

Abstract
Many types of cancers develop in the oral and maxillofacial region. Squamous cell carcinoma is the most common cancer and constitutes over 90 percent of these tumors. Malignant transformation is a genetic process, which later makes a phenotyping change at the cellular level. Some cancers such as oral squamous cell carcinomas (OSCCs) develop from pre-malignant lesions and conditions. Despite advances in the treatment of OSCC, the 5-year survival rate remains approximately 50% due to inability of early detection of OSCC and precursor lesions. Early detection of oral cancer, especially in the premalignant stage, can decrease mortality and morbidity significantly. This article reviews some clinical, histopathological features and etiopathogenesis of pre-cancerous lesions of the oral cavity and skin of face and lip vermilion. A relevant English literature search in Pubmed, Science Direct, and Google Scholar was performed from 1930 to 2015. Full text of 191 articles met the specific inclusion criteria for this review.
Key Words: Face, Head, Leukoplakia, Mouth, Neoplasm, Precancerous condition, Skin
Introduction
Different types of cancer develop in the oral and maxillofacial region. Squamous cell carcinoma is the most common cancer and constitutes over 90% of these tumors. Malignant transformation is a genetic process, which makes a phenotyping change at the cellular level (1). Some cancers such as oral squamous cell carcinomas (OSCCs) develop from pre-malignant lesions and conditions (2). Development of the oral cancer is a multistep process including genetic, epigenetic, and metabolic alterations (3). Despite advances in the treatment of OSCC, the 5-year survival rate remains approximately 50% due to inability of early detection of OSCC and precursor lesions (2). Early detection of a malignancy, especially in the pre-malignant stage, can significantly decrease mortality and morbidity (4).
From the etiological and clinicopathological aspects, tumors in the oral and maxillofacial region can be divided into two main categories: cancers of the oral cavity and cancers of the skin of face including lip vermilion.
This article reviews pre-cancerous lesions of the oral cavity and skin of face and lip vermilion with particular emphasis on etiopathogenesis.
Methods
A relevant English literature search in PubMed, Science-Direct, and Google Scholar was performed. The keywords; ‘face’, ‘head’ , ‘leukoplakia’, ‘ mouth’, ‘neoplasm’,‘ precancerous condition’, and ‘ skin’ were searched in title/abstract of publications; limited to 1930 to 2015. The inclusion criterion was all related precancerous lesions. A total of 3056 articles were found. Among them, full text of 191 articles met the specific inclusion criteria for this review.
Oral Cancer
Oral cancer is considered as the sixth most common cancer worldwide (5). In 96% of cases, the oral cancer is detected above the age of 40 (6). The ratio of males to females is 2:1 but in older ages, the ratio is nearly 1:1.The tongue, particularly the posterior-lateral surface, is the most common site in both men and women (7). Malignant lesions of the floor of mouth, soft palate and oropharynx are also common. These locations are surfaced by an unkeratinized mucosa. It is assumed that carcinogens dissolved in the saliva, have a prolonged contact with a thin mucosal barrier, so have better access to the surface epithelial squamous cells (8).
In terms of etiology, oral cancers have a strong association with tobacco use (9, 10). In addition, alcohol may involve in the tumor development by increasing oral mucosal permeability, which facilitates the passage of carcinogens such as nitrosamines. Additionally, alcohol also has effects on the cell membrane, and inhibits DNA repair (11). Plummer–Vinson syndrome (PVS) also called Paterson–Brown–Kelly syndrome, a type of iron deficiency anemia, contributes to an increased risk of oral cancer as well (12). Recently, human papillomavirus (HPV) has been suggested as another the etiologic factor for the oral and oropharyngeal cancer (13). Early genetic changes at specific chromosome sites (3p14 and 9p21) involve in malignant transformation (14). Studying the gene mutations can help in distinguishing the lesions with a higher risk of progressing to malignancy. For example, allelic loss of either 3p or 9p chromosome arms has been detected in 50% of leukoplakias which is associated with a 3-8 fold increased risk of malignant transformation (15). Moreover, the anterior oral cavity is constantly exposed to chemicals, drinks, food, infectious agents, and physical injury (16).
Leukoplakia
This term often causes confusion and controversy. According to WHO definition, leukoplakia is a white patch or plaque that cannot be characterized clinically or pathologically as any other disease (17). Smokeless tobacco is associated with developing a leukoplakia in 8.4% of cases (18). Clinically, leukoplakia presents in different views including thin, thick or homogeneous, granular or nodular and proliferative verrucous leukoplakia (19). The risk of malignant transformation significantly increases among people aged 60-70 years (20). Leukoplakias on the floor of mouth, lateral tongue, and lower lip show more dysplasia or malignant transformation (20-22). The possible risk factors for malignant transformation are female gender, idiopathic leukoplakia (in non-smokers), larger than 200 mm2, long duration, non-hemogenous type, presence of Candida species, and epithelial dysplasia (4). Location on the tongue and/or floor of mouth that oral leukoplakia with dysplasia has a higher risk of malignant transformation rate compared to oral leukoplakia without dysplasia (23). Speckled leukoplakia (white lesions or white nodular patches interspersed with erythematous regions) and erosive leukoplakia are often associated with epithelial dysplasia or carcinoma (24, 25).
Different molecular markers have been detected regarding dysplastic changes, and malignant transformation of oral leukoplakia. For example, accumulated p53 protein has been shown in 89% of oral leukoplakias, mainly in basal layers (26). Mutated TP53 in premalignant oral lesions was assumed to predict malignant progression (27). TP53 and Mdm2 are highly expressed in the leukoplakia cancer group compared to the normal group (28). Another study has reported the association between a higher expression of SMAD4 and increased rate of malignant transformation (29). Overexpressionin of cyclin D1 and p63 with increasing severity of dysplasia and also a decrease in p27 expression have been found in oral leukoplakia as well (30). Additionally, increased expression levels of metalloproteinases 1, 9, and 11, and vascular endothelial growth factor (VEGF) have been detected in dysplastic leukoplakia progressing to squamous cell carcinoma compared to those that do not (31, 32). Overexpression of the human telomerase reverse transcriptase (hTERT) associated with increased telomerase activity has been detected in oral leukoplakias as an early phenomenon in the process of carcinogenesis (33). A previous study has shown the overexpression of retinoblastoma (Rb) protein in leukoplakic lesion compared to the normal tissue (34). Increased expression level of COX-2 and Ki-67 are related to the degree of dysplasia (35).
Leukoplakia has no specific histopathological feature and is only a clinical term (36). However, histopathologic findings are hyperkeratosis with or without epithelial dysplasia (Figure 1). Epithelial dysplasia is divided into three subclassifications: mild, moderate, and severe (37). Although the presence of epithelial dysplasia is the gold standard for the detection of malignant transformation of the lesions, but there are three major problems as follows: (1) as the diagnosis is subjective it cannot be standardized; (2) not only all lesions with dysplasia do not become a malignant lesion but also some of them even regress; and (3) in some cases carcinoma develops from lesions without any previous history of epithelial dysplasia (38).
Fig 1.

A photomicrograph of a leukoplakic lesion showing mild epithelial dysplasia. (H & E, X 40
Other oral white lesions such as frictional keratoses, morsicatio buccarum are not considered as leukoplakia as they are not premalignant lesions, and are reversible after elimination of suspected etiological factors. Additionally, other oral white lesions such as candidiasis, lichen planus, leukedema should not be considered as leukoplakia as they have specific microscopic features (19).
Proliferative verrucous leukoplakia
Proliferative verrucous leukoplakia (PVL) is a rare lesion. In the early stage it is similar to conventional leukoplakia, both clinically and histopathologically (39), but in the advance stage it appears clinically as verrucous carcinoma (40). PVL is classified as a potentially malignant lesion in the oral cavity (38). In the clinic, the lesion initially develops as a focal hyperkeratosis, which gradually progresses to form an exophytic multifocal lesion (41). Therefore, it is characterized by 4 phases: 1) focal early development; 2) geographic expansion over time; 3) development of a verrucoid/warty appearance; and 4) malignant transformation. In some patients several different OSCCs can develop, therefore, PVL has been considered as a representative of the concept of field cancerization (42). PVL shows variable microscopic features. In early stages, it shows a benign hyperkeratosis. With time, it appears as a papillary and exophytic mass. In later stages the papillary proliferation exhibits downgrowth of well-differentiated squamous epithelium with blunt and broad rete ridges, which invades into the underlying lamina propria. In the final stages the invading epithelium transforms to SCC (43). There are no specific histologic criteria, therefore, diagnosis is based on the histopathologic and cilinical features, along with the behavior (44). TP53 mutaion has not been identified in PVL (18).
