

ABSTRACT
Pneumolysin (PLY), a major virulence factor of Streptococcus pneumoniae, is a pore-forming cytolysin that modulates host innate responses contributing to host defense against and pathogenesis of pneumococcal infections. Interleukin-1α (IL-1α) has been shown to be involved in tissue damage in a pneumococcal pneumonia model; however, the mechanism by which this cytokine is produced during S. pneumoniae infection remains unclear. In this study, we examined the role of PLY in IL-1α production. Although the strains induced similar levels of pro-IL-1α expression, wild-type S. pneumoniae D39, but not a deletion mutant of the ply gene (Δply), induced the secretion of mature IL-1α from host macrophages, suggesting that PLY is critical for the maturation and secretion of IL-1α during S. pneumoniae infection. Further experiments with calcium chelators and calpain inhibitors indicated that extracellular calcium ions and calpains (calcium-dependent proteases) facilitated the maturation and secretion of IL-1α from D39-infected macrophages. Moreover, we found that PLY plays a critical role in calcium influx and calpain activation, as elevated intracellular calcium levels and the degradation of the calpain substrate α-fodrin were detected in macrophages infected with D39 but not the Δply strain. These results suggested that PLY induces the influx of calcium in S. pneumoniae-infected macrophages, followed by calpain activation and subsequent IL-1α maturation and secretion.
KEYWORDS: Streptococcus pneumoniae, IL-1α, pneumolysin, calpain
INTRODUCTION
Streptococcus pneumoniae is a classic Gram-positive extracellular pathogen that is responsible for significant mortality and morbidity worldwide, causing bacterial pneumonia, otitis media, meningitis, and septicemia. Due to the severe disease burden and mortality in newborns, elderly persons, and immunocompromised patients and the increasing incidence of drug-resistant clinical isolates, it is critically important to understand the pathogenesis of pneumococcal diseases in order to develop novel therapy methods and effective vaccines (1, 2). Pneumolysin (PLY), a 53-kDa protein toxin encoded by the ply gene, is a key virulence factor of S. pneumoniae and is produced by virtually all clinical isolates. It has been regarded as a candidate for vaccine development against pneumococcal infection (3, 4). PLY is one of the cholesterol-dependent cytolysins, forming ring- or arc-shaped transmembrane pores on cholesterol-containing membranes, eventually causing cytolysis in various cells (5). The functional role of PLY has been studied, and it is recognized as a double-edged sword during host-pathogen interactions. Besides its cytotoxic function as a virulence factor, several reports demonstrated that PLY is involved in the activation of host innate immune responses, generating inflammation or contributing to the host defense against S. pneumoniae (4, 6–8).
Upon infection with S. pneumoniae, macrophages secrete a series of proinflammatory cytokines, including tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), IL-12, IL-18, IL-1α, and IL-1β (8). Several reports have shown that the Δply strain, an S. pneumoniae mutant deficient for the ply gene, is incapable of inducing the production of IL-1α, IL-1β, and IL-18 (8–12). IL-1 family cytokines, including IL-1α, IL-1β, and IL-18, have been reported to contribute to host defense and tissue damage in pneumococcal infection models in mice (13–18). Therefore, the PLY-dependent secretion of these cytokines could contribute to both host protection and pathogenesis during S. pneumoniae infection. To understand the mechanism of how these cytokine responses are induced in a PLY-dependent manner, we and others previously reported that in S. pneumoniae-infected macrophages, the maturation and secretion of IL-1β and IL-18 are dependent on the activation of caspase-1, which is induced by the assembly of inflammasomes, including the absent in melanoma 2 (AIM2) and NLR family pyrin domain-containing 3 (NLRP3) inflammasomes (9–11, 19).
IL-1α is first expressed as a 35-kDa polypeptide. Although pro-IL-1α is capable of binding to the IL-1 receptor and is biologically active (20), this cytokine is susceptible to proteolysis by certain intracellular proteases, such as granzyme B, calpain-1, elastase, and mast cell chymase, and is processed to a 17-kDa protein, which increases the possibility of secretion and enhances its biological activity severalfold (21, 22). Several studies have suggested that the maturation of IL-1α in macrophages is mediated by a calcium-dependent cysteine protease, calpain (23–28). However, the precise mechanism of PLY-dependent IL-1α secretion in S. pneumoniae-infected macrophages remains unclear. In this study, we analyzed the molecular basis for S. pneumoniae-induced IL-1α secretion, and PLY was indispensable for the activation of calcium signaling and the activation of calpain, which are required for the processing and secretion of IL-1α.
RESULTS
PLY is required for IL-1α secretion in S. pneumoniae-infected macrophages.
Macrophages were infected with the D39 and Δply strains, and the levels of IL-1α secreted into the culture supernatants were detected by an enzyme-linked immunosorbent assay (ELISA). D39 induced a high level of IL-1α secretion in infected macrophages, while the Δply strain did not (Fig. 1A). However, these strains induced comparable levels of TNF-α production (Fig. 1B). To test whether the PLY protein is sufficient to confer IL-1α-inducing activity to the Δply strain, macrophages were infected with the Δply strain and treated with different concentrations of recombinant PLY (rPLY). Although rPLY only modestly induced IL-1α production alone (Fig. 1C), stimulation with the Δply strain plus rPLY significantly facilitated IL-1α production in a dose-dependent manner. Moreover, the production of IL-1α was significantly reduced when rPLY was pretreated with cholesterol, which inhibits the pore-forming activity of PLY (Fig. 1D), suggesting that the effect of rPLY depends on its activity. These results indicated that PLY is required for the secretion of IL-1α from S. pneumoniae-infected macrophages.
