurons should respond to a given ligand in a consistent manner (for example, all DOR expressing neurons might be excited by DPDPE and inhibited by deltorphin II). We did not observe such a pattern. Second, we observed a subset of neurons that responded to one agonist but not the other. In many cases neurons that were insensitive to the first DOR agonist applied responded to the second, ruling out receptor internalization or desensitization causing the apparent differential responsiveness. Then, how can a single neuron expressing DOR respond differentially to DPDPE or deltorphin II? One possibility is that the conformation of DOR varies, leading to two stable configurations of the binding pocket. This could enable selective docking of DPDPE or deltorphin II, in which case expression of two distinct populations of receptor with these different conformations could explain our observations. There is evidence that a variety of proteins and other molecules can affect the configuration of the binding pocket. For instance, the conformation of the binding site may be influenced by whether or not the receptor is contained in a lipid raft; interacting with RGS proteins, GRKs, and other molecules; in proximity to the G protein; phosphorylated; glycosylated; ubiquitinated; or palmitoylated (Alonso and Friedman, 2013; Chini and Parenti, 2009; Gahbauer and Böckmann, 2016; Vošahlíková and Svoboda, 2011; Wisler et al., 2014; Zheng et al., 2013). DORs also have a large Na+ pocket in their inactive state that affects ligand binding and coupling to downstream signaling pathways (Appelmans et al., 1986; Fenalti et al., 2014; Strasser et al., 2015). This Na+ pocket is affected by Vm, decreasing the probability that it occupies its allosteric binding site and moving to the orthosteric ligand binding site as Vm increases (Vickery et al., 2016), therefore the local membrane electrical and ionic environments at individual receptors may influence pharmacological responses as well. The selective distribution of a variety of intracellular molecules could contribute to both the differential signaling by agonist subtype observed in some neurons, as well as the observations that an individual neuron may respond to only DPDPE or deltorphin II.
The fact that DOR1, DOR2, and MOR can predominantly activate either a GIRK or Cav2.1, both of which depend upon G protein activation (Margolis et al., 2014), make it unlikely that differences in binding account for the differences in net change in excitability observed in different VTA neurons. Rather, it seems more likely that these differences result from the sorting of subpopulations of DORs or MORs into somatodendritic domains consisting of distinct complexes of associated proteins, including ion channels, and that these domains are relatively isolated from each other (Fig. 8). This idea is consistent with evidence that GPCR complexes, including G proteins and other accessory molecules, are assembled in the endoplasmic reticulum before being delivered to the plasma membrane (Hasbi et al., 2007). One instance of differential function of MORs sorting by cellular compartment has been demonstrated in the nucleus accumbens, where morphine-induced activation of MORs causes receptor internalization in dendrites, but not in cell bodies (Haberstock-Debic et al., 2003). Further, different GPCRs, even within a single dendritic spine, that are coupled to the same primary signaling pathway can have segregated, independent downstream signaling consequences (Lur and Higley, 2015). Finally, there is accumulating evidence that other GPCRs traffic to very specific parts of the neuron, such as the axon initial segment (Bender et al., 2010). Together, these observations raise the possibility for fine control of the trafficking and signaling of opioid receptors within the somatodendritic region of VTA neurons.
Fig. 8. Hypothesis to explain how DOR subtypes and MOR independently produce either direct excitations or inhibitions in VTA neurons.