Erythroplakia
Erythroplakia is an uncommon fiery red patch, which cannot be classified as any other condition clinically, and histopathologically (17). Clinically, the lesions present as flat to slightly raised red lesions with irregular borders (40). TP53 mutation has been detected in 46% of oral erythroplakias (45). The histopathological characteristics include the lack of excess surface keratinization, some degree of dysplasia, and even carcinoma in situ or SCC (40, 46) (Figure 2).
Fig 2.

A photomicrograph showing atrophic oral epithelium with atypia, squamatization of the basal cell layer, and underlying chronic inflammation in a clinically diagnosed erythroplakia. (H & E, X 400
Verrucous hyperplasia
Oral verrucous hyperplasia (OVH) appears as a white or pink single or multifocal plaque or nodule with a verrucous or papillary surface, resembling as a large wart. This term can be used as a clinical or a histopathologic feature (47). Moderate dysplasia is predominant than mild dysplasia and is correlated with consumption of different tobacco preparations (48). Verrucous hyperplasia can develop a malignancy, mostly SCC and in a lesser number a verrucous carcinoma (49).
Histopathologic features include sharp and keratotic projections with keratin-filled invaginations without obvious fibrovascular cores (Figure 3). It never extends below that of the adjacent normal epithelium. Mild dysplasia associated with a lichenoid/interface inflammatory reaction can also be seen. In 68% of cases heavy inflammatory cell infiltration including lymphocytes, plasma cells and histiocytes can be observed (50). Lateral and downward growth, broadened and bulbous-like rete ridges are formed. If a broad-front invasion occurs, it can be designated as a verrucous carcinoma. A verrucous carcinoma can be distinguished from a verrucous hyperplasia by a peripheral buttress/shoulder and extension below the lower border of the normal epithelium (51).
Fig 3.

A photomicrograph of a clinically verrucous lesions showing epithelial proliferation. (H & E, X 40
Tobacco pouch keratosis; Smokeless tobacco keratosis; Smokeless tobacco-induced keratosis; Snuff dipper’s keratosis
This lesion is mostly occurs on the buccal or labial vestibule where the tobacco is held, however, the extension of the lesion into the adjacent gingiva and buccal mucosa has been reported (52). In the early stage, it appears as a white wrinkled lesion disappearing by stretching. In the advanced stage, the lesion exhibits as a thickened grayish white zone with folds and fissures. Most of the lesions resolve within 2-6 weeks after cessation of the habit, otherwise, an incisional biopsy should be performed (19). In the microscopic examination, hyperkeratinized and acanthotic epithelium with parakeratin chevrons can be seen. The epithelial dysplasia is not a common finding. Although a significant dysplasia or SCC may be seen, it is usually mild in degree (43).
Reverse smoking
In some countries, due to placing of the lit end of the cigarette or cigar in the mouth, reverse smoking (RS) can develop, and is associated with increased risk of malignant transformation (53). Among 497 patients with leukoplakia, 91.7% of the palatal leukoplakias were found in reverse smokers, and out of 10 oral cancer cases, 9 were located on the palate (54). Keratosis associated with reverse smoking is a precancerous lesion (55).
Histopathological findings include marked hyperorthokeratosis in 80% of cases associated with epithelial hyperplasia in 73.1% of cases. The granular cell layer is dispersed throughout the upper half of the epithelium. The presence of melanin-containing cells in the basal layer is another histological feature. Mild inflammation can be found in the lamina propria of the palatal biopsies (54, 56).
Oral submucous fibrosis
Oral submucous fibrosis (OSF) is a chronic lesion, which mostly develops in Indians (57). The possible mechanisms in the development of the lesion are increased collagen synthesis or reduced collagen degradation (58). Areca nut contact with epithelial cells induces transforming growth factor beta (TGF-β) signaling, which in turn induces inflammation and fibrosis in the underlying connective tissue. In addition, TGF-β produced by epithelial cells can diffuse into the connective tissue (59). The characteristic clinical features of OSF are burning sensation, blanching and stiffening of the oral mucosa such as the lips, tongue, and palate (58). A previous study has indicated that among 371 patients with oral cancer, 30% had OSF. Additionally, the patients with both oral cancer and OSF were younger than patients with oral cancer (45.11 vs 50.07 yr). Oral cancer with OSF was also more common in men (male: female ratio= 10:1) compared with oral cancer (male to female ratio=3.2:1). The tongue was the most common site of involvement in oral cancer-OSF group (60). Up-regulation of some cytokines such as IL-8, IL-6, IL-1, and fibroblast growth factor (FGF) has been reported in fibrosis or OSF (61). In addition, loss of heterozygosity in 23 “hotspot” loci, which controls the cell cycle has been recognized as a malignancy marker in OSF (62). Overexpression of p53 and p63 has been detected in OSF. In a study on PCNA expression status, positive expression of PCNA mainly in basal and suprabasal layers had been detected in all cases of OSF. Although, there was no statistical significant mean difference of PCNA expression in basal and suprabasal layers between OSCC and OSF, there was a statistical significant mean difference in PCNA expression in superficial layers (63).
In early stages of OSF, the microscopic examination shows a juxtra –epithelial inflammation, followed by hyalinization. Later, the atrophy of epithelium with focal para-keratosis or hyperkeratosis along with imbalance between degradation and synthesis of extracellular matrix (ECM), mainly collagen occurs. Finally, marked collagen accumulation in the lamina propria, submucosa, and superficial muscle layer can be seen (58, 64). Increased deposition of type I collagen, elevated expression of plasminogen activator inhibitot-1 (PAI-1), and tissue inhibitor metalloproteinase-1 (TIMPs) had been show in the connective tissue (65, 66).
Oral Lichen Planus and Oral Lichenoid Reaction
Oral Lichen Planus (OLP) is a chronic inflammatory disease (67). It is suggested that OLP is a T cell –mediated autoimmune disease. Induction of apoptosis of the basal cells of epithelium by CD8+ T cells is the possible mechanism of developing of OLP (68). WHO considers OLP as a precancerous lesion especially in the presence of dysplasia (69). Krutchkoff et al. criticized this opinion. According to their review, there is not sufficient document in terms of their criteria for the malignant transformation of OLP (70). Krutchkoff and Eisenberg have suggested the term lichenoid dysplasia for cases of OLP with dysplasia. They believed that some reported cases of OLP, which developed a malignancy, were lichenoid lesions with dysplasia (71, 72). These authors proposed histopathological and/or clinico-pathological diagnostic criteria; however, these criteria have not been validated (73). Additionally, both OLP and OSCC are not rare diseases; therefore, they may develop simultaneously (43). On the other hand, there are some reports of developing a malignancy in the same location of previously diagnosed as OLP (74-76). Further, strict clinical studies need to resolve the question.
Candida albicans, Hepatitis C virus (HCV) infection, and immunosuppression are considered as the possible risk factors in OLP malignant transformation. Besides, H. pylori was detected in 59.2% of OLP tissue samples in a previous study (77). Treatment with topical corticosteroids is also associated with a higher risk of developing a cancer on the OLP lesion (78). Different sites of the oral cavity have been reported as the preferred site for malignant transformation. While some studies have reported the tongue as the preferred site of malignancy (75), some others had indicated the midline of the palate, gingiva and lips (79, 80). The buccal mucosa had been reported as the highest risk site for malignant transformation (81). Interestingly, development of a second carcinoma has been indicated in 50% of the cases among them new malignancy develops in the same site of the primary tumor in 20% of the cases (82). Atrophic-erosive forms were predisposed to cancer development (79, 81), however, in some series, keratotic form (plaque) was more likely to undergo malignant transformation (75, 79, 80). Most cases of OSCC have been reported on the lateral side of tongue, however, some cases of OSCC have been found on the dorsum of tongue (75, 83).