FIG 1.
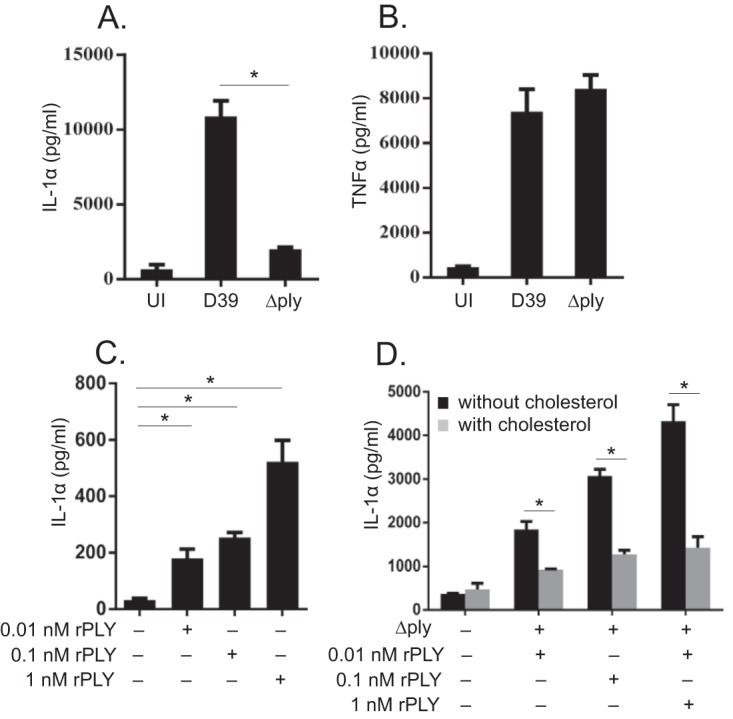
PLY is required for IL-1α secretion from S. pneumoniae-infected macrophages. Adherent PECs were infected with the D39 and Δply strains at an MOI of 1, gentamicin was added to cultures (final concentration, 100 μg/ml) 6 h later, and culture supernatants were collected after an additional 18 h of incubation (24 h after infection). (A and B) Levels of IL-1α (A) and TNF-α (B) were measured by an ELISA. UI, uninfected. (C) Macrophages were stimulated with different concentrations of rPLY (0.01 nΜ, 0.1 nM, and 1 nM). (D) Macrophages were infected with the Δply strain and treated with different concentrations of rPLY or cholesterol-treated rPLY. The levels of IL-1α in the supernatants were determined by an ELISA. Data represent the means and the standard deviations of results from triplicate assays and are representative of data from three independent experiments. Statistical significance was determined by Student's t test. *, P < 0.05.
PLY is involved in the maturation and secretion of IL-1α but not in the induction of IL-1α transcription in S. pneumoniae-infected macrophages.
It has been known that IL-1α is synthesized as a 35-kDa precursor protein (pro-IL-1α) and processed into the mature form for secretion (29). To analyze the mechanism of how PLY facilitates IL-1α secretion in S. pneumoniae-infected macrophages, we analyzed the kinetics of IL-1α mRNA expression, pro-IL-1α production in macrophages, and IL-1α secretion into the culture supernatant at different times after infection with the D39 and Δply strains. As determined by real-time reverse transcription-PCR (RT-PCR), a significant increase in IL-1α mRNA expression was observed in both D39- and Δply strain-infected macrophages by as early as 3 h after infection, and no statistical difference was found between them (Fig. 2A). Consistent with the pattern of mRNA expression, similar levels of IL-1α were detected in culture supernatants plus cell lysates of macrophages infected with the D39 and Δply strains (Fig. 2B). These results indicated that there is no significant difference between the D39 and Δply strains in their abilities to induce IL-1α expression. IL-1α levels in cell lysates decreased at later time points (12 and 24 h) of infection with D39 but not the Δply strain (Fig. 2C). The secretion of IL-1α seemed to depend on PLY, as IL-1α was detected in culture supernatants of macrophages infected with D39 but not the Δply strain (Fig. 2D). Consistently, the mature form of IL-1α was detected by Western blotting in culture supernatants after D39 infection, while the Δply strain did not induce the maturation and secretion of IL-1α (Fig. 2E). Pro-IL-1α was detected in cell lysates of both D39- and Δply strain-infected macrophages (Fig. 2E). In contrast to IL-1α, PLY deficiency did not affect the secretion of TNF-α, since the D39 and Δply strains induced similar levels of TNF-α secretion (Fig. 2F to H). Taken together, these data suggested that PLY is dispensable for IL-1α transcription and pro-IL-1α synthesis but is important for the maturation and secretion of IL-1α in S. pneumoniae-infected macrophages.
FIG 2.

PLY is involved in IL-1α maturation and secretion but not in IL-1α transcription or pro-IL-1α synthesis. Macrophages were infected with the D39 and the Δply strains at an MOI of 1. (A) Total cellular RNA was extracted at the indicated times, and the level of IL-1α mRNA expression was analyzed by real-time RT-PCR. (B to D and F to H) The levels of IL-1α and TNF-α in the culture supernatants plus cell lysates (B and F), cell lysates (C and G), or culture supernatants (D and H) were determined by an ELISA at the indicated times. Data represent the means and standard deviations of results from three independent experiments. (E) Macrophages were infected with the D39 and the Δply strains at an MOI of 1 for 24 h. The culture supernatant (Sup) and the cell lysate were prepared 24 h after infection and subjected to SDS-PAGE. IL-1α was detected by Western blotting. β-Actin was detected as a loading control. All of the experiments were repeated three times. Statistical significance was determined by Student's t test. *, P < 0.05. p. i., postinfection.