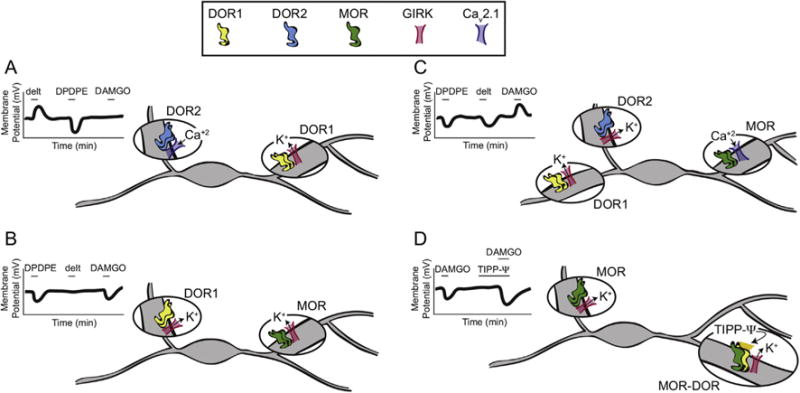
We suggest that the DOR1, DOR2, and MOR signaling observed here results from the segregation of these different receptors and their respective signaling channels to separated neural compartments. The long dendrites and bipolar geometry of many VTA neurons may facilitate separation into such relatively isolated domains. For instance, in A, B, and C, homodimers of DOR1, DOR2, or MOR are localized to separate cellular domains and signal differentially through the GIRK or Cav2.1 specifically associated with that particular opioid receptor. (D) A proposed organization of MOR homodimers and MOR-DOR heterodimers that elicit a GIRK conductance at the MOR homodimers in the absence of TIPP-Ψ. When TIPP-Ψ is bound to the MOR-DOR heterodimer, the MOR agonist acting at the heterodimer more effectively opens the GIRK, resulting in a more pronounced hyperpolarization.
4.3. The relationship between MOR and DOR in VTA neurons
The fact that there was no significant within cell relationship between the sign and magnitude of the effects of DAMGO, DPDPE, and deltorphin II indicates that MOR, DOR1, and DOR2 signal largely independently, even when they are expressed in the same neuron. The absence of a relationship between DOR subtype responses and DAMGO responses makes MOR-DOR heterodimerization an unlikely explanation for the DOR subtype differences we observed in these experiments. However, we did find evidence for interaction of DOR and MOR in a subset of neurons: the DOR selective antagonist TIPP-Ψ augmented the response to a MOR agonist and the MOR selective antagonist CTAP augmented the response to DPDPE or deltorphin II. This finding is consistent with the heterodimer concept, and with previous observations that heterodimerization leads to a DOR antagonist increasing the efficacy of a MOR agonist, and vice versa, in a cell culture model system (Gomes et al., 2000) and in spinal cord membrane (Gupta et al., 2010). While it is possible that the antagonist is enabling augmented signaling by reversing the action of an endogenous opioid, this seems unlikely since neither TIPP-Ψ nor CTAP have effects in this preparation on their own. In cell culture systems, opioid receptor heterodimers preferentially signal through G protein independent mechanisms (Rozenfeld and Devi, 2007), but the antagonist at one receptor recovers G protein signaling. This is a potential mechanism underlying the increase of K+ channel effects observed here. Heterodimer induced activation of G protein independent, electrophysiologically observable signaling in acute brain slices remains to be demonstrated.
4.4. MOR-DOR interaction and endogenous opioid peptides
Understanding MOR and DOR actions and interactions is particularly important for elucidating the function of endogenous opioid peptides. MOR and DOR are often expressed in the same cells in vivo (Erbs et al., 2015) and morphine treatment can increase heterodimer expression (Gupta et al., 2010). Three of the four known large opioid precursor peptides (preproenkephalin, preprodynorphin, and preproopioimelanocortin) can be processed to shorter opioid peptides that act at both MORs and DORs; both classic and emerging studies indicate that the majority of the opioid peptides activate all three receptors and that enkephalins, dynorphins, and β-endorphin have similar affinities for the MOR and DOR (Chang et al., 2004; Mansour et al., 1995). Because of the presumed volume transmission of peptides, if MOR and DOR are expressed on the same neurons it is likely that these endogenous ligands will activate both receptors concurrently in vivo. Yet, even though these different peptides bind to both receptors, the physiological impact of each of the different peptides acting at MOR may be different from that at DOR. Further, different opioid peptides may preferentially activate different signaling pathways at, for instance, the DOR. In the VTA, m-enk and l-enk can be cleaved from preproenkephalin contained in projections from the ventral pallidum and bed nucleus of the stria terminalis (Kalivas et al., 1993; Kudo et al., 2014), while peptides derived from preprodynorphin, potentially including l-enk, would be contained in projections from the nucleus accumbens, lateral hypothalamus, and amygdala (Fallon et al., 1985). With electron microscopy, terminals containing enkephalin have been shown to synapse onto dopamine (50–60% of postsynaptic appositions) and non-dopamine VTA neurons (Sesack and Pickel, 1992). If MORs and DORs in the same cells are trafficked to neighboring synaptic sites our data indicate that an endogenous opioid peptide that is locally released will act on both. Consequently, it is likely the biological functions of the two receptors can only be fully understood in relation to each other.
