

Abstract
Hepatic cytosolic alcohol and aldehyde dehydrogenases (ADHs and ALDHs) catalyze the biotransformation of xenobiotics (e.g., cyclophosphamide and ethanol) and vitamin A. Because age-dependent hepatic abundance of these proteins is unknown, we quantified protein expression of ADHs and ALDH1A1 in a large cohort of pediatric and adult human livers by liquid chromatography coupled with tandem mass spectrometry proteomics. Purified proteins were used as calibrators. Two to three surrogate peptides per protein were quantified in trypsin digests of liver cytosolic samples and calibrator proteins under optimal conditions of reproducibility. Neonatal levels of ADH1A, ADH1B, ADH1C, and ALDH1A1 were 3-, 8-, 146-, and 3-fold lower than the adult levels, respectively. For all proteins, the abundance steeply increased during the first year of life, which mostly reached adult levels during early childhood (age between 1 and 6 years). Only for ADH1A protein abundance in adults (age > 18 year) was ∼40% lower relative to the early childhood group. Abundances of ADHs and ALDH1A1 were not associated with sex in samples with age > 1 year compared with males. Known single nucleotide polymorphisms had no effect on the protein levels of these proteins. Quantification of ADHs and ALDH1A1 protein levels could be useful in predicting disposition and response of substrates of these enzymes in younger children.
Introduction
Age-dependent maturation of the expression or activity of drug metabolizing enzymes (DMEs) in humans is commonly observed (Hines, 2008). Although substantial data exist on the ontogeny of major microsomal DMEs, age-dependent regulation of cytosolic enzymes is not well studied. Alcohol dehydrogenase 1 isoforms (ADH1A, ADH1B, and ADH1C) and aldehyde dehydrogenase 1A1 (ALDH1A1) are high-abundant cytosolic proteins in human liver (Edenberg, 2000; Sladek, 2003) and belong to a family of NAD(P)+-dependent enzymes and primarily involved in the biotransformation of primary alcohols to aldehydes and aldehydes to weak carboxylic acids, respectively (Sladek, 2003). For example, these enzymes play major roles in the metabolism of vitamin A (Kam et al., 2012; Arnold et al., 2015), alcohol (Zakhari, 2006), and drugs such as cyclophosphamide (CP) (de Jonge et al., 2005). Specifically, ALDH1A1 is responsible for converting retinaldehyde to retinoic acid, whereas ADH enzymes convert ethanol to acetaldehyde. CP is first converted by multiple cytochrome P450 enzymes to 4-hydroxy-CP, which exists in equilibrium with its open ring tautomer, aldophosphamide. Both 4-hydroxy CP and aldophosphamide are irreversibly deactivated by ADHs and ALDH1A1 to 4-keto CP and carboxyphosphamide, respectively. The ALDH-catalyzed detoxification reaction competes with the activation reaction that converts aldophosphamide to cytotoxic phosphoramide mustard, which is responsible for the anticancer activity (de Jonge et al., 2005). Therefore, these enzymes play a major role in determining the efficacy as well as off-target toxicity of CP.
Immature levels of DMEs and transporters are often associated with reduced clearance and an increased risk of toxicity of xenobiotics in children (Hines, 2008). Although midterm fetal levels of hepatic ADH and ALDH are significantly lower relative to adults (Smith et al., 1971), ontogenic trajectories of these proteins across the entire age spectrum from newborns to adults have not been established. Such data are important to predict the effect of age on in vivo clearance of ADHs and ALDH1A1 substrates in children before clinical use. Therefore, we quantified ADH1A, ADH1B, ADH1C, and ALDH1A1 in the hepatic cytosolic (HLC) fractions isolated from a bank of tissues using our well-established proteomics methodology (Prasad et al., 2016). We then studied the effect of age, sex, race, and genetic polymorphisms on protein expression of these enzymes. Although there are multiple ADHs (Estonius et al., 1996) and ALDHs (Stewart et al., 1996) expressed in various human tissues, this study is limited to four major hepatic dehydrogenases (ADH1A, ADH1B, ADH1C, and ALDH1A1). Integration of the protein abundance data into physiologically based pharmacokinetics modeling software would be useful to predict accurate drug disposition in a pediatric population.
