

ABSTRACT
Porphyromonas gulae is an anaerobic, Gram-negative coccobacillus that has been associated with periodontal disease in companion animals. The aims of this study were to analyze the ligation of pattern recognition receptors by P. gulae and the subsequent activation of macrophages. Exposure of HEK cells transfected with Toll-like receptors (TLRs) or NOD-like receptors to P. gulae resulted in the ligation of TLR2, TLR4, and NOD2. The effects of this engagement of receptors were investigated by measuring the synthesis of nitric oxide (NO), CD86 expression, and inflammatory cytokine production by wild-type, TLR2−/−, and TLR4−/− macrophages. The addition of P. gulae to unprimed and gamma interferon (IFN-γ)-primed (M1 phenotype) macrophages significantly increased the surface expression of CD86, but only M1 macrophages produced nitric oxide. P. gulae-induced expression of CD86 on unprimed macrophages was dependent on both TLR2 and TLR4, but CD86 expression and NO production in M1 macrophages were only TLR2 dependent. P. gulae induced an increase in secretion of interleukin-1α (IL-1α), IL-1β, IL-6, IL-12p70, IL-13, tumor necrosis factor alpha (TNF-α), granulocyte colony-stimulating factor (G-CSF), monocyte chemoattractant protein 1 (MCP-1), and macrophage inflammatory protein 1α (MIP-1α) by M1 macrophages compared to that by unprimed controls. Among these cytokines, secretion of IL-6 and TNF-α by M1 macrophages was dependent on either TLR2 or TLR4. Our data indicate that TLR2 and TLR4 are important for P. gulae activation of unprimed macrophages and that activation and effector functions induced in M1 macrophages by P. gulae are mainly dependent on TLR2. In conclusion, P. gulae induces a strong TLR2-dependent inflammatory M1 macrophage response which may be important in establishing the chronic inflammation associated with periodontal disease in companion animals.
KEYWORDS: periodontal disease, companion animals, Porphyromonas gulae, pattern recognition receptors
INTRODUCTION
Companion animals, such as dogs and cats, are susceptible to periodontal disease, a chronic inflammatory disease of the tissues supporting the teeth (1). Severe forms of the disease involve destruction of the supporting tissues, including alveolar bone, eventually leading to tooth loss (2). In humans, the black-pigmented bacterium Porphyromonas gingivalis has been shown to be associated with the onset and progression of chronic periodontitis, a severe form of the disease. In companion animals (dogs and cats), the incidence of periodontal disease has been shown to increase with age, and in a North American study, 82% of dogs aged 6 to 8 years old had disease (3).
Of the bacteria isolated from subgingival plaque from companion animals, three Porphyromonas species are most frequently isolated from dogs with periodontal disease: Porphyromonas gulae, Porphyromonas salivosa, and Porphyromonas denticanis (4–7). Of these, P. gulae has been shown to be present in 92% of animals with periodontitis (1). P. gulae is an anaerobic, Gram-negative coccobacillus that exhibits some biochemical and antigenic similarities to P. gingivalis (5, 7–9). Although P. gulae is commonly isolated from dogs with periodontal disease, the disease in companion animals most likely is similar to that in humans, in that it is a polymicrobial disease in which Porphyromonas spp. play an important, immune-dysregulating function (10). Furthermore, we previously showed that of several Porphyromonas species isolated from companion animals with periodontal disease, only P. gulae possesses arginine- and lysine-specific proteinases similar to the key virulence factors of P. gingivalis (11). This finding is consistent with P. gulae playing an important role in the initiation and progression of periodontal disease in companion animals.
Dogs are commonly used as animal models of human periodontal disease, using either the ligature model or infection with P. gingivalis (12). However, limited information is available concerning the interactions between immune cells and companion animal periodontal pathogens, such as P. gulae. In humans, chronic periodontitis is characterized by an inflammatory cell infiltrate of the gingival tissue, in which macrophages are a significant component (13). Furthermore, macrophages are often associated with other chronic inflammatory conditions, indicating that they play an important role in initiating and sustaining inflammation (14, 15). Therefore, the interaction of pathogens with macrophages is an important area of investigation to understand the induction of inflammation in periodontitis.
Inflammation typically begins after interaction of host innate immune cells, such as resident tissue macrophages, with pathogen-associated molecular patterns (PAMPs) via pattern recognition receptors (PRRs), such as NOD-like receptors (NLRs) or Toll-like receptors (TLRs). Ligation of TLRs on the macrophage surface by bacterial PAMPs, such as lipopolysaccharide (LPS) or lipoproteins, leads to macrophage activation (16). This activation results in the upregulation of antimicrobial compounds, increased antigen presentation capacity, and secretion of proinflammatory cytokines and chemokines. There have been limited studies regarding the interaction of P. gulae with cells of the innate immune system, such as macrophages, and no research, to date, has investigated the activation of individual TLRs. Recent research into macrophage activation has focused on the phenotypes developed during disease as a result of the cytokine environment at the time of activation (17, 18).
The presence of gamma interferon (IFN-γ), an important cytokine present during chronic inflammatory diseases, during macrophage activation polarizes macrophages toward an M1 phenotype (pre-M1-Mϕ), and when these cells are exposed to LPS, they mature into classically activated M1 macrophages (M1-Mϕ) (19). These macrophages exhibit greater antibacterial features than those of unprimed macrophages, increasing production of microbicidal compounds, such as nitric oxide. Classical inflammatory macrophages also have a greatly increased antigen presentation capacity, with increased expression of major histocompatibility complex (MHC) class II molecules and CD86, both of which are required for the initiation of an adaptive immune response.
We recently showed that P. gulae exposure activates mouse macrophages to produce interleukin-1α (IL-1α) and tumor necrosis factor alpha (TNF-α) at concentrations equivalent to those seen with P. gingivalis W50 but induces significantly higher concentrations of IL-6 (11). Here we characterize the PRR molecules with which P. gulae interacts and analyze how that PRR activation affects macrophage maturation.
RESULTS
Porphyromonas gulae activates transfected HEK cells through TLR2, TLR4, and NOD2.
The ligation of TLRs on immune cells is essential for induction of an immune response against pathogens. The activation of different TLRs and NOD receptors by P. gulae was investigated using HEK-Blue cells transfected with either NOD1, NOD2, TLR2, TLR4, TLR5, TLR7, TLR8, or TLR9 as a surface receptor, which if ligated activated NK-κB phosphorylation, resulting in secretion of alkaline phosphatase. HEK-Blue cells were incubated with P. gulae whole cells at various multiplicities of infection (MOI) for 20 h, and then the accumulation of alkaline phosphatase in the medium was analyzed by combining the medium with HEK-Blue detection medium.
P. gulae induced the phosphorylation of NF-κB in HEK-Blue cells transfected with TLR2, TLR4, or NOD2 (Fig. 1). No other TLRs or NOD receptors were activated at a significant level (data not shown). HEK-Blue (Null1; untransfected) cells were used as a control for endogenous TLRs and NOD receptors and were not activated by P. gulae or any TLR or NOD ligand (Fig. 1).