MOR-DOR receptor interactions could also explain the unexpected physiological antagonism between MOR and DOR that has been reported. One apparent paradox is that microinjecting a MOR antagonist into the VTA produces conditioned place aversion but also increases dopamine release in the nucleus accumbens (Devine et al., 1993c; Shippenberg and Bals-Kubik, 1995). These effects suggest some endogenous peptide tone in the VTA in vivo. While such data are generally interpreted to indicate that an ongoing effect of an endogenous peptide is being reversed at MORs, our data here suggest an alternative explanation for one or both of these effects: an action of an endogenous peptide at the DOR could be augmented by the MOR antagonist for instance by causing a conformational change in a MOR-DOR heterodimer.
Behaviorally, activation of the MOR and DOR systems within the VTA often have dissimilar or even opposing effects. For instance, while injection of MOR agonists into the VTA of naïve animals produces conditioned place preference (e.g. Bals-Kubik et al., 1993; Bozarth, 1987; Nader and van der Kooy, 1997; Shippenberg and Herz, 1987), injection of DPDPE or deltorphin II does not (Mitchell et al., 2014). DOR antagonists in the VTA decrease feeding (Lamonte et al., 2002; Ragnauth et al., 1997) whereas MOR antagonists have no effect (Badiani et al., 1995). While a MOR agonist (DAMGO) decreases activity when rats are placed in an open field or elevated plus maze, a DOR agonist (Tyr-D-Ser-(O-tert-butyl)-Gly-Phe-Leu-Thr-(O-tert-butyl) BUBU) increases activity in these paradigms (Calenco-Choukroun et al., 1991). One possibility is that such behavioral differences can be accounted for by the different distributions of MOR and DOR on different VTA neurons, as we have observed here. We previously showed that KORs are selectively functionally expressed on the somatodendritic regions of VTA dopamine neurons that project to the medial prefrontal cortex and amygdala, but not those that project to the nucleus accumbens (Margolis et al., 2006a, 2008b), raising the possibility that the different MOR and DOR effects reported here also sort by projection target.
5. Conclusions
Here we provide the first direct evidence that both DOR1 and DOR2 subtype selective ligands can act differentially on individual neurons and that when both are expressed in a single neuron the postsynaptic response to one does not predict the presence or valence of the response to the other. We present functional evidence for interactions of MORs and DORs, however, these data are not consistent with either DOR subtype pharmacology being a product of MOR-DOR receptor heterodimerization or biased signaling. Together, these data indicate that DOR subtypes not only differentially control individual neurons, but also modulate different, partially overlapping, subpopulations of VTA neurons. Such differences likely account for the behavioral differences previously observed in vivo. This work reveals physiological differences between DOR1 and DOR2 ligands at the individual neuronal level that has not been observed in cell culture models, raising the important question of whether endogenous opioid peptides also show a more selective and heterogeneous response pattern in specific neural circuits. This more refined understanding of opioid receptor systems indicates that using a cellular electrophysiological approach to identifying the DOR subtype selectivity of new ligands will greatly inform therapeutic design.
Supplementary Material
Acknowledgments
We thank Joseph Driscoll, Peter Fong, Hagar Lock, Kelley Pollack, Junko Ishikawa, Mirabel Lim, and Adam Ferris for technical assistance. We also thank Toni Shippenberg, Vladimir Chefer, Gregory Hjelmstad, and Jennifer Mitchell for helpful discussions.