Materials and Methods
Materials.
Purified ADH1A, ADH1B, ADH1C, and ALDH1A1 protein standards were procured from Abnova (Walnut, CA). Synthetic heavy labeled peptides (Supplemental Table 1S) were obtained from Thermo Fisher Scientific (Rockford, IL). Ammonium bicarbonate (98% purity), bovine serum albumin (BSA) and Pierce Trypsin protease (MS-grade) were purchased from Thermo Fisher Scientific (Rockford, IL). Chloroform, ethyl ether, Optima MS-grade acetonitrile, methanol, and formic acid were purchased from Fischer Scientific (Fair Lawn, NJ).
Human Liver Cytosol Samples.
Human cytosol samples of 129 pediatric liver donors were provided by Children’s Mercy-Kansas City (Kansas City, MO). Tissues were obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland the Liver Tissue Cell Distribution System at the University of Minnesota and the University of Pittsburgh as well as Vitron (Tucson, AZ) and XenoTech LLC (Lenexa, KS). An additional 57 adult and 8 pediatric samples were available through the University of Washington School of Pharmacy liver bank. The samples were classified based on the following age categories: neonatal (0–27 days; n = 4), infancy (28–364 days; n = 17), toddler/early childhood (1 year to < 6 years; n = 30), middle childhood (6 years to < 12 years; n = 38), adolescence (12–18 years; n = 48), and adulthood (> 18 years; n = 57). The use of these samples has been classified as nonhuman subject research by the institutional review boards of the University of Washington (Seattle, WA) and Children’s Mercy-Kansas City. Procurement and storage information were described previously (Pearce et al., 2016; Prasad et al., 2016; Shirasaka et al., 2016; Boberg et al., 2017). Detailed donor demographic information is provided in Supplemental Table 2S. The HLC fraction was isolated from liver tissue samples by differential centrifugation as per established protocols (Pearce et al., 2016; Shirasaka et al., 2016).
Protein Denaturation, Reduction, Alkylation, Enrichment, and Trypsin Digestion.
The HLC samples were trypsin digested as described with few modifications (Boberg et al., 2017). Detailed method is also described in the Supplemental Material. For absolute quantification, protein standards (on column amounts, 0.032–2.04 pmol (ADH1A), 0.017–1.104 pmol (ADH1B), 0.017–1.115 pmol (ADH1C), and 0.013–0.803 pmol (ALDH1A1)) were injected to create the calibration curves.
Quantitative Analysis of ADH1A, ADH1B, ADH1C, and ALDH1A1 by LC-MS/MS.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) consisted of an Acquity (Waters Technologies, Milford, MA) LC coupled to an AB Sciex Triple Quadrupole 6500 MS system (Framingham, MA). Two to three surrogate peptides per protein were selected for the quantification of ADH1A, ADH1B, ADH1C, and ALDH1A1 protein abundance (Supplemental Table 1S) following previously published protocol (Vrana et al., 2017) (QPrOmics; www.qpromics.uw.edu/qpromics/assay/). The peptide separation was achieved on an Acquity UPLC column (HSS T3 1.8 μm, 2.1 × 100 mm, Waters). Mobile phase A (water with formic acid 0.1%; v/v) and mobile phase B (acetonitrile with formic acid 0.1%; v/v) were used with a flow rate of 0.3 ml/min in a gradient manner (Supplemental Table 1S). Multiple reaction monitoring conditions for targeted analysis of ADH1A, ADH1B, ADH1C, and ALDH1A1 proteins are shown in Supplemental Table 1S. Peak integration and quantification were performed using Analyst (Version 1.6, Mass Spectrometry Toolkit v3.3, Framingham, MA). We used a robust strategy to ensure optimum reproducibility when quantifying these proteins. For example, ion suppression was addressed by using heavy peptide. BSA was used as an exogenous protein internal standard, which was added to each sample before methanol-chloroform-water extraction and trypsin digestion to correct for protein loss during processing and digestion efficiency. To address interbatch variability, three sets of pooled representative cytosolic samples were processed each day, which served as quality controls. In total, three-step data normalization was used. First, average light peak areas for specific peptide daughter fragments were divided by corresponding average heavy peak areas. This ratio was further divided by BSA light/heavy area ratio. For each day, these data were further normalized to average quality control values. Absolute quantification of ADHs and ALDH1A1 in the pooled quality control samples was performed using the purified protein calibrators, which was used to calculate the protein abundance in each sample. The protein abundance data presented are the mean of the three analyses with standard deviation (S.D.).