FIG 1.
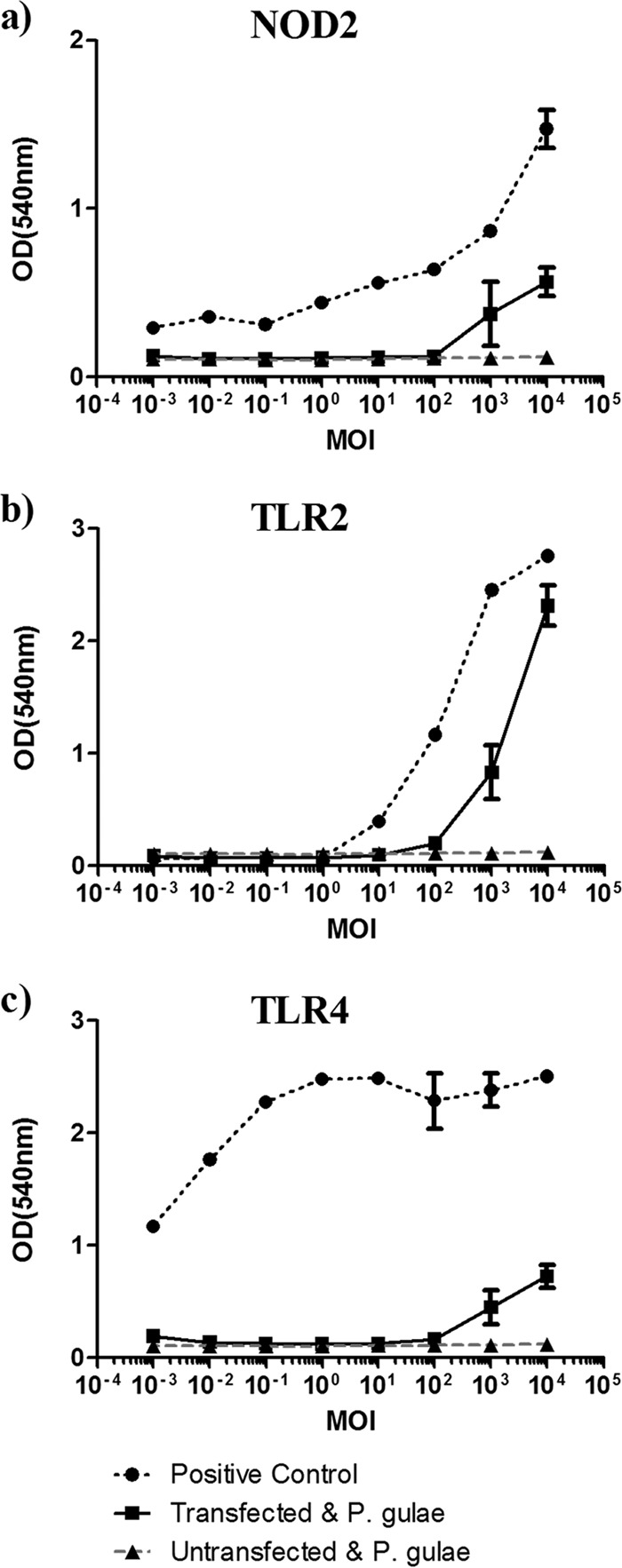
TLR activation by Porphyromonas gulae whole cells. HEK-Blue cells transfected with the pattern recognition receptor NOD2 (a), TLR2 (b), or TLR4 (c) (2 × 104 cells/well) were incubated with P. gulae whole cells at various MOI for 2 h and then washed with DMEM containing antibiotics. After 24 h, the amount of alkaline phosphatase secreted into the medium was measured as a direct correlation of NF-κB phosphorylation. The assay was repeated twice on each of three biological replicates (n = 6). HEK-Blue cells expressing NOD1, TLR5, TLR7, TLR8, or TLR9 were also tested but did not respond to whole P. gulae cells.
Porphyromonas gulae increases the expression of CD86 on M0 and M1 macrophages.
As TLR ligation is an essential step in macrophage maturation, the overall activation state of macrophages incubated with P. gulae was analyzed by the cell surface expression of CD86, which we have shown in previous studies to be strongly upregulated in activated macrophages, along with CD80, CD40, and MHC class II activation markers (20). CD86 expression was measured by flow cytometry on C57/BL6 (wild type [WT]) immortalized macrophages (iMACs), TLR2 knockout (TLR2−/−) iMACs, TLR4 knockout (TLR4−/−) iMACs, or NOD2 knockdown iMACs after incubation with P. gulae and control TLR ligands. The experiment was performed on unprimed M0-Mϕ and IFN-γ-primed pre-M1-Mϕ. Escherichia coli LPS (TLR4 ligand) and Pam3CSK4 (TLR2 ligand) were used as TLR activation controls. Following P. gulae and TLR ligand incubation, macrophages were stained with anti-mouse CD86–phycoerythrin (PE)–Cy7 antibody and analyzed by flow cytometry. P. gulae and both TLR ligands induced significant (P < 0.05) increases in CD86 expression on unprimed WT M0-Mϕ (Fig. 2a). P. gulae-induced expression of CD86 was completely abrogated on TLR2−/− and TLR4−/− unprimed M0-Mϕ (Fig. 2c and e). Reducing the expression of NOD2 on naive iMACs through small interfering RNA (siRNA) interference did not appreciably decrease the expression of CD86 (Fig. 2g).
FIG 2.

Expression of CD86 on macrophages after exposure to Porphyromonas gulae whole cells. WT iMACs, TLR2−/− iMACs, and TLR4−/− iMACs (105 cells) were incubated overnight with or without IFN-γ to prime the M1 macrophage phenotype or to give unprimed M0-Mϕ, respectively. The unprimed and M1-primed macrophages were then incubated with viable P. gulae (MOI of 180:1) for 2 h and washed with DMEM containing antibiotics. E. coli LPS and Pam3CSK4 were used as TLR4 and TLR2 control ligands, respectively. The negative control was macrophages that were not incubated with TLR ligands or P. gulae cells. After 24 h, the level of CD86 expression was measured by flow cytometry. Data are expressed as percentages of CD86-positive macrophages. The assay was repeated twice on each of three biological replicates (n = 6). (a) WT Mϕ; (b) WT M1-Mϕ; (c) TLR2−/− Mϕ; (d) TLR2−/− M1-Mϕ; (e) TLR4−/− Mϕ; (f) TLR4−/− M1-Mϕ; (g) NOD2−/− Mϕ; (h) NOD−/− M1-Mϕ. *, P < 0.05 compared to the corresponding unstimulated control.