Funding
This work was supported by the National Institutes of Health [grant number R01 DA030529 to E.B.M. and DA008863 to L.A.D.]; funds were also provided by the Wheeler Center for the Neurobiology of Addiction and the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.neuropharm.2017.06.019.
References
- Alonso V, Friedman PA. Minireview: ubiquitination-regulated G protein-coupled receptor signaling and trafficking. Mol Endocrinol. 2013;27:558–572. doi: 10.1210/me.2012-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelmans N, Carroll JA, Rance MJ, Simon EJ, Traynor JR. Sodium ions increase the binding of the antagonist peptide ICI 174864 to the delta-opiate receptor. Neuropeptides. 1986;7:139–143. doi: 10.1016/0143-4179(86)90089-2. [DOI] [PubMed] [Google Scholar]
- Badiani A, Leone P, Noel MB, Stewart J. Ventral tegmental area opioid mechanisms and modulation of ingestive behavior. Brain Res. 1995;670:264–276. doi: 10.1016/0006-8993(94)01281-l. [DOI] [PubMed] [Google Scholar]
- Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther. 1993;264:489–495. [PubMed] [Google Scholar]
- Bender KJ, Ford CP, Trussell LO. Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron. 2010;68:500–511. doi: 10.1016/j.neuron.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozarth MA. Neuroanatomical boundaries of the reward-relevant opiate-receptor field in the ventral tegmental area as mapped by the conditioned place preference method in rats. Brain Res. 1987;414:77–84. doi: 10.1016/0006-8993(87)91327-8. [DOI] [PubMed] [Google Scholar]
- Calenco-Choukroun G, Daugé V, Gacel G, Féger J, Roques BP. Opioid delta agonists and endogenous enkephalins induce different emotional reactivity than mu agonists after injection in the rat ventral tegmental area. Psychopharmacol (Berl) 1991;103:493–502. doi: 10.1007/BF02244249. [DOI] [PubMed] [Google Scholar]
- Chang KJ, Porreca F, Woods J. The Delta Receptor. Marcel Dekker, Inc; New York: 2004. [Google Scholar]
- Chini B, Parenti M. G-protein-coupled receptors, cholesterol and palmitoylation: facts about fats. J Mol Endocrinol. 2009;42:371–379. doi: 10.1677/JME-08-0114. [DOI] [PubMed] [Google Scholar]
- Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine DP, Leone P, Carlezon WA, Wise RA. Ventral mesencephalic delta opioid receptors are involved in modulation of basal mesolimbic dopamine neurotransmission: an anatomical localization study. Brain Res. 1993a;622:348–352. doi: 10.1016/0006-8993(93)90843-c. [DOI] [PubMed] [Google Scholar]
- Devine DP, Leone P, Pocock D, Wise RA. Differential involvement of ventral tegmental mu, delta and kappa opioid receptors in modulation of basal mesolimbic dopamine release: in vivo microdialysis studies. J Pharmacol Exp Ther. 1993b;266:1236–1246. [PubMed] [Google Scholar]
- Devine DP, Leone P, Wise RA. Mesolimbic dopamine neurotransmission is increased by administration of mu-opioid receptor antagonists. Eur J Pharmacol. 1993c;243:55–64. doi: 10.1016/0014-2999(93)90167-g. [DOI] [PubMed] [Google Scholar]
- Devine DP, Wise RA. Self-administration of morphine, DAMGO, and DPDPE into the ventral tegmental area of rats. J Neurosci. 1994;14:1978–1984. doi: 10.1523/JNEUROSCI.14-04-01978.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem. 2008;283:5669–5676. doi: 10.1074/jbc.M708118200. [DOI] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, Koch M, Kessler P, Hentsch D, Birling MC, Koutsourakis M, Vasseur L, Veinante P, Kieffer BL, Massotte D. A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct Funct. 2015;220:677–702. doi: 10.1007/s00429-014-0717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erspamer V, Melchiorri P, Falconieri-Erspamer G, Negri L, Corsi R, Severini C, Barra D, Simmaco M, Kreil G. Deltorphins: a family of naturally occurring peptides with high affinity and selectivity for delta opioid binding sites. Proc Natl Acad Sci U S A. 1989;86:5188–5192. doi: 10.1073/pnas.86.13.5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon JH, Leslie FM, Cone RI. Dynorphin-containing pathways in the substantia nigra and ventral tegmentum: a double labeling study using combined immunofluorescence and retrograde tracing. Neuropeptides. 1985;5:457–460. doi: 10.1016/0143-4179(85)90053-8. [DOI] [PubMed] [Google Scholar]