DNA Isolation and Genotyping.
ADH and ALDH genotyping of the control adult samples was conducted per established protocols (Prasad et al., 2014; Rasmussen-Torvik et al., 2014). Briefly, genomic DNA was extracted from liver tissues. The genotyping was performed using PGRN-SeqV1 (Gordon et al., 2016) and the Affymetrix DMET Plus Array (Santa Clara, CA) per the manufacturer’s protocol.
Statistical analysis.
For individual age categories (neonates to adults), protein abundance data were compared using Kruskal-Wallis test followed by Dunn’s multiple comparison test. Mann-Whitney test was used to analyze the effect of sex on protein abundance. A nonlinear, allosteric sigmoidal model using eq. 1 was fitted to the continuous ontogeny protein abundance data until age 18 (GraphPad Prism, San Diego, CA), as described previously (Boberg et al., 2017).
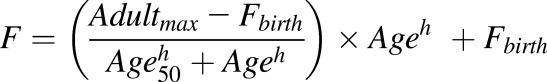 |
(1) |
Adultmax is the maximum average relative protein abundance; Age is the age in years of the subject at the time of sample collection; Age50 is the age in years at which half-maximum adult protein abundance is obtained; F is the fractional protein abundance in adult samples; Fbirth is the fractional protein abundance (of adult) at birth; and h is the exponential factor.
The goodness of model fit was evaluated by visual inspection, 95% confidence intervals (CIs) of the parameter estimates, and residual plots. Weights of 1/Y2 were used. The Pearson regression test was used to test correlation between proteins across entire population. A P value below 0.05 was considered statistically significant. The observed data were illustrated in graphs and tables using Microsoft Excel (Version 16, Redmond, WA) and GraphPad.
Results and Discussion
The dynamic detection range was established for internal standard peptides (Supplemental Table 3S). There was no saturation of signal at higher points. The lower limit of quantification for ADH1A, ADH1B, ADH1C, and ALDH1A1 was 0.12, 0.07, 0.08, and 0.07 pmol (on-column), respectively. Correlations between peak area ratios of multiple surrogate peptides of ADHs or ALDH1A1 across samples were excellent (r2 > 0.93), indicating the robustness of protein quantification by LC-MS/MS (Supplemental Fig. 1S). Neonatal levels of ADH1A, ADH1B, ADH1C, and ALDH1A1 were 3-, 8-, 146-, and 3-fold lower than the adult levels, respectively (Fig. 1). Age50 values, i.e., the age at which protein expression is 50% of maximum abundance, for ADH1A, ADH1B, ADH1C, and ALDH1A1 was 10.1, 9.3, 12.3, and 10.9 months (Fig. 2). For all these proteins, the abundance increased steeply during the first year of life, approaching adult levels during early childhood (age 1–6 years). Developmental trajectories of each of the ADH proteins displayed some unique features. For example, ADH1A protein levels in adults (>18 years) were ∼40% lower as compared with the early childhood group. ADH1B was the most abundant of all the proteins assessed in this study, with an absolute level that was significantly higher than other proteins across the entire population (Fig. 1). ADH1C demonstrated the most rapid trajectory, steeply increasing to adult levels during the first year of life.
Fig. 1.