Priming with IFN-γ to generate a pre-M1-Mϕ phenotype resulted in significantly higher levels of CD86 expression than those on unprimed M0-Mϕ, and incubation with P. gulae significantly (P < 0.05) increased CD86 expression compared to that on the pre-M1-Mϕ controls (Fig. 2b). CD86 expression on TLR4−/− but not TLR2−/− M1-Mϕ was significantly (P < 0.05) increased upon incubation with P. gulae compared to that of controls (Fig. 2d and f). Reducing the expression of NOD2 on M1 iMACs through siRNA interference did not appreciably decrease the expression of CD86 (Fig. 2h). Although the TLR4−/− macrophages exposed to P. gulae had less CD86 expression than that of the WT iMACs, the level of induction by the positive-control Pam3CSK4 ligand was correspondingly lower, indicating a lower overall responsiveness of these cells. The TLR2−/− and TLR4−/− iMACs showed increased expression only when they were incubated with E. coli LPS and Pam3CSK4, respectively, indicating the TLR specificity of the knockout iMACs.
Porphyromonas gulae induces nitric oxide production in macrophages.
Since P. gulae was able to induce the upregulation of CD86 in macrophages, functional assays were performed to quantify that activation. The ability of macrophages to produce nitric oxide in response to bacterial infection is essential for the bactericidal function of macrophages. Nitric oxide production was measured in WT, TLR2−/−, and TLR4−/− iMACs. The experiment was performed on unprimed M0-Mϕ and IFN-γ-treated pre-M1-Mϕ.
P. gulae was found not to induce nitric oxide production in unprimed M0-Mϕ, whereas the control TLR ligands E. coli LPS and Pam3CSK4 induced strong and TLR-specific responses (Fig. 3a, c, and e). However, M1-Mϕ (IFN-γ-treated pre-M1-Mϕ) incubated with P. gulae produced significantly (P < 0.05) larger amounts of nitric oxide than those in controls (Fig. 3b). The control TLR ligands induced significantly higher levels of nitric oxide than those induced by P. gulae; however, this could be attributed to the purity of the control ligands. TLR4−/− but not TLR2−/− M1-Mϕ produced significantly (P < 0.05) larger amounts of nitric oxide upon incubation with P. gulae than those seen with controls (Fig. 3d and f). The TLR2−/− and TLR4−/− iMACs responded only to E. coli LPS and Pam3CSK4, respectively, indicating that the cells were functioning correctly.
FIG 3.

Nitric oxide production by macrophages in response to Porphyromonas gulae whole cells. WT iMACs, TLR2−/− iMACs, and TLR4−/− iMACs (105 cells) were incubated overnight with or without IFN-γ to prime the M1 macrophage phenotype or to give unprimed M0-Mϕ, respectively. The unprimed and M1-primed macrophages were then incubated with viable P. gulae (MOI of 180:1) for 2 h and washed with DMEM containing antibiotics. E. coli LPS and Pam3CSK4 were used as TLR4 and TLR2 control ligands, respectively. The negative control was macrophages that were not incubated with TLR ligands or P. gulae cells. After 24 h, the level of nitric oxide expression was measured using the Griess reaction. The assay was repeated twice on each of three biological replicates (n = 6). (a) WT Mϕ; (b) WT M1-Mϕ; (c) TLR2−/− Mϕ; (d) TLR2−/− M1-Mϕ; (e) TLR4−/− Mϕ; (f) TLR4−/− M1-Mϕ; (g) NOD2−/− Mϕ; (h) NOD−/− M1-Mϕ. *, P < 0.05 compared to the corresponding unstimulated control.
Production of cytokines and chemokines by M0 and M1 polarized macrophages after incubation with P. gulae.
The maturation of M1 macrophages is characterized by the production of cytokines and chemokines which promote chronic inflammation. To investigate the cytokine response induced by P. gulae, we determined the concentrations of cytokines in the cell-free supernatants of WT, TLR2−/−, and TLR4−/− M1-Mϕ after incubation with P. gulae and TLR ligands by using a 23-plex Bioplex assay. Of the 23 cytokines and chemokines analyzed in the culture supernatant, only 14 (IL-1α, IL-1β, IL-6, IL-10, IL-12p70, IL-13, IL-17, TNF-α, KC, granulocyte colony-stimulating factor [G-CSF], RANTES, eotaxin, monocyte chemoattractant protein 1 [MCP-1], and macrophage inflammatory protein 1α [MIP-1α]) were consistently secreted by M1-Mϕ after incubation with P. gulae, E. coli LPS, or Pam3CSK4 (Fig. 4 and 5). None of the TLR ligands or P. gulae induced the secretion of IL-2, IL-3, IL-4, IL-5, IL-9, IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), or MIP-1β from M1-Mϕ (data not shown).
FIG 4.

Cytokine production by M1 macrophages in response to Porphyromonas gulae whole cells. WT iMACs, TLR2−/− iMACs, and TLR4−/− iMACs (105 cells) were incubated overnight with IFN-γ to prime the M1 macrophage phenotype. The M1-primed macrophages were then incubated with viable P. gulae (MOI of 180:1) for 2 h and washed with DMEM containing antibiotics. E. coli LPS and Pam3CSK4 were used as TLR4 and TLR2 control ligands, respectively. The negative control was IFN-γ-primed macrophages that were not incubated with TLR ligands or P. gulae cells. After 24 h, the levels of cytokines in the assay supernatants were measured using a 23-plex Bioplex assay. The assay was repeated twice on each of three biological replicates (n = 6). (a) IL-1α; (b) IL-1β; (c) IL-6; (d) IL-10; (e) IL-12p70; (f) IL-13; (g) IL-17; (h) TNF-α. *, P < 0.05 compared to the corresponding negative control.
FIG 5.

Chemokine production by M1 macrophages in response to Porphyromonas gulae whole cells. WT iMACs, TLR2−/− iMACs, and TLR4−/− iMACs (105 cells) were incubated overnight with IFN-γ to prime the M1 macrophage phenotype. The M1-primed macrophages were then incubated with viable P. gulae (MOI of 180:1) for 2 h and washed with DMEM containing antibiotics. E. coli LPS and Pam3CSK4 were used as TLR4 and TLR2 control ligands, respectively. The negative control was IFN-γ-primed macrophages that were not incubated with TLR ligands or P. gulae cells. After 24 h, the levels of chemokines in the assay supernatants were measured using a 23-plex Bioplex assay. The assay was repeated twice on each of three biological replicates (n = 6). (a) Eotaxin; (b) G-CSF; (c) KC; (d) MCP-1; (e) MIP-1α; (f) RANTES. *, P < 0.05 compared to the corresponding negative control.