The presence of a well-defined band-like infiltration of inflammatory cells dominantly lymphocytes, hydropic degeneration of epithelial basal layer, and absence of epithelial dysplasia (Figure 4) are the histopathological criteria for OLP diagnosis (84). The lesions with epithelial dysplasia should not be considered as an OLP lesion. Therefore, terms such as OLP with atypia or OLP with dysplasia should not be used. On the other hand, it is not so easy to rule out the development of epithelial dysplasia in OLP, hence the exclusion of all lesions resembling OLP with epithelial dysplasia may lead the underestimation of malignant transformation rate of OLPs (85). Infiltration of chronic inflammatory cells can be a strong risk factor for cancer development (86, 87). Inflammatory cells may produce an excess nitric oxide (NO). In addition, epithelial apoptosis, probably due to infiltration of inflammatory cells is another risk factor (88), as increased cell proliferation rate of basal epithelial cells results in cancer development (89). A previous study revealed the decreased expression levels of β-catenin, E-cadherin and EGFR in OLP compared to normal tissue (90). Additionally, down-regulation of ANXA1 protein expression was identified in OPLs compared to normal group (91). Oral Lichenoid Reactions (Lesions) (OLRs) are lesions similar to OLPs with different etiology (92). On the lateral border of the tongue, dental materials such as amalgam and composite restorations may be associated with OLR (93). Graft–vs-host disease, seen mainly in bone marrow transplant recipients, is another lichenoid reaction with the potential of developing an oral cancer. A systematic review on the malignant transformation of OLP and OLR found that 85 cases of SCC in developed in OLP lesions and 4 cases of SCC arose in OLRs. Malignant transformation rate for OLP was between 0 and 3.5% and that for OLR was 3.2% (76). A previous study detected the TP53 and Ki67 proteins in OLP and OLR in more than 80% of the cells (94).
Fig 4.

A photomicrograph of a lichen planus lesion showing acanthosis, saw-toothed-shaped rete ridges and band-like infiltration of lymphocytes immediately underlying the epithelium. (H & E, X 400
Lichenoid dysplasia
The term lichenoid dysplasia (LD) was introduced in 1985, used in cases of lichenoid stomatitis with dysplasia. Etiopathogenesis of LD is different from that of OLP. In OLP, lichenoid infiltration represents cell-mediated immune response provoked by different antigens, whereas in LD, lichenoid infiltration occurs against atypical epithelial cells (71). Lichenoid dysplasia mostly appears as an erythematous or leukoplakic area on the buccal mucosa or gingiva and is not a symmetrical lesion as can be found in OLP. Microscopic findings of these lesions consist of hyperparakeratosis or hyperorthokeratosis, epithelial dysplasia and band-like lymphocyte infiltration (Figure 5). The basal cell hyperplasia and atypia rather than degeneration is the important histological feature (95). Lack of liquefaction degeneration and intact or even hyperplastic basal cell layer is a major distinguishing characteristic of LD from OLP (71, 96).
Fig 5.

A photomicrograph showing mild epithelial dysplasia, surface hyperorthokeratosis, and lichenoid mucositis. (H & E, X 40
Epidermolysis bullosa
Epidermolysis bullosa (EB) is a heterogeneous group of inherited diseases, characterized by trauma-induced blistering of the skin and mucous membranes (97). Three major EB types are simplex, junctional and dystrophic (98). Infants with EB have generalized recurrent blistering, resulting in ulceration, pseudosyndactyly with mitten-like deformities of hands and feet, nail loss, as well as scarring or strictures of the oral mucous membrane, and esophagus (99). Oral lesions have been reported in the junctional and dystrophic forms (100). Although malignancy mostly occurs on the skin, it can also occur on the oral cavity, especially the lingual mucosa (43, 101, 102).
Chronic Discoid Lupus Erythematous
Chronic Discoid Lupus Erythematous (CDLE) is a chronic form of cutaneous lupus, which clinically presents as an erythematous, scaly and depigmented plaque (103). Head and neck area is affected in 41% of all cases (104). Oral lesions are asymmetrically distributed affecting the palate, buccal mucosa and tongue. The buccal mucosa can be affected in 15% of the patients and may transform to leukoplakia (105). The microscopic features include hyperkeratosis, degeneration of the basal layer, and subepithelial lymphocytic infiltration. Deep inflammatory infiltration, often perivascular orientation distinguishes CDLE lesions from OLPs (43) (Figure 6).
Fig 6.
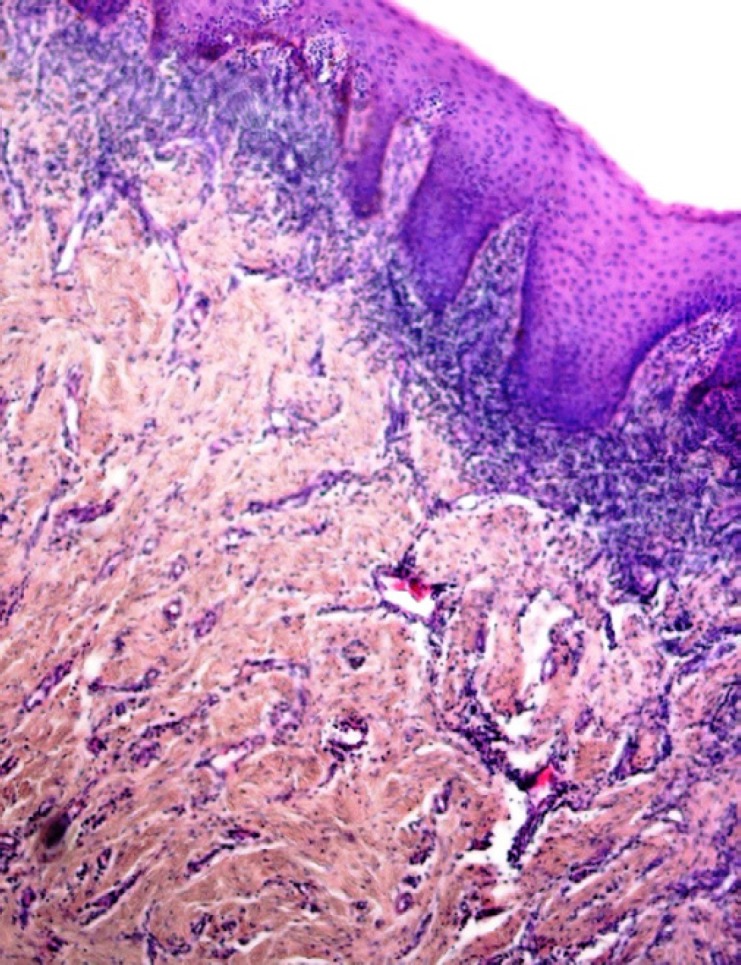
A photomicrograph of a chronic discoid lupus erythematosus lesion showing hyperparakeratosis and lichenoid pattern of inflammation. (H & E, X 40
Dyskeratosis Congenita
Dyskeratosis Congenita (DC) is a rare inherited bone marrow failure syndrome characterized by the triad of oral leukoplakia (80%), dystrophy of nails (90%), and reticular skin pigmentation (90%) (106). Mutations have been identified in TERC (telomerase RNA component), which provide a direct link between DC, telomerase, and DKC1 gene (107, 108). Malignancies develop in 10% of patients, typically in the third decade of life. The most common malignancy is SCC, and typically develops in areas of leukoplakia (109). Microscopically, oral leukoplakia in DC shows progression from hyperkeratosis to dysplasia (110). Table 1 summarizes the main characteristics of the precancerous lesions of the oral mucosa.
Table 1.
The main characteristics of the precancerous lesions of the oral mucosa
| Lesion | Gender | Age | The most prevalent site | The incidence of dysplasia or /and malignant transformation | The risk factors and possible etiological factors |
|---|---|---|---|---|---|
| Leukoplakia | Both | > 50 years | Buccal mucosa, alveolar mucosa, lower lip | 5%-19.9% | Smoking, smokeless tobacco, HPV, Candida species |
| Proliferative verrucous leukoplakia | F | >60 years | Buccal mucosa, tongue | 40-100% | Not clear may be HPV and EBV |
| Erythroplakia | M | >60 years | Floor of the mouth, lateral tongue, retromolar pad | 14-67% | Chewing tobacco, alcohol, smokeless tobacco |
| Verrucous hyperplasia | M | 40 years | Buccal mucosa ,tongue | 3-17% | Smokeless tobacco, cigarette smoking |
| Tobacco pouch keratosis | M | Any age | Buccal or labial vestibule | 0.6-2.8% | Smokeless tobacco |
| Oral submucous fibrosis | Both | 20-30 years | Fbuccal mucosa | 7-30% | Chewing areca and betel quid |
| Oral lichen planus | F | Middle age | Buccal mucosa, tongue, gingiva | 0.4-5.6% | T-cell–mediated autoimmune disease |
| Oral lichenoid reaction | F | Middle and older | Buccal mucosa, tongue | 0.71% | Dental materials |
| Lichenoid Dysplasis | No data | No data | Buccal mucosa, gingiva | 100% | Previous leukoplakia or erythroplakia |
| Epidermolysis bullosa | Both | Infants | Gingiva, buccal mucosa | Infrequently | Heredity |
| Chronic Discoid Lupus Erythematous |
F | 41 years | Palate, buccal mucosa and tongue | 13.64%, 0.5-2% | Sun exposure |
| Dyskeratosis Congenita | Both | 10 years | Tongue,buccal mucosa | 35% | Mutation of TERC gene |
The Facial and Vermilion Lip Cancer
Actinic cheilitis
Actinic cheilitis (AC) is a chronic inflammatory lesion (111). In the clinical examination, the lesion is characterized by the darkening of the lip and atrophy of the vermilion border at the borders. Over time, scaly areas develop and become thick by extending to the wet line of the lip. Chronic focal ulcers as well as leukoplakic lesions can occur (112). AC may transform into SCC (112), but SCC arising on AC rarely metastasizes to cervical lymph nodes. A malignancy develops in patients older than 50 years of age who use tobacco and are exposed to the sun chronically (113).