Age-dependent abundance of ADH1A, ADH1B, ADH1C, and ALDH1A1 in human liver cytosol samples. Age classification: neonatal (0–27 days), infancy (28–364 days), toddler/early childhood (1 year to <6 years), middle childhood (6 years to < 12 years), adolescence (12– 18 years) and adulthood (>18 years). The number of subjects in each age category are indicated in parentheses in the x-axis of categorical data. Dot plots are displayed with mean protein abundance as the horizontal line together with S.D. Representative pie chart is showing change in protein abundance of ADHs and ALDH1A1 from neonatal to adulthood. For all proteins, the abundance steeply increases during the first 2 years of life, reaching adult levels during early childhood. ADH1A protein abundance in adults (>18 years) is ∼39% lower compared with children and adolescents. *, ** and *** represents P values < 0.05, < 0.01, and < 0.001, respectively.
Fig. 2.

Continuous age-dependent abundance of ADH1A, ADH1B, ADH1C, and ALDH1A1 in human liver cytosol samples. Table presents fitted values of abundance at birth (E0), average adult abundance (Adult_max), and 50% protein abundance is observed (Age50). Additional parameters are reported in Supplemental Table 4S. Only protein expression data until age 18 were used to calculate above parameters. ND, not defined; SE, standard error; CI, confidence intervals.
There was no significant effect of sex on ADHs and ALDH1A1 abundance (Supplemental Fig. 2S). Although we detected single nucleotide polymorphisms in ADH1A (rs1826909, rs6811453, rs7684674, and rs12512110), ADH1B (rs1229983 and rs1229984), ADH1C (rs283413 and rs1789915), and ALDH1A1 (rs13959) in several samples, none were significantly associated with protein expression (Supplemental Fig. 3S). ADH1B levels were significantly correlated with ADH1A (r2 = 0.81) and ALDH1A1 (r2 = 0.77) (Supplemental Fig. 4S). We did not find any significant effect of ethnicity on abundance of ADHs and ALDH1A1 (data not shown). Based on principle component analysis as shown in Supplemental Fig. 5S, we did not observe clustering of expression pattern based on source of samples.
A comprehensive analysis of the developmental trajectories of ADH1A, ADH1B, ADH1C, and ALDH1A1 protein abundance was performed in human liver samples obtained from donors across the entire pediatric and adult population spectrum for the first time using sensitive and robust LC-MS/MS analysis. Our results are consistent with the limited existing human data on the developmental changes of ADHs and ALDH1A1 expression in fetal, infant, and adult liver samples (Smith et al., 1971). As a novel finding, we report ontogenic trajectories of these proteins (Fig. 2), e.g., Age50 for ADH1A, ADH1B, ADH1C, and ALDH1A1 was around 10–11 months, respectively. Like human data, mRNA expression of different Aldh enzymes in mouse was detected at lower levels in fetuses than in adults (Alnouti and Klaassen, 2008). In contrast to a previous study in adults (Smith et al., 1971) that reports low to nondetectable ADH1A levels, we detected ADH1A protein levels in all of our samples, which is likely due to better sensitivity of LC-MS/MS over the semiquantitative starch gel electrophoresis methodology. The sensitivity of the novel, validated, high-throughput LC-MS/MS methods presented here may also have utility for precision diagnosis and therapeutics in the context of cancer chemotherapy, because these proteins (especially ALDH1A1) are often upregulated in various types of cancers (Khoury et al., 2012; Xu et al., 2015). Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells (Liu et al., 2016).
ADH, ALDH, cytochrome P450 2E1 (CYP2E1), and catalase are the primary enzymes involved in ethanol disposition. Seven ADH genes, i.e., ADH1A, ADH1B, ADH1C, ADH4, ADH5, ADH6, and ADH7, are expressed in humans (Edenberg, 2007), but the closely related isoforms (ADH1A, ADH1B, and ADH1C), which can form homodimers or heterodimers, account for most of the ethanol-oxidizing capacity in the liver (Lee et al., 2006). In contrast, ALDH1A1 functions downstream of ADHs in the oxidative metabolism of excess retinol, and alteration of ALDH1A1 function correlates with retinol toxicity primarily due to its accumulation (Molotkov and Duester, 2003). Our data are important for predicting the effect of age on such sequential metabolism. Furthermore, ADHs and ALDH1A1 play an important role in the biotransformation of CP, which is a cornerstone of curative chemotherapy regimens in over 50% of newly diagnosed pediatric cancer patients (McCune et al., 2009). ALDH1A1 determines both toxicity and efficacy of CP. Pediatric patients with lower CP clearance and those who produce significant quantities of inactive metabolites are at greater risk of recurrence after current chemotherapy regimens for B-cell non-Hodgkin’s lymphoma (Yule et al., 2004). Decreased conversion of 4-hydroxy CP to its inactive metabolite due to lower levels of hepatic ADHs and ALDH1A could potentially result in off-target liver toxicity in younger pediatric patients.