The positive-control ligands E. coli LPS and Pam3CSK4 induced significant levels (P < 0.01) of IL-1α, IL-1β, IL-6, IL-10, IL-12p70, IL-13, IL-17, and TNF-α in M1-Mϕ compared to those in the IFN-γ-primed pre-M1-Mϕ cells (Fig. 4). The TLR2−/− and TLR4−/− M1-Mϕ produced the above cytokines only in response to E. coli LPS and Pam3CSK4, respectively, indicating that these cells were responding as expected. P. gulae whole cells induced significant (P < 0.01) levels of IL-1α, IL-1β, IL-6, IL-12p70, and TNF-α in M1-Mϕ compared to those in the pre-M1-Mϕ cells (Fig. 4). Of these cytokines, P. gulae induced very high levels of IL-1β, IL-6, and TNF-α. There was no significant production of IL-10 or IL-17A in response to P. gulae in M1-Mϕ (Fig. 4). The induction of IL-6 and TNF-α secretion by P. gulae was completely abrogated in TLR2−/− and TLR4−/− M1-Mϕ cells (Fig. 4c and h). However, the secretion of IL-1α, IL-1β, IL-12p70, and IL-13 by M1-Mϕ cells in response to P. gulae was independent of either TLR2 or TLR4 expression (Fig. 4).
Similar to the production of cytokines by macrophages, various chemokines were expressed by WT M1-Mϕ in response to the control ligands. The positive-control ligands E. coli LPS and Pam3CSK4, in a TLR-specific manner, induced significant (P < 0.05) levels of eotaxin, G-CSF, KC, MCP-1, MIP-1α, and RANTES in M1-Mϕ compared to those in the pre-M1-Mϕ cells (Fig. 5). In all cases, the deletion of either TLR2 or TLR4 resulted in a corresponding decrease in the production of chemokines in response to the corresponding control TLR ligand (Fig. 5). P. gulae whole cells induced significant (P < 0.05) levels of G-CSF, MCP-1, and MIP-1α, but not RANTES, KC, or eotaxin, in M1-Mϕ compared to those in the pre-M1-Mϕ cells (Fig. 5). The induction of G-CSF secretion by P. gulae was reduced in TLR2−/− and TLR4−/− M1-Mϕ cells (Fig. 5b), and MCP-1 and MIP-1α levels were reduced only in TLR4−/− M1-Mϕ cells (Fig. 5d and e).
To compare the immune response induced by P. gulae to that induced by E. coli, we performed a cytokine bead array assay (Fig. 6). We observed that despite the significant NOD2, TLR2, and TLR4 responses observed in this study, the overall synthesis of TNF-α, IL-6, IL-1β, and IL-10 induced by P. gulae was significantly lower than that induced by E. coli.
FIG 6.

Comparison of cytokine production levels of M1 macrophages in response to P. gulae and E. coli. WT iMACs, TLR2−/− iMACs, and TLR4−/− iMACs (105 cells) were incubated overnight with IFN-γ to prime the M1 macrophage phenotype. The M1-primed macrophages were then incubated with viable P. gulae or E. coli (MOI of 180:1) for 2 h and washed with DMEM containing antibiotics. E. coli LPS and Pam3CSK4 were used as TLR4 and TLR2 control ligands, respectively. The negative control was IFN-γ-primed macrophages that were not incubated with TLR ligands or P. gulae cells. After 24 h, the levels of cytokines in the assay supernatants were measured using an 8-plex Bioplex assay. The assay was repeated twice on each of three biological replicates (n = 6). (a) TNF-α; (b) IL-1β; (c) IL-6; (d) IL-10. *, P < 0.05 compared to the corresponding negative control.
DISCUSSION
We have shown that P. gulae cells interact with NOD2, TLR2, and TLR4 in order to activate NF-κB. No details have been published regarding the presence of lipopeptides/lipoproteins (TLR2 ligands) or the LPS structure (TLR4 ligand) of P. gulae, but given the antigenic similarities observed with P. gingivalis, some conclusions may be drawn. After the addition of P. gulae to HEK293 cells transfected with a range of TLRs and NLRs, the strongest NF-κB response observed was through TLR2, the receptor for lipopeptides and lipoproteins (21). The TLR2 response to P. gingivalis, the bacterium implicated in human chronic periodontitis, is also the strongest TLR response and is considered to be essential for disease progression (22, 23). However, the P. gingivalis TLR2 ligand is a topic of much research (23–26). It was initially thought that the modified lipid A of P. gingivalis was signaling through TLR2 (25, 27, 28). However, the discovery of a copurifying lipopeptide in LPS preparations and a recent analysis of the P. gingivalis-induced TLR2 activity implicate lipoproteins as the main TLR2 ligand (23, 24, 26). It is possible that homologues of the lipoproteins in P. gingivalis are responsible for the majority of the TLR2 activity in P. gulae. Furthermore, it has also been shown that P. gingivalis fimbriae signal through TLR2 (29–31), and considering the similarities between P. gulae and P. gingivalis fimbriae (8, 9), it is possible that P. gulae fimbriae also contribute to this TLR2 activity.
One of the major TLR ligands in bacteria is the canonical hexa-acylated biphosphorylated lipid A of the Enterobacteriaceae (32, 33). P. gingivalis, as well as other bacterial species, is able to deacylate and dephosphorylate lipid A, leading to tetra- and penta-acylated monophosphate lipid A (25, 28, 34). Both deacylation and dephosphorylation are well known to alter the immunogenicity of lipid A, with tetra-acylated lipid A being completely inert (35). The weaker TLR4 activity than TLR2 activity suggests that P. gulae may be able to modify its lipid A structure in a way similar to that of P. gingivalis. For P. gingivalis, this modification has been attributed to environmental regulation of deacylases and phosphatases; in particular, the presence of low iron in the growth medium has been shown to result in an altered lipid A structure (28). This has been proposed to be a mechanism by which P. gingivalis can escape detection of the immune system as well as dysregulate the immune response to other bacteria in the plaque biofilm (28, 36).
The NOD2 activity observed in this study indicates that peptidoglycan motifs (muramyl dipeptide [MDP] for NOD2) are potentially exposed on the cell surface, to an extent, or released into the medium. Given that peptidoglycan is covered by a lipid bilayer in Gram-negative bacteria and that whole cells were added to the HEK293 cells, it is possible that damage or leakage of cellular components resulted in the observed NOD2 activity. Furthermore, Gram-negative bacteria are known to secrete soluble forms of peptidoglycan, ranging in size from 20 to 200 kDa, as well as peptidoglycan subunits (37, 38). Regardless of the source, it is important to consider the influence of NOD2 ligation in interpreting results generated by use of knockout macrophages. It has been shown that P. gingivalis ligates both NOD1 and NOD2 in transient-transfection luciferase assays with oral epithelial cells and endothelial cells (39, 40). Although we were unable to confirm NOD1 signaling in P. gulae, a closely related organism, it is possible that given the low activities of the P. gingivalis NOD1 and NOD2 ligands observed by Okugawa et al. (39), any NOD1-ligating ability of P. gulae may have been below the detection threshold of the HEK-Blue NOD1 reporter cells. Experiments to investigate the activity of purified peptidoglycan from P. gulae in both a HEK reporter assay and immune cells are required to confirm this activity.