- Farias M, Jackson KE, Yoshishige D, Caffrey JL. Cardiac enkephalins interrupt vagal bradycardia via delta 2-opioid receptors in sinoatrial node. Am J Physiol Heart Circ Physiol. 2003;284:H1693–H1701. doi: 10.1152/ajpheart.00730.2002. [DOI] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of δ-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci. 2006;26:2788–2797. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita W, Gomes I, Devi LA. Heteromers of μ-δ opioid receptors: new pharmacology and novel therapeutic possibilities. Br J Pharmacol. 2015;172:375–387. doi: 10.1111/bph.12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita W, Gomes I, Dove LS, Prohaska D, McIntyre G, Devi LA. Molecular characterization of eluxadoline as a potential ligand targeting mu-delta opioid receptor heteromers. Biochem Pharmacol. 2014;92:448–456. doi: 10.1016/j.bcp.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahbauer S, Böckmann RA. Membrane-mediated Oligomerization of G Protein coupled receptors and its implications for GPCR function. Front Physiol. 2016;7:494. doi: 10.3389/fphys.2016.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Gupta A, Saldanha SA, Saldanha AS, Negri A, Pinello CE, Eberhart C, Roberts E, Filizola M, Hodder P, Devi LA. Identification of a μ-δ opioid receptor heteromer-biased agonist with antinociceptive activity. Proc Natl Acad Sci U S A. 2013;110:12072–12077. doi: 10.1073/pnas.1222044110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A. 2004;101:5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Ijzerman AP, Ye K, Maillet EL, Devi LA. G protein-coupled receptor heteromerization: a role in allosteric modulation of ligand binding. Mol Pharmacol. 2011;79:1044–1052. doi: 10.1124/mol.110.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Trapaidze N, Nagy V, Devi LA. Heterodimerization of mu and delta opioid receptors: a role in opiate synergy. J Neurosci. 2000;20:RC110. doi: 10.1523/JNEUROSCI.20-22-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Mulder J, Gomes I, Rozenfeld R, Bushlin I, Ong E, Lim M, Maillet E, Junek M, Cahill CM, Harkany T, Devi LA. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal. 2010;3:ra54. doi: 10.1126/scisignal.2000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberstock-Debic H, Wein M, Barrot M, Colago EE, Rahman Z, Neve RL, Pickel VM, Nestler EJ, von Zastrow M, Svingos AL. Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. J Neurosci. 2003;23:4324–4332. doi: 10.1523/JNEUROSCI.23-10-04324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond DL, Wang H, Nakashima N, Basbaum AI. Differential effects of intrathecally administered delta and mu opioid receptor agonists on formalin-evoked nociception and on the expression of Fos-like immunoreactivity in the spinal cord of the rat. J Pharmacol Exp Ther. 1998;284:378–387. [PubMed] [Google Scholar]
- Hasbi A, Nguyen T, Fan T, Cheng R, Rashid A, Alijaniaram M, Rasenick MM, O’Dowd BF, George SR. Trafficking of preassembled opioid mu-delta heterooligomer-Gz signaling complexes to the plasma membrane: coregulation by agonists. Biochemistry. 2007;46:12997–13009. doi: 10.1021/bi701436w. [DOI] [PubMed] [Google Scholar]