Histopathologically, AC presents a variety of changes including varying degrees of keratosis, epithelial hyperplasia or atrophy, solar elastosis, and the presence or absence of dysplasia (111). The most important aspect is the keratinocyte atypia, which gradually occurs in the epithelium. The number of mast cells increases compared to normal samples (114). Mast cells play a crucial role in inflammation, and contribute to the defense against tumor development as well as its invasion (115,116). However, some previous studies indicated that mast cells could promote ECM degradation and tumor progression (114). CD1a-positive Langerhans cells and mast cells were found in the lamina propria and epithelium of AC, respectively. CD1a-positive Langerhans cells were assumed to have a protective role against transforming into SCC, but the role of mast cells in AC has not yet been defined (117).
Actinic keratosis
Actinic keratosis (AK) is a cutaneous neoplasm composed of transformed keratinocytes as the result of chronic UV exposure (118), specifically; UV-B radiation which causes mutation in the p53 gene. Other etiological factors include fair skin, light colored eyes, male gender, older age, and increased sun exposure (119). Many of the AK lesions are asymptomatic, usually as an erythematous papules or plaques on sun-exposed areas. It may regress or progress to an invasive SCC. AK is the early form of squamous cell carcinoma in situ (120). Some risk factors have been known such as skin type, the amount of photo damage and a history of immunosuppression (121). Some clinical features such as induration, inflammation, diameter larger than 1cm, rapid enlargement, bleeding, erythema and ulceration suggest an increased risk for malignant transformation (122). The length of time to progress to SCC is 24.6 months (123), and the rate of metastasis is quite low, only in 1–2% of cases (124). The strong expression of Keratin-14 in spinous and granular layers of SCC tissue developed from AK, is probably a prognostic factor for tumor progression of AK (125).
The histopathological features of AK include an atrophic or acanthotic or even normal thickness of the epidermis. The acanthotic variant is characterized by elongated rete ridges. Atypical keratinocyte is the pathognomonic feature, which begins within the basal layer cells. In advanced stages, keratinocyte atypia extends above the basal layer (81) (Figure 7). Elastosis formation associated with the infiltration of mast cells can be seen in AK lesions. It has been postulated that mast cells stimulate elastosis via activation of fibroblasts to secrete elastin and proteases such as matrix metalloproteinase (MMP) (126). In human both ultraviolet and infrared radiation may stimulate mast cell proliferation. In addition, keratinocytes or fibroblasts can produce chemotactic factors for mast cells (127). The number of T–cells and Langerhans cells significantly increases in inflamed AK and decreases by progression to SCC. These findings may indicate that progression changes from benign to malignant lesions are associated with an inflammatory response (128).
Fig 7.
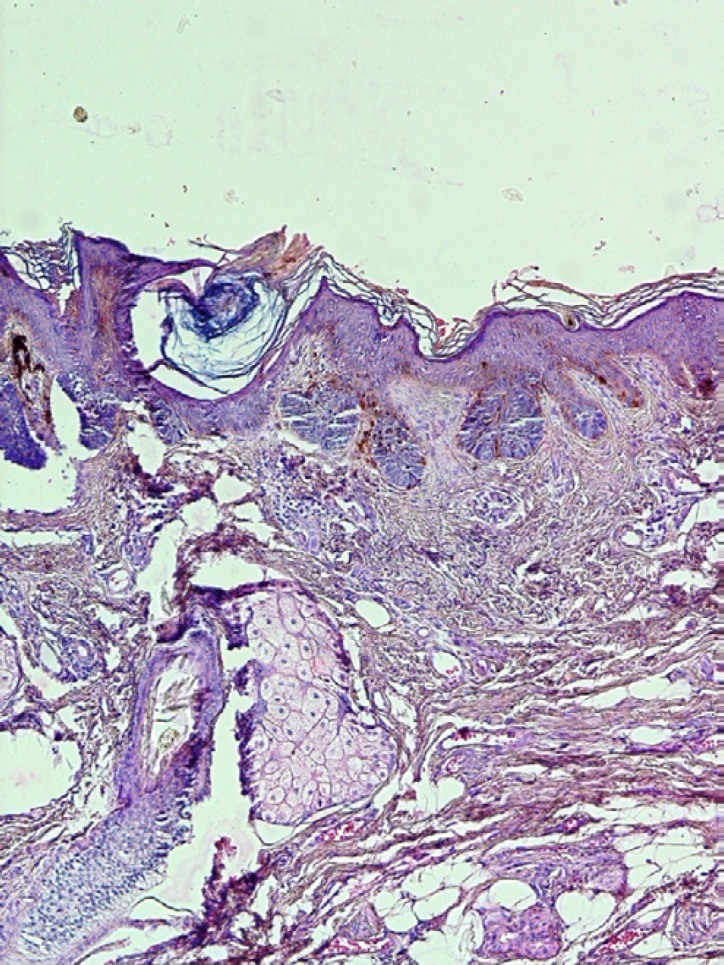
A photomicrograph of an actinic keratosis showing surface hyperkeratosis and acanthosis. (H & E, X 40
Epidermolysis bullosa
Developing a malignancy within chronic skin wounds and long-term scars is very common event in recessive dystrophic EB. Scarring and repetitive tissue stress lead to malignant changes in EB patients. Development of SCC is correlated to the severity and extent of ulceration and scarring (97). A diminished immunological status due to malnourishment can be considered as the pathogenesis in junctional EB cases (129). The patients with severe dystrophic type are at risk of developing a malignancy at younger age than junctional type. The most common age of developing SCC for dystrophic type is 20-25 years and for junctional type is 28-70 years. For both types, the patients have the risk of developing multiple primary SCCs (97). Mutation of p53 has been detected in EB related SCC (130). Mutation of the COL7A1 gene, encoding type VII collagen, has been suggested as the etiologic factor for dystrophic form of the disease (131). Large melanocytic nevi are other findings in EB, which arise in the sites of previous bullae or erosions. These lesions have been reported in children and are susceptible to develop melanoma (132). The risk of melanoma has been reported in patients with the recessive dystrophic form (97). There is also an increased risk of occurrence of multiple keratoacanthomas (133), and squamous cell carcinomas in individuals with junctional EB (134).
Chronic Discoid Lupus Erythematous (CDLE)
SCC and less commonly BCC arise from lesions of CDLE. The mean interval between initiation of the DLE lesion to the appearance of SCC is 30.8 years (135). A long history of DLE in association with bleeding and ulceration of nodules can be considered as progression to SCC (136). Ultraviolet light or radiation used for treatment of DLE in the early 20th century was the etiological factor for malignant transformation (137). Hyperkeratosis, and follicular plugging, vacuolar degeneration of the basal cell layer of epidermis and patchy dermal lymphocytic infiltration are the histopathological characteristics of CDLE (138).
Chronic Inflammation
There are some reports of SCC developing in areas of cicatrizing dermatoses, such as Marjolin’s ulcer, and inherited dermatoses like epidermolysis bullosa (139,140). Chronic irritation and infection, and repeated trauma are suggested as etiological factors. Therefore, repeated healing, and toxins released by the damaged tissue may cause the cell mutation (141,142).
Some other precancerous skin lesions which develop a malignant melanoma are as follows:
Dysplastic Nevi
Dysplastic Nevi (DN) is clinically acquired melanocytic lesions similar to malignant melanoma. Patients with DN have multiple moles but the majorities are not dysplastic (143). Some of the genetic mutations such as CDKN2A and CDK4 may play role in the pathogenesis of DN (144, 145). DN lesions are associated with overexpression of pheomelanin, which may cause DNA damage and tumour progression (146). Junctional proliferation of single nests of melanocytes often with bridging between nests, melanocytes with large pleomorphic nuclei and lymphohistiocytic infiltrate within the epidermis are histopathological characteristics of DN (147).
Congenital Melanocytic Nevus
Congenital Melanocytic Nevus (CMN) is one of the most common lesions in newborn infant (148). In the clinic, they are round to oval shaped lesions with brown to dark brown color (149). The cause of CMN is not clear but defects in the migration or maturation of melanocytes in the embryo are hypothesized. Characteristic histopathology includes the infiltration of melanocytes into the reticular dermis, and around skin appendages such as follicles and sweat glands. The surgical removal of CMN decreases the risk for development of melanoma (148).