We have further shown that M0-Mϕ and pre-M1-Mϕ cells (primed with IFN-γ) exposed to P. gulae upregulate the expression of CD86 and that the subsequent M1-Mϕ cells produce nitric oxide and secrete several inflammatory cytokines, particularly IL-1β, IL-6, and TNF-α. This indicates that the cells of the innate immune system recognize P. gulae and potentially target and kill the bacterium as well as initiating an adaptive immune response. The ability to stimulate macrophages to produce nitric oxide is interesting, as studies have shown that the systemic nitric oxide level is elevated in dogs diagnosed with periodontitis (41). The source of this nitric oxide is unknown, and while there is no direct evidence that macrophages are responsible for the elevated nitric oxide, they may play some role given the results presented here. While studies of P. gingivalis have reported the ability to persist within macrophages, none have investigated this in “inflammatory” (M1) macrophages. We recently showed that P. gingivalis is unable to survive in IFN-γ-treated M1 macrophages (42), and given that we have also demonstrated that M0 “naive” macrophages produce negligible levels of nitric oxide after exposure to P. gulae (this study) or P. gingivalis (20), it is feasible that Porphyromonas species may be unable to persist inside activated M1-Mϕ producing large amounts of microbicidal compounds. Recent thinking on antigen presentation has elevated dendritic cells to the eminent status of antigen-presenting cells, and they undoubtedly play an important role (43, 44). However, the recent focus on dendritic cells has detracted from the role that macrophages perform as antigen-presenting cells (45). The expression of CD86 and other costimulatory molecules is essential for the engagement of T lymphocytes, with subsequent clonal selection and expansion (46). Therefore, the expression of CD86 on M1 macrophages in response to P. gulae, combined with nitric oxide production, indicates that these cells can play an important role in controlling infection at the infection site and can stimulate the adaptive arm of the immune system by interacting with infiltrating lymphocytes.
Inflammatory cytokines are important in establishing and maintaining a chronic inflammatory state. Exposure of pre-M1 macrophages to P. gulae resulted in the production of significant amounts of IL-1β, IL-6, TNF-α, G-CSF, MCP-1, and MIP-1α. IL-1β, IL-6, and TNF-α are extremely important cytokines involved in the pathogenesis of human chronic periodontitis (47). Together these cytokines activate inflammatory macrophages to produce more inflammatory mediators, amplifying the immune response. TNF-α, IL-6, and IL-1β are able to upregulate the expression of RANKL on resident gingival cells (48). This results in the differentiation of macrophages into osteoclasts, leading to resorption of the alveolar bone (49). IL-1β and IL-6 direct the maturation of naive T cells toward a Th17 phenotype that is often observed in periodontitis (50, 51). These inflammatory cytokines also act on gingival cells, inducing the production of matrix metalloproteinases that contribute to the degradation of gingival tissue (52–54). While these are inferences based on the data accumulated from P. gingivalis, the similarities to the immune response shown by our data for P. gulae suggest that P. gulae may invoke a similar pathology in companion animals. The inclusion of E. coli whole cells in a cytokine bead array puts the immune response to P. gulae into some context. E. coli induced significantly larger amounts of the cytokines TNF-α, IL-6, and IL-1β than those induced by P. gulae. It is known that Porphyromonas species contain penta- and tetra-acylated lipid A, whereas E. coli contains the more active hexa-acylated lipid A; hence, the disparity in cytokine induction may be attributable to the difference in lipid A structures. The acylation state of P. gulae lipid A is not yet known, and characterization of this important TLR ligand would allow further inferences to be made.
The results of the assays performed with knockout macrophages suggest that some functions of macrophages, such as nitric oxide production and CD86 expression, are very dependent on specific TLR engagement. The deletion of TLR2 but not TLR4 abolished the synthesis of nitric oxide and expression of CD86 after exposure to P. gulae, and the production of IL-6 and TNF-α was decreased, but not completely reduced, in the absence of either TLR2 or TLR4. This indicates that the P. gulae TLR4 ligand, presumably lipid A, is unable to produce nitric oxide or induce CD86 expression but is able to induce some cytokine secretion in the absence of TLR2 (lipopeptide/lipoprotein) signaling in M1-Mϕ cells. We previously showed that a preparation of P. gingivalis LPS was unable to induce nitric oxide production or CD86 expression in murine bone marrow-derived macrophages but was able to induce significant levels of inflammatory cytokines (20). In that study, we attributed this to TLR2-activating lipopeptides contaminating the preparation; the corroborating knockout macrophage data in this paper suggest that TLR4 ligation, even weak ligation, may be more important for cytokine production than for nitric oxide production and CD86 expression.
The ability of macrophages to signal via NOD2 in response to P. gulae must also be taken into account. It is possible that signaling via NOD2 may be enough to induce the production of cytokines in the absence of TLR2, but the signal may not be strong enough to induce nitric oxide production or CD86 upregulation. Considering that NOD2 signaling recruits p38 and c-Jun N-terminal kinase (JNK) to signal through AP-1 as well as NF-κB (31), there may be a compensatory mechanism by which cytokines can be produced in the absence of TLR2 or TLR4. Knockdown of NOD2 expression in C57/BL6 iMACs reduced the synthesis of NO and expression of CD86 in response to the positive-control ligand muramyl dipeptide. However, synthesis of NO and expression of CD86 in response to P. gulae cells did not appreciably decrease in the absence of NOD2 expression. Considering the strength of the TLR2 and TLR4 signals in response to most Gram-negative bacteria and the synergy that is often observed between these TLRs, it is not surprising that silencing of NOD2 did not have a significant effect on macrophage activation. However, knockout of NOD2 may result in a more substantial effect, as we were not able to completely reduce the activation of macrophages by muramyl dipeptide, suggesting that some residual NOD2 expression remained in these cells. In conclusion, we have found that P. gulae whole cells activate immune cells predominantly via TLR2, TLR4, and NOD2. This activation induces an M1 macrophage state after priming with IFN-γ, characterized by production of nitric oxide and upregulation of CD86. These macrophages further produce an inflammatory cytokine profile that, in a chronic disease state, would maintain inflammation in the gingival tissue. We found that the absence of TLR2 or TLR4 reduced the production of nitric oxide and CD86; however, some compensation in cytokine production was observed. Based on these results, macrophages, particularly those with inflammatory phenotypes, may play an important role in establishing and maintaining chronic inflammatory periodontal disease in companion animals.
MATERIALS AND METHODS
Growth of Porphyromonas gulae.
P. gulae (ATCC 51700) obtained from the Melbourne Dental School culture collection was maintained on horse blood agar (HBA) supplemented with 10% (vol/vol) horse blood and 10 μg/ml menadione in an MK3 anaerobic workstation (Don Whitley Scientific Limited, NSW, Australia) at 37°C, with a gas composition of 5% (vol/vol) H2, 10% (vol/vol) CO2 in N2 (BOC Gases Australia, NSW, Australia). For assays, P. gulae was grown in Todd-Hewitt broth (THB) supplemented with hemin (10 μg/ml), menadione (1 μg/ml), and cysteine (500 μg/ml) under the same culture conditions as those described above for 48 h, to an optical density at 650 nm (OD650) of ∼1.0 (late exponential growth phase). Culture purity was routinely monitored by Gram staining.