- Iegorova O, Fisyunov A, Krishtal O. G-protein-independent modulation of P-type calcium channels by mu-opioids in Purkinje neurons of rat. Neurosci Lett. 2010;480:106–111. doi: 10.1016/j.neulet.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi H, Rhee JS, Akaike N. Regional difference of high voltage-activated Ca2+ channels in rat CNS neurones. Neuroreport. 1995;6:1621–1624. doi: 10.1097/00001756-199508000-00008. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Takemori AE, Sultana M, Portoghese PS, Bowen WD, Mosberg HI, Porreca F. Differential antagonism of opioid delta antinociception by [D-Ala2,Leu5,Cys6]enkephalin and naltrindole 5′-isothiocyanate: evidence for delta receptor subtypes. J Pharmacol Exp Ther. 1991;257:1069–1075. [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Churchill L, Klitenick MA. GABA and enkephalin projection from the nucleus accumbens and ventral pallidum to the ventral tegmental area. Neuroscience. 1993;57:1047–1060. doi: 10.1016/0306-4522(93)90048-k. [DOI] [PubMed] [Google Scholar]
- Khaimova E, Kandov Y, Israel Y, Cataldo G, Hadjimarkou MM, Bodnar RJ. Opioid receptor subtype antagonists differentially alter GABA agonist-induced feeding elicited from either the nucleus accumbens shell or ventral tegmental area regions in rats. Brain Res. 2004;1026:284–294. doi: 10.1016/j.brainres.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Korotkova TM, Brown RE, Sergeeva OA, Ponomarenko AA, Haas HL. Effects of arousal- and feeding-related neuropeptides on dopaminergic and GABAergic neurons in the ventral tegmental area of the rat. Eur J Neurosci. 2006;23:2677–2685. doi: 10.1111/j.1460-9568.2006.04792.x. [DOI] [PubMed] [Google Scholar]
- Korotkova TM, Sergeeva OA, Eriksson KS, Haas HL, Brown RE. Excitation of ventral tegmental area dopaminergic and nondopaminergic neurons by orexins/hypocretins. J Neurosci. 2003;23:7–11. doi: 10.1523/JNEUROSCI.23-01-00007.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreil G, Barra D, Simmaco M, Erspamer V, Erspamer GF, Negri L, Severini C, Corsi R, Melchiorri P. Deltorphin, a novel amphibian skin peptide with high selectivity and affinity for delta opioid receptors. Eur J Pharmacol. 1989;162:123–128. doi: 10.1016/0014-2999(89)90611-0. [DOI] [PubMed] [Google Scholar]
- Kudo T, Konno K, Uchigashima M, Yanagawa Y, Sora I, Minami M, Watanabe M. GABAergic neurons in the ventral tegmental area receive dual GABA/enkephalin-mediated inhibitory inputs from the bed nucleus of the stria terminalis. Eur J Neurosci. 2014;39:1796–1809. doi: 10.1111/ejn.12503. [DOI] [PubMed] [Google Scholar]
- Lamonte N, Echo JA, Ackerman TF, Christian G, Bodnar RJ. Analysis of opioid receptor subtype antagonist effects upon mu opioid agonist-induced feeding elicited from the ventral tegmental area of rats. Brain Res. 2002;929:96–100. doi: 10.1016/s0006-8993(01)03382-0. [DOI] [PubMed] [Google Scholar]
- Lur G, Higley MJ. Glutamate receptor modulation is restricted to synaptic microdomains. Cell Rep. 2015;12:326–334. doi: 10.1016/j.celrep.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Hoversten MT, Taylor LP, Watson SJ, Akil H. The cloned mu, delta and kappa receptors and their endogenous ligands: evidence for two opioid peptide recognition cores. Brain Res. 1995;700:89–98. doi: 10.1016/0006-8993(95)00928-j. [DOI] [PubMed] [Google Scholar]