Nevous sebaceous (of Jadassohn)
Nevous sebaceous (of Jadassohn) is a hamartoma of the epidermis and presents from birth which has the potential to develop to BCC. Nearly all Nevous sebaceous (of Jadassohn) can be found on scalp, forehead or face. Clinically, the lesions appear as smooth yellowish, hairless patches (150). Prevention excision is suggested (151). Table 2 summarizes the main characteristics of the precancerous lesions of the skin of the face and lip.
Table 2.
The main characteristics of the precancerous lesions of the skin of the face and lip
| Lesion | Gender | Age | The most prevalent site | The incidence of dysplasia or /and malignant transformation | The risk factors and possible etiological factors |
|---|---|---|---|---|---|
| Actinic cheilitis | M | Middle age | Lower lip | 62.07%,16.9% | UV, tobacco, alcohol |
| Actinic keratosis | Both | >40 years | Hand, wrist, and arm | Up to 20% | UV |
| Epidermolysis bullosa | Both | Infants | Exteremities | 76.5% | Heredity |
| Chronic Discoid Lupus Erythemato | F | 41 years | Scalp, ears, lips and nose | 3.3% | Sun exposure |
| Dysplastic Nevi | Both | 30-40 years | Scalp,breast and buttocks | 5.7-19.7% | Genetic mutation, environmental factors |
| Congenital Melanocytic Nevi | Both | Infants | Mouth, palms and soles | 04-10% | Congenial |
Discussion
Recently, tumor progression models have been made for a few tumors. Genetic pathway correlation helps to construct these models. Not always a benign squamous hyperplasia progresses to a malignancy, therefore, some genetic alterations develop cancers (152). Identification of early genetic alterations, tumor suppressor genes and proto-oncogenes provides necessary information in cancer treatment. Close observation of cases with dysplasia/neoplasia has an impact on patient’s life but there is always a limitation due to the clinical differences between inflammatory benign lesions and true dysplastic/neoplastic changes. Slaughter proposed the concept of field cancerization in 1953 (153). According to this hypothesis, the entire epithelium of upper aerodigestive tract is exposed to carcinogens, therefore, there is a higher incidence of multiple genetic alterations to cause cancer development. In the oral cavity, some etiological factors have been identified. Tobacco smoking and alcohol consumption play important roles in oral cancer. Components of cigarette smoke, including nicotine can stimulate the proliferation of various normal and cancerous cells (154) by increasing the levels of both growth factors such as VEGF, VEGF-C, TGF-b, and growth factor receptors like VEGFR-2, PDGFR, HGFR and EGFR. Nicotine also has anti-apoptotic effect via activation of PKC, PKA and NFkB, and down-regulation of the p53 tumor suppressor protein (155). In a study, elevated dysfunctional p53 was found in heavy smokers. Moreover, Ras (Rat sarcoma) mutation has been demonstrated in tobacco chewers (156). Ethanol becomes oxidized into acetaldehyde, which is a carcinogen. Marked levels of acetaldehyde can be detected in saliva after taking ethanol. The oral microbiota may contribute in cancer development due to acetaldehyde production (157). For example, Candida albicans has been found in the histological sections of leukoplakia invading the upper epithelium. Variants of human papillomaviruses 16 and 18 are important co-factors, especially in cancers of the tonsils (158). HPV infection inhibits p53 tumor suppressor gene expression, the most well studied mutated gene in the oral pre-malignant lesions (159). Angiogenesis has a pivotal role in carcinogenesis. VEGF is not the only factor involved in angiogenesis. Other factors such as ET axis, Galactin-1 and -3 also contribute in the control of angiogenesis and growth of cancer (86,87, 160). Angiogenesis has been detected in oral pre-malignant lesions, which persist during progression of carcinogenesis. Increased level of VEGF was found in oral pre-malignant conditions (161). Hyperactivity of the EGFR/ERK, and PI3K/AKT/mTOR signaling pathways has been found in OSCC and premalignant cell lines (162). Amplification of proto-oncogene, cyclin D1, is another finding in head and neck squamous cell carcinoma (HNSCC) (156). Laminin-5ᵧ2 positivity distinguishes truly oral premalignant lesions from those that are not (163). Podoplanin expression can be used as a predictor of the risk of cancer development in oral precancerous lesions (164).
Skin cancer is the most common human cancer. Basal cell carcinoma (BCC) is the most common skin cancer and the most frequently diagnosed cancer in humans. While BCCs are believed to arise de novo, cutaneous SCCs arise via a multistep process. Keratinocytes gradually acquire new phenotypic characteristics, which lead to aggressive behavior. Molecular and cytogenetic studies on AK provide supporting evidence that AK is a precursor of SCC. Preventing skin cancer with early diagnosis of precancerous skin lesions has a great impact on cancer treatment. Chronic exposure to UV radiation is the major etiologic factor. UV radiation causes p53 genetic mutation, which is a key regulatory molecule in the cellular response to UV radiation. UV radiation leads to migration of Langerhans cells to the draining lymph node, thereby reducing Langerhans cell number in the skin. UV radiation also inhibits the response of mast cells, cytotoxic T cells, and memory T cells. DNA of HPVs has been found in 52% of BCCs, and 41% of AKs (159) which may be another etiologic factor.
Conclusion
Oral cancer and skin cancer are very common with high mortality rate, therefore, the knowledge about precancerous lesions and their behavior has a crucial impact on patients’ life. Extra oral and intraoral examination of the head and neck region of the patients has a crucial impact on identifying changes affecting the oral mucosa and skin. Careful observation of the clinical and histological changes, along with diagnosis at earlier stages can give lower morbidity and mortality.
Acknowledgements
There has been no financial source for this study. The author declares that there is no conflict of interests.
References
- 1.Epstein JB, Zhang L, Rosin M. Advances in the diagnosis of oral premalignant and malignant lesions. J Can Dent Assoc. 2002;68(10):617–21. [PubMed] [Google Scholar]
- 2.Silverman S Jr, Gorsky M, Lozada F. Oral leukoplakia and malignant transformation A follow-up study of 257 patients. Cancer. 1984;53(3):563–8. doi: 10.1002/1097-0142(19840201)53:3<563::aid-cncr2820530332>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 3.Lippman SM, Hong WK. Molecular markers of the risk of oral cancer. N Engl J Med. 2001;344(17):1323–6. doi: 10.1056/NEJM200104263441710. [DOI] [PubMed] [Google Scholar]
- 4.van der Waal I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009;45(4-5):317–23. doi: 10.1016/j.oraloncology.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 5.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 6.Myers JN, Elkins T, Roberts D, Byers RM. Squamous cell carcinoma of the tongue in young adults: increasing incidence and factors that predict treatment outcomes. Otolaryngol Head and Neck Surg. 2000;122(1):44–51. doi: 10.1016/S0194-5998(00)70142-2. [DOI] [PubMed] [Google Scholar]
- 7.Rhodus NL. Oral cancer and precancer: improving outcomes. Compend Contin Educ Dent. 2009;30(8) 486-8, 90-4, 96-8 passim; quiz 504, 20. [PubMed] [Google Scholar]