Generation of macrophage phenotypes and activation assays.
Cell culture reagents were obtained from Sigma-Aldrich Pty. Ltd. (NSW, Australia) unless otherwise specified. Immortalized macrophages (derived from bone marrows of C57BL/6, TLR2−/−, and TLR4−/− mice) were the gift of Eicke Latz (University of Bonn, Germany). Mammalian cells were routinely grown in complete Dulbecco's minimal essential medium (DMEM), consisting of DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS), 20 mM l-glutamine, 10 mM sodium pyruvate, and 100 U-100 μg penicillin-streptomycin. All cells were grown at 37°C in a 5% (vol/vol) CO2 atmosphere in a Heracell 150 incubator (Thermo Fisher Scientific, NSW, Australia). Macrophages (Mϕ) were primed as previously described (20). Briefly, Mϕ cells were incubated overnight in complete DMEM supplemented with 10 ng/ml of IFN-γ (Cell Signaling Technologies, MA) to generate pre-M1-Mϕ. To activate the macrophages, the pre-M1-Mϕ or nonpolarized M0-Mϕ were then incubated with either 10 ng/ml Escherichia coli LPS, 10 ng/ml Pam3CSK4 (Invivogen, CA), or P. gulae cells (MOI of 180:1) in antibiotic-free DMEM; an MOI of 180:1 was chosen based on our previous studies, which showed this to be optimal for stimulation (11). After 2 h of incubation, the Mϕ cells were washed (twice at 400 × g for 10 min each) in DMEM containing antibiotics and then further incubated for 16 h in complete DMEM.
To silence NOD2 expression, the plasmid psirna-mNOD2 (Invivogen) was transfected into iMACs by use of the Lipofectamine 3000 reagent according to the manufacturer's instructions. The plasmid encoding siRNA was transfected at a concentration of 250 ng per 104 cells, a concentration found to be effective at reducing activation by muramyl dipeptide (MDP) while maximizing cell health.
TLR activation assay.
Human embryonic kidney 293-Blue cells (HEK-Blue; Invivogen) stably transfected with NOD1, NOD2, TLR2, TLR4, TLR5, TLR7, TLR8, or TLR9 were grown in complete DMEM supplemented with Normocin (100 μg/ml) and various combinations of Zeocin, blasticidin, and hygromycin, depending on the cell type, according to the manufacturer's (Invivogen) specifications. For assays, 2 × 104 cells (150 μl) were seeded into each well of 96-well plates (Sigma-Aldrich), and 10-fold serial dilutions of the appropriate TLR ligand or P. gulae cells at various MOI were added in 50 μl of complete DMEM. After 20 h of incubation at 37°C in 5% (vol/vol) CO2, 50 μl of supernatant was mixed with 150 μl of HEK-Blue detection medium (Invivogen), and the plates were incubated at 37°C until color development occurred. Developed plates were read on a Victor3 1420 multilabel counter (PerkinElmer, MA) to measure the OD540.
Nitric oxide assay.
Nitric oxide was measured using a Griess reagent kit (Life Technologies, NSW, Australia) according to the manufacturer's instructions. Briefly, the day after macrophage activation with a TLR ligand, 150 μl of the Mϕ supernatant was combined with 130 μl of distilled water (dH2O), 10 μl of N-(1-naphthyl)ethylenediamine dihydrochloride (1 mg/ml), and 10 μl of sulfanilic acid (1.0 mM). A standard curve was generated using 2-fold serial dilutions of a 100 μM nitrite standard solution (100 μM to 1.56 μM). The reaction was allowed to proceed for 30 min at room temperature, and then the absorbance (OD550) was measured on a Victor3 1420 multilabel counter (PerkinElmer).
Flow cytometry analysis of surface marker expression.
M0-Mϕ or M1-Mϕ from the activation assay were collected using a 23G syringe, washed in 2 ml phosphate-buffered saline (PBS) containing 0.1% (wt/vol) bovine serum albumin (BSA; Sigma-Aldrich) (PBS-BSA buffer), and incubated with an anti-mouse CD16/CD32 antibody (clone 2.4G2; BD Biosciences, NJ) for 20 min on ice. Cells were washed in 5 ml PBS-BSA buffer prior to incubation with a 1:1,000 dilution of anti-mouse CD86–PE–Cy7 antibody for 30 min on ice (clone GL1; BD Biosciences, NSW, Australia). Cells were washed twice in 5 ml PBS-BSA buffer and then analyzed on a model FC500 flow cytometer (Beckman Coulter, NSW, Australia). The flow cytometer was equipped with an argon ion laser operating at an excitation wavelength of 488 nm and a red solid-state diode laser operating at 635 nm. The fluorescence was measured through a 755-nm filter (PE-Cy7; FL5). The data were analyzed using FlowJo software V7.0 (Tree Star, OR). Forward and side scatter properties were used to acquire a total of 30,000 cells and to gate out the cell debris.
Cytokine bead array analysis of cell culture supernatant.
Cell culture supernatants from M0-Mϕ and M1-Mϕ activation assays were analyzed for cytokines by using the Bioplex Pro mouse cytokine 23-plex assay (Bio-Rad Pty. Ltd., NSW, Australia). The 23-plex assay measures IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12p40, IL-12p70, IL-13, IL-17A, eotaxin, G-CSF, GM-CSF, IFN-γ, KC, MCP-1, MIP-1α, MIP-1β, RANTES, and TNF-α. The assay was performed according to the manufacturer's instructions. Briefly, 50 μl of beads was added to the assay plate and washed 2 times with 100 μl wash buffer. The samples were added in a volume of 50 μl and incubated with the beads for 1 h at room temperature with no light and constant mixing at 300 rpm on an MX4 micromixer (FinePCR, South Korea). Beads were then washed 3 times with 100 μl of wash buffer, using a Bioplex ProII wash station (Bio-Rad Pty. Ltd.), and 25 μl/well of biotinylated anti-cytokine detection antibody was added. Plates were then incubated at room temperature with no light and constant mixing at 300 rpm for 1 h. Wells were then washed 3 times with 100 μl of wash buffer, using a Bioplex ProII wash station, before bound biotin-labeled anti-cytokine antibody was detected by the addition of 25 μl of streptavidin-PE. Plates were incubated at room temperature with no light and constant mixing at 300 rpm for 10 min. Beads were then washed 3 times with 100 μl of wash buffer, using a Bioplex ProII wash station, and the beads were resuspended in 125 μl of assay buffer before reading the assay on a Bioplex 200 system (Bio-Rad Pty. Ltd.).
Statistical analysis.