- Margolis EB, Coker AR, Driscoll JR, Lemaître AI, Fields HL. Reliability in the identification of midbrain dopamine neurons. PLoS One. 2010;5:e15222. doi: 10.1371/journal.pone.0015222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Fields HL, Hjelmstad GO, Mitchell JM. Delta-opioid receptor expression in the ventral tegmental area protects against elevated alcohol consumption. J Neurosci. 2008a;28:12672–12681. doi: 10.1523/JNEUROSCI.4569-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Fujita W, Fields HL. Direct bidirectional μ-opioid control of midbrain dopamine neurons. J Neurosci. 2014;34:14707–14716. doi: 10.1523/JNEUROSCI.2144-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci U S A. 2006a;103:2938–2942. doi: 10.1073/pnas.0511159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006b;577:907–924. doi: 10.1113/jphysiol.2006.117069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Mitchell JM, Ishikawa J, Hjelmstad GO, Fields HL. Midbrain dopamine neurons: projection target determines action potential duration and dopamine D(2) receptor inhibition. J Neurosci. 2008b;28:8908–8913. doi: 10.1523/JNEUROSCI.1526-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Toy B, Himmels P, Morales M, Fields HL. Identification of rat ventral tegmental area GABAergic neurons. PLoS One. 2012;7:e42365. doi: 10.1371/journal.pone.0042365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattia A, Vanderah T, Mosberg HI, Porreca F. Lack of antinociceptive cross-tolerance between [D-Pen2, D-Pen5]enkephalin and [D-Ala2]deltorphin II in mice: evidence for delta receptor subtypes. J Pharmacol Exp Ther. 1991;258:583–587. [PubMed] [Google Scholar]
- Mitchell JM, Margolis EB, Coker AR, Allen DC, Fields HL. Intra-VTA deltorphin, but not DPDPE, induces place preference in ethanol-drinking rats: distinct DOR-1 and DOR-2 mechanisms control ethanol consumption and reward. Alcohol Clin Exp Res. 2014;38:195–203. doi: 10.1111/acer.12246. [DOI] [PubMed] [Google Scholar]
- Mosberg HI, Hurst R, Hruby VJ, Gee K, Akiyama K, Yamamura HI, Galligan JJ, Burks TF. Cyclic penicillamine containing enkephalin analogs display profound delta receptor selectivities. Life Sci. 1983;33(Suppl. 1):447–450. doi: 10.1016/0024-3205(83)90538-6. [DOI] [PubMed] [Google Scholar]
- Nader K, van der Kooy D. Deprivation state switches the neurobiological substrates mediating opiate reward in the ventral tegmental area. J Neurosci. 1997;17:383–390. doi: 10.1523/JNEUROSCI.17-01-00383.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW. Incomplete cross tolerance and multiple mu opioid peptide receptors. Trends Pharmacol Sci. 2001;22:67–70. doi: 10.1016/s0165-6147(00)01616-3. [DOI] [PubMed] [Google Scholar]
- Philippart F, Destreel G, Merino-Sepúlveda P, Henny P, Engel D, Seutin V. Differential somatic Ca2+ channel profile in midbrain dopaminergic neurons. J Neurosci. 2016;36:7234–7245. doi: 10.1523/JNEUROSCI.0459-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portoghese PS, Nagase H, MaloneyHuss KE, Lin CE, Takemori AE. Role of spacer and address components in peptidomimetic delta opioid receptor antagonists related to naltrindole. J Med Chem. 1991;34:1715–1720. doi: 10.1021/jm00109a027. [DOI] [PubMed] [Google Scholar]
- Provasi D, Boz MB, Johnston JM, Filizola M. Preferred supramolecular organization and dimer interfaces of opioid receptors from simulated self-association. PLoS Comput Biol. 2015;11:e1004148. doi: 10.1371/journal.pcbi.1004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragnauth A, Ruegg H, Bodnar RJ. Evaluation of opioid receptor subtype antagonist effects in the ventral tegmental area upon food intake under deprivation, glucoprivic and palatable conditions. Brain Res. 1997;767:8–16. doi: 10.1016/s0006-8993(97)00539-8. [DOI] [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. Receptor heterodimerization leads to a switch in signaling: beta-arrestin2-mediated ERK activation by mu-delta opioid receptor heterodimers. FASEB J. 2007;21:2455–2465. doi: 10.1096/fj.06-7793com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller PW, Weltrowska G, Nguyen TM, Wilkes BC, Chung NN, Lemieux C. TIPP[psi]: a highly potent and stable pseudopeptide delta opioid receptor antagonist with extraordinary delta selectivity. J Med Chem. 1993;36:3182–3187. doi: 10.1021/jm00073a020. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Pickel VM. Dual ultrastructural localization of enkephalin and tyrosine hydroxylase immunoreactivity in the rat ventral tegmental area: multiple substrates for opiate-dopamine interactions. J Neurosci. 1992;12:1335–1350. doi: 10.1523/JNEUROSCI.12-04-01335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shippenberg TS, Bals-Kubik R. Involvement of the mesolimbic dopamine system in mediating the aversive effects of opioid antagonists in the rat. Behav Pharmacol. 1995;6:99–106. [PubMed] [Google Scholar]
- Shippenberg TS, Herz A. Place preference conditioning reveals the involvement of D1-dopamine receptors in the motivational properties of mu- and kappa-opioid agonists. Brain Res. 1987;436:169–172. doi: 10.1016/0006-8993(87)91571-x. [DOI] [PubMed] [Google Scholar]
- Sofuoglu M, Portoghese PS, Takemori AE. Differential antagonism of delta opioid agonists by naltrindole and its benzofuran analog (NTB) in mice: evidence for delta opioid receptor subtypes. J Pharmacol Exp Ther. 1991;257:676–680. [PubMed] [Google Scholar]
- Sofuoglu M, Portoghese PS, Takemori AE. 7-Benzylidenenaltrexone (BNTX): a selective delta 1 opioid receptor antagonist in the mouse spinal cord. Life Sci. 1993;52:769–775. doi: 10.1016/0024-3205(93)90240-4. [DOI] [PubMed] [Google Scholar]
- Strasser A, Wittmann HJ, Schneider EH, Seifert R. Modulation of GPCRs by monovalent cations and anions. Naunyn Schmiedeb Arch Pharmacol. 2015;388:363–380. doi: 10.1007/s00210-014-1073-2. [DOI] [PubMed] [Google Scholar]
- Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9:321–353. doi: 10.1016/0361-9230(82)90145-9. [DOI] [PubMed] [Google Scholar]
- van Rijn RM, Whistler JL. The delta(1) opioid receptor is a heterodimer that opposes the actions of the delta(2) receptor on alcohol intake. Biol Psychiatry. 2009;66:777–784. doi: 10.1016/j.biopsych.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickery ON, Machtens JP, Tamburrino G, Seeliger D, Zachariae U. Structural mechanisms of voltage Sensing in G Protein-coupled receptors. Structure. 2016;24:997–1007. doi: 10.1016/j.str.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vošahlíková M, Svoboda P. The influence of monovalent cations on trimeric G protein G(i)1α activity in HEK293 cells stably expressing DOR-G(i)1α (Cys(351)-Ile(351)) fusion protein. Physiol Res. 2011;60:541–547. doi: 10.33549/physiolres.932096. [DOI] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Buchta WC, Riegel AC. CRF-R2 and the heterosynaptic regulation of VTA glutamate during reinstatement of cocaine seeking. J Neurosci. 2014;34:10402–10414. doi: 10.1523/JNEUROSCI.0911-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Wisler JW, Xiao K, Thomsen AR, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki PA, Bilsky EJ, Vanderah TW, Lai J, Evans CJ, Porreca F. Opioid receptor types and subtypes: the delta receptor as a model. Annu Rev Pharmacol Toxicol. 1996;36:379–401. doi: 10.1146/annurev.pa.36.040196.002115. [DOI] [PubMed] [Google Scholar]
- Zhang TA, Placzek AN, Dani JA. In vitro identification and electrophysiological characterization of dopamine neurons in the ventral tegmental area. Neuropharmacology. 2010;59:431–436. doi: 10.1016/j.neuropharm.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Loh HH, Law PY. Posttranslation modification of G protein-coupled receptor in relationship to biased agonism. Methods Enzymol. 2013;522:391–408. doi: 10.1016/B978-0-12-407865-9.00018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.