- 8.Natarajan E, Eisenberg E. Contemporary concepts in the diagnosis of oral cancer and precancer. Dent Clin North Am. 2011;55(1):63–88. doi: 10.1016/j.cden.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Chen JK, Katz RV, Krutchkoff DJ. Intraoral squamous cell carcinoma Epidemiologic patterns in Connecticut from 1935 to 1985. Cancer. 1990;66(6):1288–96. doi: 10.1002/1097-0142(19900915)66:6<1288::aid-cncr2820660632>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 10.Picascia DD, Robinson JK. Actinic cheilitis: a review of the etiology, differential diagnosis, and treatment. J Am Acad Dermatol. 1987;17(2 Pt 1):255–64. doi: 10.1016/s0190-9622(87)70201-1. [DOI] [PubMed] [Google Scholar]
- 11.Maserejian NN, Joshipura KJ, Rosner BA, Giovannucci E, Zavras AI. Prospective study of alcohol consumption and risk of oral premalignant lesions in men. Cancer Epidemiol Biomarkers Prev. 2006;15(4):774–81. doi: 10.1158/1055-9965.EPI-05-0842. [DOI] [PubMed] [Google Scholar]
- 12.Ahlbom HE. Simple achlorhydric anaemia, plummer-vinson syndrome, and carcinoma of the mouth, pharynx, and oesophagus in women: observations at Radiumhemmet, Stockholm. Br Med J. 1936;2 (3945):331–3. doi: 10.1136/bmj.2.3945.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akhter M, Ali L, Hassan Z, Khan I. Association of human papilloma virus infection and oral squamous cell carcinoma in Bangladesh. J Health, Popul Nutr. 2013;31(1):65–9. doi: 10.3329/jhpn.v31i1.14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao L, Lee JS, Fan YH, Ro JY, Batsakis JG, Lippman S. Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med. 1996;2(6):682–5. doi: 10.1038/nm0696-682. [DOI] [PubMed] [Google Scholar]
- 15.Rosin MP, Cheng X, Poh C, Lam WL, Huang Y, Lovas J, et al. Use of allelic loss to predict malignant risk for low-grade oral epithelial dysplasia. Clin Cancer Res. 2000;6(2):357–62. [PubMed] [Google Scholar]
- 16.Paliga A, Mai KT. Squamous cell carcinomas of the anterior oral cavity are commonly associated with simplex (or differentiated) oral intraepithelial neoplasia: clinical and pathologic significance. Int J Surg Pathol. 2014;22(3):231–40. doi: 10.1177/1066896913512866. [DOI] [PubMed] [Google Scholar]
- 17.Kramer IR, Lucas RB, Pindborg JJ, Sobin LH. Definition of leukoplakia and related lesions: an aid to studies on oral precancer. Oral Surg Oral Med Oral Pathol. 1978;46(4):518–39. [PubMed] [Google Scholar]
- 18.Agbor MA, Azodo CC, Tefouet TS. Smokeless tobacco use, tooth loss and oral health issues among adults in Cameroon. Afr Health Sci. 2013;13(3):785–90. doi: 10.4314/ahs.v13i3.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neville BW, Day TA. Oral cancer and precancerous lesions. CA Cancer J Clin. 2002;52(4):195–215. doi: 10.3322/canjclin.52.4.195. [DOI] [PubMed] [Google Scholar]
- 20.Waldron CA, Shafer WG. Leukoplakia revisited A clinicopathologic study 3256 oral leukoplakias. Cancer. 1975;36(4):1386–92. doi: 10.1002/1097-0142(197510)36:4<1386::aid-cncr2820360430>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 21.Pogrel MA. Sublingual keratosis and malignant transformation. J Oral Pathol. 1979;8(3):176–8. doi: 10.1111/j.1600-0714.1979.tb01824.x. [DOI] [PubMed] [Google Scholar]
- 22.Kramer IR, El-Labban N, Lee KW. The clinical features and risk of malignant transformation in sublingual keratosis. Br Dent J. 1978;144(6):171–80. doi: 10.1038/sj.bdj.4804055. [DOI] [PubMed] [Google Scholar]
- 23.Cowan CG, Gregg TA, Napier SS, McKenna SM, Kee F. Potentially malignant oral lesions in northern Ireland: a 20-year population-based perspective of malignant transformation. Oral Dis. 2001;7(1):18–24. [PubMed] [Google Scholar]
- 24.Pindborg JJ, Renstrup G, Poulsen HE, Silverman S Jr. Studies In Oral Leukoplakias V Clinical and Histologic Signs of Malignancy. Acta Odontol Scand. 1963;21:407–14. doi: 10.3109/00016356309028203. [DOI] [PubMed] [Google Scholar]
- 25.Banoczy J. Follow-up studies in oral leukoplakia. J Maxillofac Surg. 1977;5(1):69–75. doi: 10.1016/s0301-0503(77)80079-9. [DOI] [PubMed] [Google Scholar]
- 26.Lippman SM, Shin DM, Lee JJ, Batsakis JG, Lotan R, Tainsky MA, Hittleman WN, Hong WK. p53 and retinoid chemoprevention of oral carcinogenesis. Cancer Res. 1995;55(1):16–9. [PubMed] [Google Scholar]
- 27.Graveland AP, Bremmer JF, de Maaker M, Brink A, Cobussen P, Zwart M, Braakhuis BJ, Bloemena E, van der Waal I, et al. Molecular screening of oral precancer. Oral Oncol. 2013;49(12):1129–35. doi: 10.1016/j.oraloncology.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Cui JJ, Han XL, Wang WM. Expression and significance of p53 and mdm2 in patients with leukoplakia cancer. Asian Pac J Trop Med. 2013;6(10):831–4. doi: 10.1016/S1995-7645(13)60147-9. [DOI] [PubMed] [Google Scholar]
- 29.Xia RH, Song XM, Wang XJ, Li J, Mao L. The combination of SMAD4 expression and histological grade of dysplasia is a better predictor for the malignant transformation of oral leukoplakia. PloS One. 2013;8(6):e66794. doi: 10.1371/journal.pone.0066794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramasubramanian A, Ramani P, Sherlin HJ, Premkumar P, Natesan A, Thiruvengadam C. Immunohistochemical evaluation of oral epithelial dysplasia using cyclin-D1, p27 and p63 expression as predictors of malignant transformation. J Nat Sci Biol Med. 2013;4(2):349–58. doi: 10.4103/0976-9668.117011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan RC, Macabeo-Ong M, Shiboski CH, Dekker N, Ginzinger DG, Wong DT, et al. Overexpression of matrix metalloproteinase-1 and -9 mRNA is associated with progression of oral dysplasia to cancer. Clin Cancer Res. 2004;10(19):6460–5. doi: 10.1158/1078-0432.CCR-04-0656. [DOI] [PubMed] [Google Scholar]
- 32.Arora S, Kaur J, Sharma C, Mathur M, Bahadur S, Shukla NK, et al. Stromelysin 3, Ets-1, and vascular endothelial growth factor expression in oral precancerous and cancerous lesions: correlation with microvessel density, progression, and prognosis. Clin Cancer Res. 2005;11(6):2272–84. doi: 10.1158/1078-0432.CCR-04-0572. [DOI] [PubMed] [Google Scholar]
- 33.Luzar B, Poljak M, Marin IJ, Eberlinc A, Klopcic U, Gale N. Human telomerase catalytic subunit gene re-expression is an early event in oral carcinogenesis. Histopathology. 2004;45(1):13–9. doi: 10.1111/j.1365-2559.2004.01892.x. [DOI] [PubMed] [Google Scholar]
- 34.Thomas S, Balan A, Balaram P. The expression of retinoblastoma tumor suppressor protein in oral cancers and precancers: A clinicopathological study. Dent Res J. 2015;12(4):307–14. doi: 10.4103/1735-3327.161427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sinanoglu A, Soluk-Tekkesin M, Olgac V. Cyclooxygenase-2 and Ki67 Expression in Oral Leukoplakia: a Clinicopathological Study. J Oral Maxillofac Res. 2015;6(2):e3. doi: 10.5037/jomr.2015.6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shafer WG, Waldron CA. A clinical and histopathologic study of oral leukoplakia. Surg Gynecol Obstet. 1961;112:411–20. [PubMed] [Google Scholar]
- 37.Bouquot JE, Whitaker SB. Oral leukoplakia--rationale for diagnosis and prognosis of its clinical subtypes or "phases". Quintessence Int. 1994;25(2):133–40. [PubMed] [Google Scholar]
- 38.Schepman KP, van der Meij EH, Smeele LE, van der Waal I. Malignant transformation of oral leukoplakia: a follow-up study of a hospital-based population of 166 patients with oral leukoplakia from The Netherlands. Oral Oncol. 1998;34(4):270–5. [PubMed] [Google Scholar]
- 39.Shopper TP, Brannon RB, Stalker WH. Proliferative verrucous leukoplakia: an aggressive form of oral leukoplakia. J Dent Hyg. 2004;78(3):7. [PubMed] [Google Scholar]