The allelic variants ADH1B*2 (rs1229984), ADH1B*3 (rs1229984), and ADH1C*1 are associated with higher rates of ethanol biotransformation with turnover rates of 350, 300, and 90 minute−1, respectively, compared with ADH1B*1, ADH1C*2, and ADH1A (turnover rates < 40 minute−1) (Edenberg, 2007). Different combinations of these isoforms and genotypes determine alcohol metabolizing capacity in humans. In our study, we found seven subjects heterozygous for ADH1B*2 (rs1229984; 48A>G), which was not associated with any change in ADH1B protein abundance. Because ADH1B*2 is associated with reduced risk of alcoholism (Muramatsu et al., 1995) and reduced risk of migraine (Garcia-Martin et al., 2010), this single nucleotide polymorphisms likely affects substrate affinity (Km) without any effect on protein abundance and Vmax. rs283413 encodes a variant of the ADH1C gene that leads to a truncated alcohol dehydrogenase protein, which is associated with Parkinson disease (Buervenich et al., 2005). This variant is relatively rare, and only one donor in our liver banks was observed to carry this gene variation.
In human hepatocytes, bile acids can induce ADH1A and ADH1B expression by activation of the nuclear receptor, farnesoid X receptor (FXR) (Langhi et al., 2013). Epigenetic modifications such as DNA methylation, histone modifications, noncoding RNAs, nucleosome positioning, and chromatin remodeling could control age-dependent changes in DMEs (Zhong and Leeder, 2013). A strong correlation between miRNA expression and age is known for miR-34a, miR-200a, and miR-200b, which are associated with regulation of some DMEs (Rieger et al., 2013). Additional studies would be needed to elucidate whether these mechanisms are associated with hepatic ADH and ALDH1A1 expression.
In summary, the age-dependent protein abundance data may be useful for predicting hepatic detoxification of ADH and ALDH1A1 substrates. Because ADHs and ALDHs are also expressed in other tissues (Arnold et al., 2015), the validated LC-MS/MS methods can be used to characterize extrahepatic levels of these proteins. Once available, integration of hepatic and extrahepatic levels of these proteins into physiologically based pharmacokinetics software platforms can be used to predict first-in children dosing of new chemical entities with primary alcohol and aldehyde groups.
Abbreviations
- ADH
alcohol dehydrogenases
- ALDH
aldehyde dehydrogenase
- CP
cyclophosphamide
- DME
drug-metabolizing enzyme
- HLC
human liver cytosol
- LC-MS/MS
liquid chromatography coupled with tandem mass spectrometry
Authorship Contributions
Participated in research design: Bhatt, Leeder, and Prasad.
Conducted experiments: Bhatt, Gaedigk, Pearce, and Prasad.
Performed data analysis: Bhatt, Gaedigk, and Prasad.
Wrote or contributed to the writing of the manuscript: Bhatt, Gaedigk, Pearce, Leeder, and Prasad.