Data were analyzed by two-way analysis of variance (ANOVA) with the Bonferroni posttest and presented as means ± standard deviations (SD) (GraphPad Prism V5.0). Differences with P values of <0.05 were considered statistically significant. Unless otherwise stated, the data presented are representative of three biological replicates, each performed in duplicate (n = 6).
ACKNOWLEDGMENTS
This work was supported by the Australian Government, Department of Industry, Innovation and Science.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Senhorinho GNA, Nakano V, Liu C, Song Y, Finegold SM, Avila-Campos MJ. 2011. Detection of Porphyromonas gulae from subgingival biofilms of dogs with and without periodontitis. Anaerobe 17:257–258. doi: 10.1016/j.anaerobe.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Oliver RC, Brown LJ. 1993. Periodontal diseases and tooth loss. Periodontol 2000 2:117–127. doi: 10.1111/j.1600-0757.1993.tb00224.x. [DOI] [PubMed] [Google Scholar]
- 3.Larsen J. 2010. Oral products and dental disease. Compend Contin Educ Vet 32:E4. [PubMed] [Google Scholar]
- 4.Dahlén G, Charalampakis G, Abrahamsson I, Bengtsson L, Falsen E. 2012. Predominant bacterial species in subgingival plaque in dogs. J Periodont Res 47:354–364. doi: 10.1111/j.1600-0765.2011.01440.x. [DOI] [PubMed] [Google Scholar]
- 5.Fournier D, Mouton C, Lapierre P, Kato T, Okuda K, Ménard C. 2001. Porphyromonas gulae sp. nov., an anaerobic, gram-negative coccobacillus from the gingival sulcus of various animal hosts. Int J Syst Evol Microbiol 51:1179–1189. doi: 10.1099/00207713-51-3-1179. [DOI] [PubMed] [Google Scholar]
- 6.Senhorinho GN, Nakano V, Liu C, Song Y, Finegold SM, Avila-Campos MJ. 2012. Occurrence and antimicrobial susceptibility of Porphyromonas spp. and Fusobacterium spp. in dogs with and without periodontitis. Anaerobe 18:381–385. doi: 10.1016/j.anaerobe.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 7.Hardham J, Dreier K, Wong J, Sfintescu C, Evans RT. 2005. Pigmented-anaerobic bacteria associated with canine periodontitis. Vet Microbiol 106:119–128. doi: 10.1016/j.vetmic.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Hamada N, Takahashi Y, Watanabe K, Kumada H, Oishi Y, Umemoto T. 2008. Molecular and antigenic similarities of the fimbrial major components between Porphyromonas gulae and P. gingivalis. Vet Microbiol 128:108–117. doi: 10.1016/j.vetmic.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 9.Oishi Y, Watanabe K, Kumada H, Ishikawa E, Hamada N. 2012. Purification and characterization of a novel secondary fimbrial protein from Porphyromonas gulae. J Oral Microbiol 4:1–7. doi: 10.3402/jom.v4i0.19076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hajishengallis G, Darveau RP, Curtis MA. 2012. The keystone-pathogen hypothesis. Nat Rev Microbiol 10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenzo JC, O'Brien-Simpson NM, Orth RK, Mitchell HL, Dashper SG, Reynolds EC. 2016. Porphyromonas gulae has virulence and immunological characteristics similar to those of the human periodontal pathogen Porphyromonas gingivalis. Infect Immun 84:2575–2585. doi: 10.1128/IAI.01500-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graves DT, Kang J, Andriankaja O, Wada K, Rossa C Jr. 2012. Animal models to study host-bacteria interactions involved in periodontitis. Front Oral Biol 15:117–132. doi: 10.1159/000329675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moskow BS, Polson AM. 1991. Histologic studies on the extension of the inflammatory infiltrate in human periodontitis. J Clin Periodontol 18:534–542. doi: 10.1111/j.1600-051X.1991.tb00086.x. [DOI] [PubMed] [Google Scholar]
- 14.Magnusson MK, Wick MJ. 2011. Intestinal dendritic cell and macrophage subsets: tipping the balance to Crohn's disease? Eur J Microbiol Immunol (Bp) 1:19–24. doi: 10.1556/EuJMI.1.2011.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore KJ, Sheedy FJ, Fisher EA. 2013. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat Rev Immunol 4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 17.Benoit M, Desnues B, Mege J-L. 2008. Macrophage polarization in bacterial infections. J Immunol 181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 18.Martinez FO, Helming L, Gordon S. 2009. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 19.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. 2006. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol 80:1298–1307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holden JA, Attard TJ, Laughton KM, Mansell A, O'Brien-Simpson NM, Reynolds EC. 2014. Porphyromonas gingivalis lipopolysaccharide weakly activates M1 and M2 polarized mouse macrophages but induces inflammatory cytokines. Infect Immun 82:4190–4203. doi: 10.1128/IAI.02325-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schenk M, Belisle JT. 2009. TLR2 looks at lipoproteins. Immunity 31:847–849. doi: 10.1016/j.immuni.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Burns E, Bachrach G, Shapira L, Nussbaum G. 2006. Cutting edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol 177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 23.Jain S, Coats SR, Chang AM, Darveau RP. 2013. A novel class of lipoprotein lipase-sensitive molecules mediates TLR2 activation by Porphyromonas gingivalis. Infect Immun 81:1277–1286. doi: 10.1128/IAI.01036-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asai Y, Hashimoto M, Fletcher HM, Miyake K, Akira S, Ogawa T. 2005. Lipopolysaccharide preparation extracted from Porphyromonas gingivalis lipoprotein-deficient mutant shows a marked decrease in Toll-like receptor 2-mediated signaling. Infect Immun 73:2157–2163. doi: 10.1128/IAI.73.4.2157-2163.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darveau RP, Pham TT, Lemley K, Reife RA, Bainbridge BW, Coats SR, Howald WN, Way SS, Hajjar AM. 2004. Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both Toll-like receptors 2 and 4. Infect Immun 72:5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimoto M, Asai Y, Ogawa T. 2004. Separation and structural analysis of lipoprotein in a lipopolysaccharide preparation from Porphyromonas gingivalis. Int Immunol 16:1431–1437. doi: 10.1093/intimm/dxh146. [DOI] [PubMed] [Google Scholar]
- 27.Kocgozlu L, Elkaim R, Tenenbaum H, Werner S. 2009. Variable cell responses to P. gingivalis lipopolysaccharide. J Dent Res 88:741–745. doi: 10.1177/0022034509341166. [DOI] [PubMed] [Google Scholar]