Inflammasome activation is not involved in IL-1α secretion in S. pneumoniae-infected macrophages.
It was reported previously that several inflammasome activators induce the maturation and secretion of IL-1α in a manner dependent on inflammasome components, including ASC (apoptosis-associated speck-like protein containing a CARD [caspase activation and recruitment domain]) and caspase-1 (30–33). We previously reported that in S. pneumoniae-infected macrophages, the AIM2 and NLRP3 inflammasomes play a critical role in caspase-1 activation and the subsequent processing and secretion of IL-18 and IL-1β (11). Based on these assumptions, we hypothesized that S. pneumoniae induces IL-1α production through inflammasome activation. To test this possibility, macrophages from wild-type (WT), caspase-1−/−, and ASC−/− mice were infected with S. pneumoniae D39, and IL-1α secretion was assessed. Comparable levels of IL-1α were detected in WT, caspase-1−/−, and ASC−/− macrophages (Fig. 3A), and the levels of TNF-α were also similar among them (Fig. 3B). IL-1β production was almost completely abrogated in caspase-1−/− and ASC−/− macrophages, confirming that these inflammasome proteins are essential for this response (Fig. 3C). Data from Western blot analysis also suggested that there was no significant difference in the mature form of IL-1α in culture supernatants from caspase-1−/− and ASC−/− macrophages compared to WT macrophages (Fig. 3D). Our previous report demonstrated that S. pneumoniae induction of inflammasome activation requires the phagocytosis of bacterial cells by host macrophages (11). In this study, macrophages were pretreated with cytochalasin B, which abrogates phagocytosis by blocking actin polymerization, and mature IL-1α and IL-1β secreted into culture supernatant were detected by an ELISA (Fig. 3E) and Western blotting (Fig. 3F). Cytochalasin B treatment markedly reduced the secretion and maturation of IL-1α and IL-1β, suggesting that S. pneumoniae-induced IL-1α production also requires bacterial uptake by host macrophages. Taken together, these results indicated that inflammasome activation is dispensable but that bacterial entry into macrophages is important for the secretion of IL-1α in S. pneumoniae-infected macrophages, and it can thus be speculated that S. pneumoniae triggers at least two different pathways after being engulfed by macrophages: one is inflammasome activation, which is required for IL-1β processing, and another is an inflammasome-independent pathway involved in IL-1α processing.
FIG 3.

Inflammasomes are not involved in IL-1α secretion. Macrophages from WT, caspase-1−/−, and ASC−/− mice were infected with D39 at an MOI of 1. (A to C) The levels of IL-1α, TNF-α, and IL-1β in the culture supernatants were detected by an ELISA. (D) In addition, Western blotting was carried out to detect pro-IL-1α in the cell lysate and mature IL-1α in the supernatant 24 h after infection. (E) Macrophages were treated with 10 μM cytochalasin B 30 min prior to infection, and the level of IL-1α production was measured 24 h after infection. (F) Furthermore, the mature forms of IL-1α and IL-1β were detected by Western blotting in the culture supernatant. β-Actin was detected as a loading control. All of the experiments were repeated three times. Statistical significance was determined by using one-way ANOVA. *, P < 0.05. DMSO, dimethyl sulfoxide.
Calpain activation is required for IL-1α secretion in S. pneumoniae-infected macrophages.
Calpain is a calcium-dependent cysteine protease, which has been shown to process pro-IL-1α into the mature form in response to various stimuli (23, 24, 34). Intracellular bacteria, such as Mycobacterium tuberculosis and Listeria monocytogenes, have been suggested to induce the secretion of mature IL-1α from host macrophages in a calpain-dependent manner (26, 27). We hypothesized that in S. pneumoniae-infected macrophages, PLY might participate in calpain activation, thereby facilitating IL-1α secretion. The effects of calpain inhibitors {calpain inhibitor III, calpain inhibitor IV, and EST [(2S,3S)-trans-epoxysuccinyl-l-leucylamido-3-methylbutane ethyl ester]} on IL-1α secretion were therefore examined, and all the inhibitors markedly inhibited IL-1α secretion in D39-infected macrophages. In addition, the intracellular calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM) inhibited IL-1α secretion (Fig. 4A). Meanwhile, these reagents did not affect TNF-α secretion in S. pneumoniae-infected macrophages (Fig. 4B). These results indicated that calpain is involved in IL-1α secretion during S. pneumoniae infection. α-Fodrin is a 240-kDa cytoskeletal protein, and its cleavage into a 145-kDa fragment is indicative of calpain activation (25). Next, we investigated the degradation of α-fodrin in S. pneumoniae-infected macrophages. As shown in Fig. 4C, various calpain inhibitors and BAPTA-AM significantly reduced the degradation of α-fodrin. These results were consistent with the reduction of the level of IL-1α secretion. In addition, the calpain-cleaved fragment of α-fodrin was detected in the culture supernatant after infection with D39 but was almost not detected in that of Δply strain-infected macrophages (Fig. 4D), suggesting that PLY-expressing S. pneumoniae induces calpain activation in host macrophages. Thus, it appeared that calpain is involved in IL-1α secretion from D39-infected macrophages and that PLY plays a critical role in calpain activation.