- 40.Summerlin DJ. Precancerous and cancerous lesions of the oral cavity. Dermatol Clin. 1996;14(2):205–23. doi: 10.1016/s0733-8635(05)70351-x. [DOI] [PubMed] [Google Scholar]
- 41.Hansen LS, Olson JA, Silverman S Jr. Proliferative verrucous leukoplakia A long-term study of thirty patients. Oral Surg Oral Med Oral Pathol. 1985;60(3):285–98. doi: 10.1016/0030-4220(85)90313-5. [DOI] [PubMed] [Google Scholar]
- 42.Bagan J, Scully C, Jimenez Y, Martorell M. Proliferative verrucous leukoplakia: a concise update. Oral Dis. 2010;16(4):328–32. doi: 10.1111/j.1601-0825.2009.01632.x. [DOI] [PubMed] [Google Scholar]
- 43.Neville B, Dam D, Allen C, Bouquot J. Oral and Maxillofacial Pathology. 3rd ed. . China: Saunders Elsevier; 2009. [Google Scholar]
- 44.Barnes L EJ, Reichart P, Sidransky D. World Health Organization Classification of Tumours.Pathology and Genetics of Head and Neck Tumours. Lyon: IARC Press; 2005. [Google Scholar]
- 45.Qin GZ, Park JY, Chen SY, Lazarus P. A high prevalence of p53 mutations in pre-malignant oral erythroplakia. Int J Cancer. 1999;80(3):345–8. doi: 10.1002/(sici)1097-0215(19990129)80:3<345::aid-ijc2>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 46.Shafer WG, Waldron CA. Erythroplakia of the oral cavity. Cancer. 1975;36(3):1021–8. doi: 10.1002/1097-0142(197509)36:3<1021::aid-cncr2820360327>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 47.Wang YP, Chen HM, Kuo RC, Yu CH, Sun A, Liu BY, et al. Oral verrucous hyperplasia: histologic classification, prognosis, and clinical implications. J Oral Pathol Med. 2009;38(8):651–6. doi: 10.1111/j.1600-0714.2009.00790.x. [DOI] [PubMed] [Google Scholar]
- 48.Hazarey VK, Ganvir SM, Bodhade AS. Verrucous hyperplasia: A clinico-pathological study. J Oral Maxillofac Pathol. 2011;15(2):187–91. doi: 10.4103/0973-029X.84492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsue SS, Wang WC, Chen CH, Lin CC, Chen YK, Lin LM. Malignant transformation in 1458 patients with potentially malignant oral mucosal disorders: a follow-up study based in a Taiwanese hospital. J Oral Pathol Med. 2007;36(1):25–9. doi: 10.1111/j.1600-0714.2006.00491.x. [DOI] [PubMed] [Google Scholar]
- 50.Shear M, Pindborg JJ. Verrucous hyperplasia of the oral mucosa. Cancer. 1980;46(8):1855–62. doi: 10.1002/1097-0142(19801015)46:8<1855::aid-cncr2820460825>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 51.Woolgar JA, Triantafyllou A. Squamous cell carcinoma and precursor lesions: clinical pathology. Periodontol 2000. 2011;57(1):51–72. doi: 10.1111/j.1600-0757.2011.00389.x. [DOI] [PubMed] [Google Scholar]
- 52.Somatunga LC, Sinha DN, Sumanasekera P, Galapatti K, Rinchen S, Kahandaliyanage A, et al. Smokeless tobacco use in Sri Lanka. Indian J Cancer. 2012;49(4):357–63. doi: 10.4103/0019-509X.107729. [DOI] [PubMed] [Google Scholar]
- 53.Ortiz GM, Pierce AM, Wilson DF. Palatal changes associated with reverse smoking in Filipino women. Oral Dis. 1996;2(3):232–7. doi: 10.1111/j.1601-0825.1996.tb00230.x. [DOI] [PubMed] [Google Scholar]
- 54.Pindborg JJ, Mehta FS, Gupta PC, Daftary DK, Smith CJ. Reverse smoking in Andhra Pradesh, India: a study of palatal lesions among 10,169 villagers. Br J Cancer. 1971;25(1):10–20. doi: 10.1038/bjc.1971.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Axell T, Pindborg JJ, Smith CJ, van der Waal I. Oral white lesions with special reference to precancerous and tobacco- related lesions: conclusions of an international symposium held in Uppsala, Sweden, May 18-21 1994. International Collaborative Group on Oral White Lesions. J Oral Pathol Med. 1996;25(2):49–54. doi: 10.1111/j.1600-0714.1996.tb00191.x. [DOI] [PubMed] [Google Scholar]
- 56.Ramulu C, Raju MV, Venkatarathnam G, Reddy CR. Nicotine stomatitis and its relation to carcinoma of the hard palate in reverse smokers of chuttas. J Dent Res. 1973;52(4):711–8. doi: 10.1177/00220345730520041201. [DOI] [PubMed] [Google Scholar]
- 57.Pindborg JJ. Is submucous fibrosis a precancerous condition in the oral cavity? Int Dental J. 1972;22(4):474–80. [PubMed] [Google Scholar]
- 58.Tilakaratne WM, Klinikowski MF, Saku T, Peters TJ, Warnakulasuriya S. Oral submucous fibrosis: review on aetiology and pathogenesis. Oral Oncol. 2006;42(6):561–8. doi: 10.1016/j.oraloncology.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 59.Khan I, Kumar N, Pant I, Narra S, Kondaiah P. Activation of TGF-beta pathway by areca nut constituents: a possible cause of oral submucous fibrosis. PloS One. 2012;7(12):e51806. doi: 10.1371/journal.pone.0051806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaturvedi P, Vaishampayan SS, Nair S, Nair D, Agarwal JP, Kane SV, et al. Oral squamous cell carcinoma arising in background of oral submucous fibrosis: a clinicopathologically distinct disease. Head Neck. 2013;35(10):1404–9. doi: 10.1002/hed.23143. [DOI] [PubMed] [Google Scholar]
- 61.Haque MF, Meghji S, Khitab U, Harris M. Oral submucous fibrosis patients have altered levels of cytokine production. J Oral Pathol Med. 2000;29(3):123–8. doi: 10.1034/j.1600-0714.2000.290304.x. [DOI] [PubMed] [Google Scholar]
- 62.Angadi PV, Rao SS. Areca nut in pathogenesis of oral submucous fibrosis: revisited. Oral Maxillofac Surg. 2011;15(1):1–9. doi: 10.1007/s10006-010-0219-8. [DOI] [PubMed] [Google Scholar]
- 63.Keshav R, Narayanappa U. Expression of Proliferating Cell Nuclear Antigen (PCNA) in Oral Submucous Fibrosis: An Immunohistochemical Study. J Clin Diagn Res. 2015;9 (5):Zc20–3. doi: 10.7860/JCDR/2015/13046.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rajalalitha P, Vali S. Molecular pathogenesis of oral submucous fibrosis--a collagen metabolic disorder. J Oral Pathol Med. 2005;34(6):321–8. doi: 10.1111/j.1600-0714.2005.00325.x. [DOI] [PubMed] [Google Scholar]
- 65.Kaur J, Rao M, Chakravarti N, Mathur M, Shukla NK, Sanwal BD, et al. Co-expression of colligin and collagen in oral submucous fibrosis: plausible role in pathogenesis. Oral Oncol. 2001;37(3):282–7. doi: 10.1016/s1368-8375(00)00121-4. [DOI] [PubMed] [Google Scholar]
- 66.Shieh DH, Chiang LC, Shieh TY. Augmented mRNA expression of tissue inhibitor of metalloproteinase-1 in buccal mucosal fibroblasts by arecoline and safrole as a possible pathogenesis for oral submucous fibrosis. Oral Oncol. 2003;39(7):728–35. doi: 10.1016/s1368-8375(03)00101-5. [DOI] [PubMed] [Google Scholar]
- 67.Ebrahimi M, Nylander K, van der Waal I. Oral lichen planus and the p53 family: what do we know? J Oral Pathol Med. 2011;40(4):281–5. doi: 10.1111/j.1600-0714.2010.00979.x. [DOI] [PubMed] [Google Scholar]
- 68.Eversole LR. Immunopathogenesis of oral lichen planus and recurrent aphthous stomatitis. Semin Cutan Med Surg. 1997;16(4):284–94. doi: 10.1016/s1085-5629(97)80018-1. [DOI] [PubMed] [Google Scholar]
- 69.Holmstrup P. The controversy of a premalignant potential of oral lichen planus is over. Oral Surg Oral Med Oral Pathol. 1992;73(6):704–6. doi: 10.1016/0030-4220(92)90014-h. [DOI] [PubMed] [Google Scholar]
- 70.Krutchkoff DJ, Cutler L, Laskowski S. Oral lichen planus: the evidence regarding potential malignant transformation. J Oral Pathol. 1978;7(1):1–7. doi: 10.1111/j.1600-0714.1978.tb01879.x. [DOI] [PubMed] [Google Scholar]
- 71.Krutchkoff DJ, Eisenberg E. Lichenoid dysplasia: a distinct histopathologic entity. Oral Surg Oral Med Oral Pathol. 1985;60(3):308–15. doi: 10.1016/0030-4220(85)90315-9. [DOI] [PubMed] [Google Scholar]