Footnotes
This work is primarily funded by grant from National Institutes of Health (NIH) Eunice Kennedy Shriver National Institute of Child Health and Human Development (Grant R01.HD081299-02). The National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland is funded by the NIH Contract HHSN275200900011C, reference number, N01-HD-9-0011, and the Liver Tissue Cell Distribution System is funded by NIH Contract N01-DK-7-0004/HHSN267200700004C.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Alnouti Y, Klaassen CD. (2008) Tissue distribution, ontogeny, and regulation of aldehyde dehydrogenase (Aldh) enzymes mRNA by prototypical microsomal enzyme inducers in mice. Toxicol Sci 101:51–64. [DOI] [PubMed] [Google Scholar]
- Arnold SL, Kent T, Hogarth CA, Schlatt S, Prasad B, Haenisch M, Walsh T, Muller CH, Griswold MD, Amory JK, et al. (2015) Importance of ALDH1A enzymes in determining human testicular retinoic acid concentrations. J Lipid Res 56:342–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boberg M, Vrana M, Mehrotra A, Pearce RE, Gaedigk A, Bhatt DK, Leeder JS, Prasad B. (2017) Age-dependent absolute abundance of hepatic carboxylesterases (CES1 and CES2) by LC-MS/MS proteomics: application to PBPK modeling of oseltamivir in vivo pharmacokinetics in infants. Drug Metab Dispos 45:216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buervenich S, Carmine A, Galter D, Shahabi HN, Johnels B, Holmberg B, Ahlberg J, Nissbrandt H, Eerola J, Hellström O, et al. (2005) A rare truncating mutation in ADH1C (G78Stop) shows significant association with Parkinson disease in a large international sample. Arch Neurol 62:74–78. [DOI] [PubMed] [Google Scholar]
- de Jonge ME, Huitema AD, Rodenhuis S, Beijnen JH. (2005) Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet 44:1135–1164. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ. (2000) Regulation of the mammalian alcohol dehydrogenase genes. Prog Nucleic Acid Res Mol Biol 64:295–341. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ.(2007) The genetics of alcohol metabolism: role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res Health 30:5–13. [PMC free article] [PubMed] [Google Scholar]
- Estonius M, Svensson S, Höög JO. (1996) Alcohol dehydrogenase in human tissues: localisation of transcripts coding for five classes of the enzyme. FEBS Lett 397:338–342. [DOI] [PubMed] [Google Scholar]
- García-Martín E, Martínez C, Serrador M, Alonso-Navarro H, Navacerrada F, Agúndez JA, Jiménez-Jiménez FJ. (2010) Alcohol dehydrogenase 2 genotype and risk for migraine. Headache 50:85–91. [DOI] [PubMed] [Google Scholar]
- Gordon AS, Fulton RS, Qin X, Mardis ER, Nickerson DA, Scherer S. (2016) PGRNseq: a targeted capture sequencing panel for pharmacogenetic research and implementation. Pharmacogenet Genomics 26:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines RN. (2008) The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther 118:250–267. [DOI] [PubMed] [Google Scholar]
- Kam RKT, Deng Y, Chen Y, Zhao H. (2012) Retinoic acid synthesis and functions in early embryonic development. Cell Biosci 2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury T, Ademuyiwa FO, Chandrasekhar R, Jabbour M, Deleo A, Ferrone S, Wang Y, Wang X. (2012) Aldehyde dehydrogenase 1A1 expression in breast cancer is associated with stage, triple negativity, and outcome to neoadjuvant chemotherapy. Mod Pathol 25:388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhi C, Pedraz-Cuesta E, Haro D, Marrero PF, Rodríguez JC.(2013) Regulation of human class I alcohol dehydrogenases by bile acids. J Lipid Res 54:2475–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SL, Chau GY, Yao CT, Wu CW, Yin SJ. (2006) Functional assessment of human alcohol dehydrogenase family in ethanol metabolism: significance of first-pass metabolism. Alcohol Clin Exp Res 30:1132–1142. [DOI] [PubMed] [Google Scholar]