FIG 4.
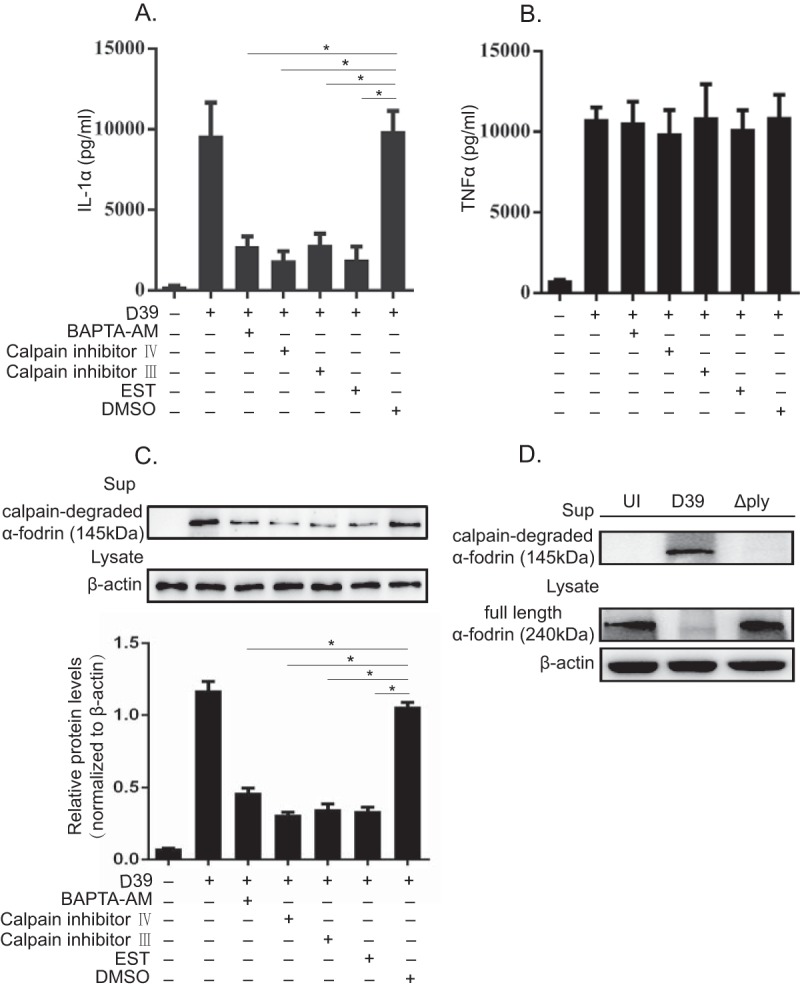
Calpain inhibitors markedly inhibit IL-1α secretion from D39-infected macrophages. (A and B) Macrophages were infected with D39 at an MOI of 1. Subsequently, cells were treated with 20 μM BAPTA-AM and various calpain inhibitors 3 h after infection in the presence of gentamicin, and the culture was continued for a further 21 h. The culture supernatant was collected, and the levels of IL-1α (A) and TNF-α (B) were measured by an ELISA. (C) In addition, the proteolytic fragment of α-fodrin was detected in the culture supernatant by Western blotting. β-Actin was detected as a loading control. (D) Macrophages were infected with the D39 and Δply strains at an MOI of 1 for 6 h and cultured in the presence of gentamicin for 18 h. The full-length form and the proteolytic fragment of α-fodrin were detected in the cell lysate and the culture supernatant, respectively, by Western blotting. The activity of calpain was monitored by measuring the amount of the degraded fragment of α-fodrin. β-Actin was utilized as a loading control. All of the experiments were repeated three times. Tests for statistical significance were performed by using one-way ANOVA followed by the Bonferroni test. *, P < 0.05.
Infection with PLY-expressing S. pneumoniae increases intracellular Ca2+ levels in host macrophages.
The activity of calpain is calcium dependent, and an elevation of intracellular calcium levels triggers the activation of calpain (24, 25). The chelation of extracellular Ca2+ using EDTA and EGTA-Mg2+ significantly decreased D39-induced IL-1α secretion, while TNF-α secretion was not affected, suggesting that Ca2+ is involved in calpain activation (Fig. 5A and B). After the addition of EDTA and EGTA-Mg2+, the degradation of α-fodrin was significantly reduced, which was consistent with the reduction of the level of IL-1α secretion (Fig. 5C). We also found that there was a significant reduction in IL-1α secretion in the culture supernatants of macrophages infected with D39 in calcium-free medium (Fig. 5D). Furthermore, Δply strain-infected macrophages were treated with the calcium ionophore A23187, and this treatment significantly enhanced the secretion of IL-1α, verifying that the elevation of intracellular Ca2+ levels is sufficient for the secretion of IL-1α in S. pneumoniae-infected macrophages (Fig. 5E). Next, we analyzed the kinetics of cytosolic Ca2+ levels in macrophages infected with the D39 or Δply strain using the Ca2+ indicator Fluo-4. Infection of Fluo-4-loaded macrophages with the WT D39 strain resulted in a significant increase in the fluorescence intensity. In contrast, the Δply strain hardly increased the fluorescence intensity (Fig. 5F). Taken together, these data suggested that PLY contributes to the increased intracellular Ca2+ levels in S. pneumoniae-infected macrophages, followed by calpain activation and IL-1α secretion.