- 28.Al-Qutub MN, Braham PH, Karimi-Naser LM, Liu X, Genco Ca Darveau RP. 2006. Hemin-dependent modulation of the lipid A structure of Porphyromonas gingivalis lipopolysaccharide. Infect Immun 74:4474–4485. doi: 10.1128/IAI.01924-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asai Y, Ohyama Y, Gen K, Ogawa T. 2001. Bacterial fimbriae and their peptides activate human gingival epithelial cells through Toll-like receptor 2. Infect Immun 69:7387–7395. doi: 10.1128/IAI.69.12.7387-7395.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harokopakis E, Hajishengallis G. 2005. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur J Immunol 35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- 31.Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, Lin X. 2007. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol 8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- 32.Freudenberg MA, Merlin T, Gumenscheimer M, Kalis C, Landmann R, Galanos C. 2001. Role of lipopolysaccharide susceptibility in the innate immune response to Salmonella typhimurium infection: LPS, a primary target for recognition of Gram-negative bacteria. Microb Infect 3:1213–1222. doi: 10.1016/S1286-4579(01)01481-2. [DOI] [PubMed] [Google Scholar]
- 33.Freudenberg MA, Tchaptchet S, Keck S, Fejer G, Huber M, Schütze N, Beutler B, Galanos C. 2008. Lipopolysaccharide sensing an important factor in the innate immune response to Gram-negative bacterial infections: benefits and hazards of LPS hypersensitivity. Immunobiology 213:193–203. doi: 10.1016/j.imbio.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 34.Sawada N, Ogawa T, Asai Y, Makimura Y, Sugiyama A. 2007. Toll-like receptor 4-dependent recognition of structurally different forms of chemically synthesized lipid As of Porphyromonas gingivalis. Clin Exp Immunol 148:529–536. doi: 10.1111/j.1365-2249.2007.03346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flad H-D, Loppnow H, Rietschel ET, Ulmer AJ. 1993. Agonists and antagonists for lipopolysaccharide-induced cytokines. Immunobiology 187:303–316. doi: 10.1016/S0171-2985(11)80346-3. [DOI] [PubMed] [Google Scholar]
- 36.Coats SR, Jones JW, Do CT, Braham PH, Bainbridge BW, To TT, Goodlett DR, Ernst RK, Darveau RP. 2009. Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4′-phosphatase activities. Cell Microbiol 11:1587–1599. doi: 10.1111/j.1462-5822.2009.01349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horcajo P, de Pedro MA, Cava F. 2012. Peptidoglycan plasticity in bacteria: stress-induced peptidoglycan editing by noncanonical d-amino acids. Microb Drug Resist 18:306–313. doi: 10.1089/mdr.2012.0009. [DOI] [PubMed] [Google Scholar]
- 38.Takada H, Uehara A. 2006. Enhancement of TLR-mediated innate immune responses by peptidoglycans through NOD signaling. Curr Pharm Des 12:4163–4172. doi: 10.2174/138161206778743510. [DOI] [PubMed] [Google Scholar]
- 39.Okugawa T, Kaneko T, Yoshimura A, Silverman N, Hara Y. 2010. NOD1 and NOD2 mediate sensing of periodontal pathogens. J Dent Res 89:186–191. doi: 10.1177/0022034509354843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan M, Liu J, Ouyang X. 2015. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-kappaB pathway. J Periodont Res 50:189–196. doi: 10.1111/jre.12192. [DOI] [PubMed] [Google Scholar]
- 41.Nemec A, Verstraete FJM, Jerin A, Šentjurc M, Kass PH, Petelin M, Pavlica Z. 2013. Periodontal disease, periodontal treatment and systemic nitric oxide in dogs. Res Vet Sci 94:542–544. doi: 10.1016/j.rvsc.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 42.Lam RS, O'Brien-Simpson NM, Holden JA, Lenzo JC, Fong SB, Reynolds EC. 2016. Unprimed, M1 and M2 macrophages differentially interact with Porphyromonas gingivalis. PLoS One 11:e0158629. doi: 10.1371/journal.pone.0158629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gordon S. 2007. The macrophage: past, present and future. Eur J Immunol 37(Suppl 1):S9–S17. doi: 10.1002/eji.200737638. [DOI] [PubMed] [Google Scholar]
- 44.Cutler CW, Jotwani R. 2004. Antigen-presentation and the role of dendritic cells in periodontitis. Periodontol 2000 35:135–157. doi: 10.1111/j.0906-6713.2004.003560.x. [DOI] [PubMed] [Google Scholar]
- 45.Hume DA. 2008. Macrophages as APC and the dendritic cell myth. J Immunol 181:5829–5835. doi: 10.4049/jimmunol.181.9.5829. [DOI] [PubMed] [Google Scholar]
- 46.Hugues S. 2010. Dynamics of dendritic cell-T cell interactions: a role in T cell outcome. Semin Immunol 32:227–238. doi: 10.1007/s00281-010-0211-2. [DOI] [PubMed] [Google Scholar]
- 47.Yucel-Lindberg T, Båge T. 2013. Inflammatory mediators in the pathogenesis of periodontitis. Expert Rev Mol Med 15:e7. doi: 10.1017/erm.2013.8. [DOI] [PubMed] [Google Scholar]
- 48.Hormdee D, Nagasawa T, Kiji M, Yashiro R, Kobayashi H, Koshy G, Noguchi K, Nitta H, Ishikawa I. 2005. Protein kinase-A-dependent osteoprotegerin production on interleukin-1 stimulation in human gingival fibroblasts is distinct from periodontal ligament fibroblasts. Clin Exp Immunol 142:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartold PM, Cantley MD, Haynes DR. 2010. Mechanisms and control of pathologic bone loss in periodontitis. Periodontol 2000 53:55–69. doi: 10.1111/j.1600-0757.2010.00347.x. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi K, Azuma T, Motohira H, Kinane DF, Kitetsu S. 2005. The potential role of interleukin-17 in the immunopathology of periodontal disease. J Clin Periodontol 32:369–374. doi: 10.1111/j.1600-051X.2005.00676.x. [DOI] [PubMed] [Google Scholar]
- 51.Vernal R, Dutzan N, Chaparro A, Puente J, Antonieta Valenzuela M, Gamonal J. 2005. Levels of interleukin-17 in gingival crevicular fluid and in supernatants of cellular cultures of gingival tissue from patients with chronic periodontitis. J Clin Periodontol 32:383–389. doi: 10.1111/j.1600-051X.2005.00684.x. [DOI] [PubMed] [Google Scholar]
- 52.Kwan Tat S, Padrines M, Théoleyre S, Heymann D, Fortun Y. 2004. IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev 15:49–60. doi: 10.1016/j.cytogfr.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 53.Weber A, Wasiliew P, Kracht M. 2010. Interleukin-1 (IL-1) pathway. Sci Signal 3:cm1. doi: 10.1126/scisignal.3105cm1. [DOI] [PubMed] [Google Scholar]
- 54.Dong W, Xiang J, Li C, Cao Z, Huang Z. 2009. Increased expression of extracellular matrix metalloproteinase inducer is associated with matrix metalloproteinase-1 and -2 in gingival tissues from patients with periodontitis. J Periodont Res 44:125–132. doi: 10.1111/j.1600-0765.2008.01105.x. [DOI] [PubMed] [Google Scholar]