- Liu X, Wang L, Cui W, Yuan X, Lin L, Cao Q, Wang N, Li Y, Guo W, Zhang X, et al. (2016) Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells. Oncotarget 7:58516–58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCune JS, Salinger DH, Vicini P, Oglesby C, Blough DK, Park JR. (2009) Population pharmacokinetics of cyclophosphamide and metabolites in children with neuroblastoma: a report from the Children’s Oncology Group. J Clin Pharmacol 49:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molotkov A, Duester G. (2003) Genetic evidence that retinaldehyde dehydrogenase Raldh1 (Aldh1a1) functions downstream of alcohol dehydrogenase Adh1 in metabolism of retinol to retinoic acid. J Biol Chem 278:36085–36090. [DOI] [PubMed] [Google Scholar]
- Muramatsu T, Wang ZC, Fang YR, Hu KB, Yan H, Yamada K, Higuchi S, Harada S, Kono H. (1995) Alcohol and aldehyde dehydrogenase genotypes and drinking behavior of Chinese living in Shanghai. Hum Genet 96:151–154. [DOI] [PubMed] [Google Scholar]
- Pearce RE, Gaedigk R, Twist GP, Dai H, Riffel AK, Leeder JS, Gaedigk A. (2016) Developmental expression of CYP2B6: a comprehensive analysis of mRNA expression, protein content and bupropion hydroxylase activity and the impact of genetic variation. Drug Metab Dispos 44:948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B, Evers R, Gupta A, Hop CE, Salphati L, Shukla S, Ambudkar SV, Unadkat JD. (2014) Interindividual variability in hepatic organic anion-transporting polypeptides and P-glycoprotein (ABCB1) protein expression: quantification by liquid chromatography tandem mass spectroscopy and influence of genotype, age, and sex. Drug Metab Dispos 42:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad B, Gaedigk A, Vrana M, Gaedigk R, Leeder JS, Salphati L, Chu X, Xiao G, Hop C, Evers R, et al. (2016) Ontogeny of hepatic drug transporters as quantified by LC-MS/MS proteomics. Clin Pharmacol Ther 100:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen-Torvik LJ, Stallings SC, Gordon AS, Almoguera B, Basford MA, Bielinski SJ, Brautbar A, Brilliant MH, Carrell DS, Connolly JJ, et al. (2014) Design and anticipated outcomes of the eMERGE-PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin Pharmacol Ther 96:482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger JK, Klein K, Winter S, Zanger UM. (2013) Expression variability of absorption, distribution, metabolism, excretion-related microRNAs in human liver: influence of nongenetic factors and association with gene expression. Drug Metab Dispos 41:1752–1762. [DOI] [PubMed] [Google Scholar]
- Shirasaka Y, Chaudhry AS, McDonald M, Prasad B, Wong T, Calamia JC, Fohner A, Thornton TA, Isoherranen N, Unadkat JD, et al. (2016) Interindividual variability of CYP2C19-catalyzed drug metabolism due to differences in gene diplotypes and cytochrome P450 oxidoreductase content. Pharmacogenomics J 16:375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sládek NE. (2003) Human aldehyde dehydrogenases: potential pathological, pharmacological, and toxicological impact. J Biochem Mol Toxicol 17:7–23. [DOI] [PubMed] [Google Scholar]
- Smith M, Hopkinson DA, Harris H. (1971) Developmental changes and polymorphism in human alcohol dehydrogenase. Ann Hum Genet 34:251–271. [DOI] [PubMed] [Google Scholar]
- Stewart MJ, Malek K, Crabb DW. (1996) Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J Investig Med 44:42–46. [PubMed] [Google Scholar]
- Vrana M, Whittington D, Nautiyal V, Prasad B. (2017) Database of optimized proteomic quantitative methods for human drug disposition-related proteins for applications in physiologically based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol 6:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Chai S, Wang P, Zhang C, Yang Y, Yang Y, Wang K. (2015) Aldehyde dehydrogenases and cancer stem cells. Cancer Lett 369:50–57. [DOI] [PubMed] [Google Scholar]
- Yule SM, Price L, McMahon AD, Pearson ADJ, Boddy AV. (2004) Cyclophosphamide metabolism in children with non-Hodgkin’s lymphoma. Clin Cancer Res 10:455–460. [DOI] [PubMed] [Google Scholar]
- Zakhari S. (2006) Overview: how is alcohol metabolized by the body? Alcohol Res Health 29:245–254. [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Leeder JS.(2013) Epigenetic regulation of ADME-related genes: focus on drug metabolism and transport. Drug Metab Dispos 41:1721–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]