FIG 5.

Involvement of elevated intracellular calcium concentrations in IL-1α secretion. (A and B) Macrophages were infected with D39 and/or the Δply strain at an MOI of 1. Subsequently, cells were treated with 10 mM EDTA and 10 mM EGTA plus 0.7 mM Mg2+ 3 h after infection in the presence of gentamicin, and the culture was continued for a further 21 h. The culture supernatants were collected, and the levels of IL-1α (A) and TNF-α (B) were measured. (C) The full-length form and the proteolytic fragment of α-fodrin were detected by Western blotting in the cell lysate and the culture supernatant. β-Actin was utilized as a loading control. (D) Alternatively, macrophages were infected with D39 for 6 h and cultured in calcium-free (Ca2+-free) or calcium-containing (+Ca2+) RPMI 1640 medium. HBSS, Hanks' balanced salt solution. (E) Similarly, 10 μM the reagent A23187 was added 3 h after infection in the presence of gentamicin, and the culture was continued for a further 21 h. The culture supernatants were collected, and the level of IL-1α was measured by an ELISA. Data represent the means and the standard deviations of results from triplicate assays and are representative of data from three independent experiments. (F) Macrophages were infected with the D39 and Δply strains at an MOI of 1. The intracellular calcium concentration was monitored every 3 h after infection for 24 h by using a Fluo-4 NW calcium assay kit (Invitrogen) according to the manufacturer's instructions. All of the experiments were repeated three times. Statistical significance was determined by Student's t test. *, P < 0.05.
DISCUSSION
In the present study, we elucidated the role of PLY in IL-1α secretion in S. pneumoniae-infected macrophages. We found that the secretion of the mature form of IL-1α in macrophages upon S. pneumoniae infection is critically dependent on PLY. It was also suggested that PLY is critical for the increases in intracellular calcium levels and calpain activation in S. pneumoniae-infected macrophages, which are responsible for the processing and secretion of mature IL-1α. Importantly, proinflammatory cytokines that are produced in a PLY-dependent manner contribute not only to host defense but also to pathogenesis in pneumococcal infections, and IL-1α has been suggested to have a role in tissue damage in a model of pneumococcal pneumonia. Accordingly, this study revealed the mechanism of S. pneumoniae-induced IL-1α production, which would explain, at least in part, how PLY mediates immunopathology.
Previous studies have shown that S. pneumoniae induced the AIM2 and NLRP3 inflammasomes, leading to caspase-1 activation and the subsequent maturation and secretion of IL-1β and IL-18. It has also been reported that inflammasome-activating stimuli triggered the secretion of IL-1α (30–33). Gross et al. showed that caspase-1 contributed to the processing and secretion of IL-1α independently of its proteolytic activity, but IL-1α secretion was not universally inflammasome dependent, as several particulate NLRP3 activators induced it in the absence of NLRP3, caspase-1, or ASC (30). Zheng et al. proposed that IL-1 receptor 2 (IL-1R2) interacts with pro-IL-1α, which prevents IL-1α maturation by calpain, and that caspase-1 cleaves IL-1R2 to resolve this inhibition (33). However, Dewamitta et al. reported that the maturation of IL-1α in macrophages infected by Listeria monocytogenes is independent of caspase-1 and ASC (26). Our study also showed that the maturation and secretion of IL-1α were normally observed in caspase-1−/− and ASC−/− macrophages infected with S. pneumoniae, indicating that the inflammasome is dispensable for IL-1α secretion. Hence, it appears that S. pneumoniae is capable of activating not only inflammasomes but also an inflammasome-independent pathway that leads to the maturation and secretion of IL-1α.
Since PLY is a pore-forming toxin, one simple interpretation for PLY-dependent IL-1α production is that it mediates calcium influx by forming pores on the cell membrane of host cells, resulting in calpain activation. As it lacks a typical signal sequence, PLY is not a secreted protein and is released after cell wall degradation, for example, by autolysis. Moreover, in this study, phagocytosis of bacteria by host macrophages was involved in S. pneumoniae-induced IL-1α production. We and others previously suggested that S. pneumoniae cells internalized by macrophages underwent rapid death and released PLY, thereby destabilizing phagosomal membranes in a PLY-dependent manner (11, 35). Interestingly, phagosomal destabilization by particulate NLRP3 activators has been proposed to facilitate the production of IL-1α independently of the inflammasome machinery (30). From those observations, it is also conceivable that PLY released from internalized pneumococci promotes IL-1α production through the destabilization of phagosomal membranes. Therefore, multiple PLY-dependent mechanisms, including the formation of Ca2+-permeable pores, inflammasome activation, and phagosomal destabilization, could contribute to IL-1α production in response to S. pneumoniae, and further study is needed to clarify the mechanism of PLY-dependent IL-1α production during pneumococcal infection.
Calpains have been defined as intracellular calcium-dependent cysteine proteases, which participate in a variety of signal transduction pathways (25). Several reports have shown that the calcium-dependent activation of calpain is required for the processing of pro-IL-1α, with special regard to bacterial infection, such as infections by Listeria monocytogenes and Mycobacterium tuberculosis (26, 27). Here we found that this process also similarly exists in macrophages upon S. pneumoniae infection. These bacteria have virulence factors involved in membrane permeabilization, which are the type VII secretion system ESX-1 of M. tuberculosis, listeriolysin O of L. monocytogenes, and PLY of S. pneumoniae. It is noteworthy that all these virulence factors play a critical role in IL-1α maturation and secretion upon infection by each bacterial pathogen. Taken together, IL-1α maturation and secretion, dependent on calcium ion influx, and calpain activation might be common outcomes of bacterial infections, accompanied by membrane disruption, especially with phagosomal membrane destabilization.
In conclusion, data from our study clearly indicated that in S. pneumoniae-infected macrophages, PLY was not involved in IL-1α mRNA expression and pro-IL-1α synthesis, but PLY was critically involved in the maturation and secretion of IL-1α. PLY caused an elevation of the intracellular calcium level, calpain activation, and, finally, the maturation and secretion of IL-1α. These findings provide further insight into the interactions between PLY and the host cellular response to S. pneumoniae infection.
MATERIALS AND METHODS
Reagents.
rPLY was purchased from Fitzgerald (USA). Cholesterol was purchased from Sigma-Aldrich (St. Louis, MO). EDTA and EGTA-MgCl2 were purchased from Beyotime (Beijing, China). BAPTA-AM and A23187 were purchased from Sigma-Aldrich (St. Louis, MO). The calpain inhibitors carbobenzoxy-valyl-phenylalanial (calpain inhibitor III), (2S,3S)-trans-epoxysuccinyl-l-leucylamido-3-methylbutane ethyl ester (EST), and N-benzyloxycarbonyl-l-leucyl-l-leucyl-l-tyrosylfluoromethyl ketone (calpain inhibitor IV) were obtained from Calbiochem (Darmstadt, Germany). Cytochalasin B was purchased from Solarbio (Beijing, China).
Mice.
Female C57BL/6 mice were purchased from the Chongqing Academy of Chinese Materia Medical, China. Caspase-1−/− mice and mice deficient for apoptosis-associated speck-like protein containing a CARD (ASC−/− mice) on a C57BL/6 background were kindly provided by Feng Shao from the NIBS (National Institute of Biological Sciences, Beijing, China). The animals were maintained under specific-pathogen-free conditions and used at 7 to 9 weeks of age. All animal experiments were approved by the Ethics Committee of Southwest University and in compliance with laboratory animal care principles of the National Institutes of Health, China.
Bacteria.
Streptococcus pneumoniae D39 (serotype 2) and the ply gene deletion mutant (Δply) were kindly provided by Kohsuke Tsuchiya (Kanazawa University, Japan). Bacteria were grown overnight on tryptic soy agar (Hope Biotech, Qingdao, China) with 5% (vol/vol) defibrinated sheep blood at 37°C and 5% CO2. Colonies were inoculated into Todd-Hewitt broth (Hope Biotech, Qingdao, China) supplemented with 0.5% yeast extract (THY), grown until the mid-logarithmic phase (optical density at 600 nm [OD600] = 0.5), and centrifuged at 6,000 × g for 15 min. The bacterial pellet was suspended in phosphate-buffered saline (PBS) and stocked at −80°C. The concentration was determined by colony counting on blood agar plates.
Macrophages.
Mice were injected intraperitoneally with 3 ml of thioglycolate medium (Eiken, Japan), and peritoneal exudate cells (PECs) were obtained by peritoneal lavage 3 days later. PECs were washed and suspended in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and incubated at a density of 2.5 × 105 cells/well in 48-well tissue culture microplates at 37°C plus 5% CO2. After incubation for 2 h, nonadherent cells were removed, and adherent cells were used as macrophages. More than 95% of the adherent PECs were F4/80+ macrophages, as determined by flow cytometry.
ELISA.
Macrophages were infected with S. pneumoniae D39 and/or the Δply mutant at a multiplicity of infection (MOI) of 1 for 6 h, 100 μg/ml gentamicin (Beyotime, Beijing, China) was then added to the cultures, and the cultures were incubated for an additional 18 h. In one experiment, macrophages were infected with the D39 and Δply strains, and culture supernatants were collected 6, 12, and 24 h after incubation. Meanwhile, cells were lysed in PBS containing 1% Triton X-100 at each time point. The lysate was centrifuged to prepare the cleared cell lysate. In the experiments to determine the effects of 10 mM EDTA, 10 mM EGTA plus 0.7 mM Mg2+, 20 μM BAPTA-AM, 10 μM A23187, and various calpain inhibitors, the reagents were added 3 h after infection in the presence of gentamicin, and the culture was continued for a further 21 h. In addition, macrophages were treated with 10 μM cytochalasin B 30 min prior to infection, and cytokine production was measured 24 h after infection. Levels of cytokines in all the culture supernatants and cell lysates mentioned above were determined by a two-site sandwich ELISA. ELISA kits for IL-1α and TNF-α were purchased from eBioscience (San Diego, CA).
Western blot analysis.
Macrophages were infected with the D39 and Δply strains at an MOI of 1 for 6 h, and gentamicin (100 μg/ml) was added. The culture supernatants were collected 24 h after infection, and the cells were lysed with radioimmunoprecipitation assay (RIPA) buffer (Beyotime, China). The supernatants and the cell lysates were subjected to SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane by electroblotting. The membranes were immunoblotted with anti-IL-1α and anti-IL-1β (R&D Systems, Minneapolis, MN), streptavidin-horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Cwbio, Beijing, China), anti-α-fodrin monoclonal antibody (Abcam, Cambridge, UK), and HRP-conjugated goat anti-rabbit IgG(H+L) antibody (Beyotime, China). β-Actin was employed as a loading control for the cell lysates and was detected by mouse anti-β-actin antibody (Beyotime, China) and HRP-conjugated goat anti-mouse IgG antibody (Cwbio, Beijing, China).
Real-time RT-PCR.
Macrophages were infected with the D39 and Δply strains at an MOI of 1 for 3, 6, and 9 h. Total RNA was extracted by using the RNA Pre-Pure kit (Tiangen, Beijing, China). Total RNA (0.2 μg) was treated with RNase-free DNase (Tiangen) and subjected to reverse transcription (RT) using a 250R iSCRIPT advanced cDNA synthesis kit (Bio-Rad). Quantitative real-time RT-PCR was performed on a Bio-Rad CFX 96 instrument using SsoFast Eva Green Super-Mix (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. Primers for quantitative real-time RT-PCR were as follows: 5′-CTCTAGAGCACCATGCTACAGAC-3′ (forward) and 5′-TGGAATCCAGGGGAAACACTG-3′ (reverse) for IL-1α and 5′-TGGAATCCTGTGGCATCCATGAAAC-3′ (forward) and 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′ (reverse) for β-actin.
Measurement of intracellular calcium levels.
Macrophages were plated at 1.25 × 105 cells/well in 96-well black microplates (Invitrogen) for 2 h and infected with the D39 and Δply strains at an MOI of 1. The intracellular calcium concentration was monitored every 3 h after infection for 24 h by using a Fluo-4 NW calcium assay kit (Invitrogen) according to the manufacturer's instructions.
Statistical analysis.
For comparisons between two groups, Student's t test was used. For multigroup comparisons, analysis of variance (ANOVA) and the Bonferroni post hoc test were used. Statistical significance was determined as a P value of <0.05.
ACKNOWLEDGMENTS
We thank Feng Shao from the NIBS (National Institute of Biological Sciences, Beijing, China) for providing the caspase-1−/− and ASC−/− mice.
This work was supported by the National Natural Science Foundation of China (grant 31400762), MEXT/JSPS KAKENHI grant number JP 26460523, the Chongqing Science and Technology Commission (grants cstc2015jcyjBX0108, cstc2015shmszx80010, and cstc2015shmszx80022), the earmarked fund for the China Agriculture Research System (CARS-38), the Fundamental Research Funds for the Central Universities (grant XDJK2015B002), and the National University Students Innovation Projects (grant 201610635020).
We declare no conflicts of interest.
REFERENCES
- 1.Henriques-Normark B, Tuomanen EI. 2013. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med 3:a010215. doi: 10.1101/cshperspect.a010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tuomanen EI, Austrian R, Masure HR. 1995. Pathogenesis of pneumococcal infection. N Engl J Med 332:1280–1284. doi: 10.1056/NEJM199505113321907. [DOI] [PubMed] [Google Scholar]
- 3.Kadioglu A, Weiser JN, Paton JC, Andrew PW. 2008. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol 6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- 4.Marriott HM, Mitchell TJ, Dockrell DH. 2008. Pneumolysin: a double-edged sword during the host-pathogen interaction. Curr Mol Med 8:497–509. doi: 10.2174/156652408785747924. [DOI] [PubMed] [Google Scholar]
- 5.Alouf JE. 2000. Cholesterol-binding cytolytic protein toxins. Int J Med Microbiol 290:351–356. doi: 10.1016/S1438-4221(00)80039-9. [DOI] [PubMed] [Google Scholar]
- 6.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A 100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houldsworth S, Andrew PW, Mitchell TJ. 1994. Pneumolysin stimulates production of tumor necrosis factor alpha and interleukin-1 beta by human mononuclear phagocytes. Infect Immun 62:1501–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shoma S, Tsuchiya K, Kawamura I, Nomura T, Hara H, Uchiyama R, Daim S, Mitsuyama M. 2008. Critical involvement of pneumolysin in production of interleukin-1alpha and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infect Immun 76:1547–1557. doi: 10.1128/IAI.01269-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Petrilli V, Andrew PW, Kadioglu A, Lavelle EC. 2010. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog 6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, Holmes A, Trendelenburg G, Heimesaat MM, Bereswill S, van der Linden M, Tschopp J, Mitchell TJ, Suttorp N, Opitz B. 2011. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol 187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 11.Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, Yamamoto T, Fernandes-Alnemri T, Yang R, Hernandez-Cuellar E, Dewamitta SR, Xu Y, Qu H, Alnemri ES, Mitsuyama M. 2011. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J Immunol 187:4890–4899. doi: 10.4049/jimmunol.1100381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fang R, Hara H, Sakai S, Hernandez-Cuellar E, Mitsuyama M, Kawamura I, Tsuchiya K. 2014. Type I interferon signaling regulates activation of the absent in melanoma 2 inflammasome during Streptococcus pneumoniae infection. Infect Immun 82:2310–2317. doi: 10.1128/IAI.01572-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kafka D, Ling E, Feldman G, Benharroch D, Voronov E, Givon-Lavi N, Iwakura Y, Dagan R, Apte RN, Mizrachi-Nebenzahl Y. 2008. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol 20:1139–1146. doi: 10.1093/intimm/dxn071. [DOI] [PubMed] [Google Scholar]
- 14.Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. 2005. Lung NF-kappaB activation and neutrophil recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. J Immunol 175:7530–7535. doi: 10.4049/jimmunol.175.11.7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, Van Furth AM. 2003. IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J Immunol 170:4724–4730. doi: 10.4049/jimmunol.170.9.4724. [DOI] [PubMed] [Google Scholar]
- 16.Rijneveld AW, Florquin S, Branger J, Speelman P, Van Deventer SJ, van der Poll T. 2001. TNF-alpha compensates for the impaired host defense of IL-1 type I receptor-deficient mice during pneumococcal pneumonia. J Immunol 167:5240–5246. doi: 10.4049/jimmunol.167.9.5240. [DOI] [PubMed] [Google Scholar]
- 17.Lauw FN, Branger J, Florquin S, Speelman P, van Deventer SJ, Akira S, van der Poll T. 2002. IL-18 improves the early antimicrobial host response to pneumococcal pneumonia. J Immunol 168:372–378. doi: 10.4049/jimmunol.168.1.372. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell AJ, Yau B, McQuillan JA, Ball HJ, Too LK, Abtin A, Hertzog P, Leib SL, Jones CA, Gerega SK, Weninger W, Hunt NH. 2012. Inflammasome-dependent IFN-gamma drives pathogenesis in Streptococcus pneumoniae meningitis. J Immunol 189:4970–4980. doi: 10.4049/jimmunol.1201687. [DOI] [PubMed] [Google Scholar]
- 19.Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG, Camilli A, Kadioglu A, Dubyak GR, Pearlman E. 2015. Neutrophil IL-1beta processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J Immunol 194:1763–1775. doi: 10.4049/jimmunol.1401624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, Azam T, Kim S, Dinarello CA. 2013. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol 4:391. doi: 10.3389/fimmu.2013.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Luthi AU, Reeves EP, McElvaney NG, Medema JP, Lavelle EC, Martin SJ. 2011. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol Cell 44:265–278. doi: 10.1016/j.molcel.2011.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garlanda C, Dinarello CA, Mantovani A. 2013. The interleukin-1 family: back to the future. Immunity 39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi Y, Yamamoto K, Saido T, Kawasaki H, Oppenheim JJ, Matsushima K. 1990. Identification of calcium-activated neutral protease as a processing enzyme of human interleukin 1 alpha. Proc Natl Acad Sci U S A 87:5548–5552. doi: 10.1073/pnas.87.14.5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carruth LM, Demczuk S, Mizel SB. 1991. Involvement of a calpain-like protease in the processing of the murine interleukin 1 alpha precursor. J Biol Chem 266:12162–12167. [PubMed] [Google Scholar]
- 25.Goll DE, Thompson VF, Li H, Wei W, Cong J. 2003. The calpain system. Physiol Rev 83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 26.Dewamitta SR, Nomura T, Kawamura I, Hara H, Tsuchiya K, Kurenuma T, Shen Y, Daim S, Yamamoto T, Qu H, Sakai S, Xu Y, Mitsuyama M. 2010. Listeriolysin O-dependent bacterial entry into the cytoplasm is required for calpain activation and interleukin-1 alpha secretion in macrophages infected with Listeria monocytogenes. Infect Immun 78:1884–1894. doi: 10.1128/IAI.01143-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang R, Xi C, Sita DR, Sakai S, Tsuchiya K, Hara H, Shen Y, Qu H, Fang R, Mitsuyama M, Kawamura I. 2014. The RD1 locus in the Mycobacterium tuberculosis genome contributes to the maturation and secretion of IL-1alpha from infected macrophages through the elevation of cytoplasmic calcium levels and calpain activation. Pathog Dis 70:51–60. doi: 10.1111/2049-632X.12075. [DOI] [PubMed] [Google Scholar]
- 28.England H, Summersgill HR, Edye ME, Rothwell NJ, Brough D. 2014. Release of interleukin-1alpha or interleukin-1beta depends on mechanism of cell death. J Biol Chem 289:15942–15950. doi: 10.1074/jbc.M114.557561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arend WP, Palmer G, Gabay C. 2008. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- 30.Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, Quadroni M, Drexler SK, Tschopp J. 2012. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 31.Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, Contassot E, Bachmann MF, French LE, Oxenius A, Kundig TM. 2011. Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci U S A 108:18055–18060. doi: 10.1073/pnas.1109176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keller M, Ruegg A, Werner S, Beer HD. 2008. Active caspase-1 is a regulator of unconventional protein secretion. Cell 132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 33.Zheng Y, Humphry M, Maguire JJ, Bennett MR, Clarke MC. 2013. Intracellular interleukin-1 receptor 2 binding prevents cleavage and activity of interleukin-1alpha, controlling necrosis-induced sterile inflammation. Immunity 38:285–295. doi: 10.1016/j.immuni.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar V, Everingham S, Hall C, Greer PA, Craig AW. 2014. Calpains promote neutrophil recruitment and bacterial clearance in an acute bacterial peritonitis model. Eur J Immunol 44:831–841. doi: 10.1002/eji.201343757. [DOI] [PubMed] [Google Scholar]
- 35.Koppe U, Hogner K, Doehn JM, Muller HC, Witzenrath M, Gutbier B, Bauer S, Pribyl T, Hammerschmidt S, Lohmeyer J, Suttorp N, Herold S, Opitz B. 2012. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J Immunol 188:811–817. doi: 10.4049/jimmunol.1004143. [DOI] [PubMed] [Google Scholar]