- 72.Eisenberg E. Oral lichen planus: a benign lesion. J Oral Maxillofac Surg. 2000;58(11):1278–85. doi: 10.1053/joms.2000.16629. [DOI] [PubMed] [Google Scholar]
- 73.Gandolfo S, Richiardi L, Carrozzo M, Broccoletti R, Carbone M, Pagano M, et al. Risk of oral squamous cell carcinoma in 402 patients with oral lichen planus: a follow-up study in an Italian population. Oral Oncol. 2004;40(1):77–83. doi: 10.1016/s1368-8375(03)00139-8. [DOI] [PubMed] [Google Scholar]
- 74.Gándara-Reya JM FM, Vilab PG, Carrióna AB, Suárez Peñarandac, Garcia Garciad JMA. Malignant transformation of oral lichen planus in lingual location: report of a case. Oral Oncol Extra. 2004;40(1):1–4. [Google Scholar]
- 75.Irani S. Squamous Cell Carcinoma arising in Oral Lichen Planus: A case report. DJH. 2010;1(2):1–6. [Google Scholar]
- 76.Fitzpatrick SG, Hirsch SA, Gordon SC. The malignant transformation of oral lichen planus and oral lichenoid lesions: a systematic review. J Am Dent Assoc. 2014;145(1):45–56. doi: 10.14219/jada.2013.10. [DOI] [PubMed] [Google Scholar]
- 77.Irani S Monsef Esfahani A, Sabeti Sh, Bidari Zerehpoush F. Detection of Helicobacter pylori in Oral Lichen Planus and Oral Lichenoid Reaction. Avicenna J Dent Res. 2014;6(2):e23213. [Google Scholar]
- 78.Gonzalez-Moles MA, Scully C. Vesiculo-erosive oral mucosal disease--management with topical corticosteroids: (2) Protocols, monitoring of effects and adverse reactions, and the future. J Dent Res. 2005;84(4):302–8. doi: 10.1177/154405910508400402. [DOI] [PubMed] [Google Scholar]
- 79.Lanfranchi-Tizeira HE, Aguas SC, Sano SM. Malignant transformation of atypical oral lichen planus: a review of 32 cases. Med Oral. 2003;8(1):2–9. [PubMed] [Google Scholar]
- 80.Mignogna MD, Lo Muzio L, Lo Russo L, Fedele S, Ruoppo E, Bucci E. Clinical guidelines in early detection of oral squamous cell carcinoma arising in oral lichen planus: a 5-year experience. Oral Oncol. 2001;37(3):262–7. doi: 10.1016/s1368-8375(00)00096-8. [DOI] [PubMed] [Google Scholar]
- 81.Rajentheran R, McLean NR, Kelly CG, Reed MF, Nolan A. Malignant transformation of oral lichen planus. Eur J Surg Oncol. 1999;25(5):520–3. doi: 10.1053/ejso.1999.0689. [DOI] [PubMed] [Google Scholar]
- 82.Mignogna MD, Fedele S, Lo Russo L, Mignogna C, de Rosa G, Porter SR. Field cancerization in oral lichen planus. Eur J Surg Oncol. 2007;33(3):383–9. doi: 10.1016/j.ejso.2006.09.028. [DOI] [PubMed] [Google Scholar]
- 83.Coombes D, Cascarini L, Booth PW. Carcinoma of the midline dorsum of the tongue. Br J Oral Maxillofac Surg. 2008;46(6):485–6. doi: 10.1016/j.bjoms.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 84.Larsson A, Warfvinge G. Malignant transformation of oral lichen planus. Oral Oncol. 2003;39(6):630–1. doi: 10.1016/s1368-8375(03)00051-4. [DOI] [PubMed] [Google Scholar]
- 85.Gonzalez-Moles MA, Scully C, Gil-Montoya JA. Oral lichen planus: controversies surrounding malignant transformation. Oral Dis. 2008;14(3):229–43. doi: 10.1111/j.1601-0825.2008.01441.x. [DOI] [PubMed] [Google Scholar]
- 86.Irani S, Salajegheh A, Smith RA, Lam AK. A review of the profile of endothelin axis in cancer and its management. Crit Rev Oncol/ Hematol. 2014;89(2):314–21. doi: 10.1016/j.critrevonc.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 87.Irani S, Salajegheh A, Gopalan V, Smith RA, Lam AK. Expression profile of endothelin 1 and its receptor endothelin receptor A in papillary thyroid carcinoma and their correlations with clinicopathologic characteristics. Ann Diagn Pathol. 2014;18(2):43–8. doi: 10.1016/j.anndiagpath.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 88.Mignogna MD, Fedele S, Lo Russo L, Lo Muzio L, Bucci E. Immune activation and chronic inflammation as the cause of malignancy in oral lichen planus: is there any evidence ? Oral Oncol. 2004;40(2):120–30. doi: 10.1016/j.oraloncology.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 89.Taniguchi Y, Nagao T, Maeda H, Kameyama Y, Warnakulasuriya KA. Epithelial cell proliferation in oral lichen planus. Cell Prolif. 2002;35 (Suppl 1):103–9. doi: 10.1046/j.1365-2184.35.s1.11.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ebrahimi M, Boldrup L, Wahlin YB, Coates PJ, Nylander K. Decreased expression of the p63 related proteins beta-catenin, E-cadherin and EGFR in oral lichen planus. Oral Oncol. 2008;44(7):634–8. doi: 10.1016/j.oraloncology.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 91.Nomura H, Uzawa K, Yamano Y, Fushimi K, Nakashima D, Kouzu Y, et al. Down-regulation of plasma membranous Annexin A1 protein expression in premalignant and malignant lesions of the oral cavity: correlation with epithelial differentiation. J Cancer Res Clin Oncol. 2009;135(7):943–9. doi: 10.1007/s00432-008-0530-z. [DOI] [PubMed] [Google Scholar]
- 92.van der Waal I. Oral lichen planus and oral lichenoid lesions; a critical appraisal with emphasis on the diagnostic aspects. Med Oral Patol Oral Cir Bucal. 2009;14(7):E310–4. [PubMed] [Google Scholar]
- 93.Ismail SB, Kumar SK, Zain RB. Oral lichen planus and lichenoid reactions: etiopathogenesis, diagnosis, management and malignant transformation. J Oral Sci. 2007;49(2):89–106. doi: 10.2334/josnusd.49.89. [DOI] [PubMed] [Google Scholar]
- 94.Acay RR, Felizzola CR, de Araujo N, de Sousa SO. Evaluation of proliferative potential in oral lichen planus and oral lichenoid lesions using immunohistochemical expression of p53 and Ki67. Oral Oncol. 2006;42(5):475–80. doi: 10.1016/j.oraloncology.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 95.Muller S. Oral manifestations of dermatologic disease: a focus on lichenoid lesions. Head Neck Pathol. 2011;5(1):36–40. doi: 10.1007/s12105-010-0237-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hiremath SK, Kale AD, Charantimath S. Oral lichenoid lesions: Clinico-pathological mimicry and its diagnostic implications. Indian J Dent Res. 2011(22.6):827. doi: 10.4103/0970-9290.94679. [DOI] [PubMed] [Google Scholar]
- 97.Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol. 2009;60(2):203–11. doi: 10.1016/j.jaad.2008.09.035. [DOI] [PubMed] [Google Scholar]
- 98.Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008;58(6):931–50. doi: 10.1016/j.jaad.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 99.Sawamura D, Nakano H, Matsuzaki Y. Overview of epidermolysis bullosa. J Dermatol. 2010;37(3):214–9. doi: 10.1111/j.1346-8138.2009.00800.x. [DOI] [PubMed] [Google Scholar]
- 100.Reichart PA. Oral precancerous conditions--an overview. Mund Kiefer Gesichtschir. 2003;7(4):201–7. doi: 10.1007/s10006-003-0483-y. [DOI] [PubMed] [Google Scholar]
- 101.Wright JT, Fine JD, Johnson LB. Oral soft tissues in hereditary epidermolysis bullosa. Oral Surg Oral Med Oral Pathol. 1991;71(4):440–6. doi: 10.1016/0030-4220(91)90426-d. [DOI] [PubMed] [Google Scholar]
- 102.Martinez L, Goodman P, Crow WN. Squamous cell carcinoma of the maxillary sinus and palate in epidermolysis bullosa: CT demonstration. J Comput Assist Tomogr. 1992;16(2):317–9. doi: 10.1097/00004728-199203000-00027. [DOI] [PubMed] [Google Scholar]
- 103.Gupta U, Barman KD, Saify K. Squamous cell carcinoma complicating an untreated chronic discoid lupus erythematosus (CDLE) lesion in a black female. J Dermatol. 2005;32(12):1010–3. doi: 10.1111/j.1346-8138.2005.tb00892.x. [DOI] [PubMed] [Google Scholar]
- 104.Aviles Izquierdo JA, Cano Martinez N, Lazaro Ochaita P. Epidemiological characteristics of patients with cutaneous lupus erythematosus. Actas Dermosifiliogra. 2014;105(1):69–73. doi: 10.1016/j.ad.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 105.Schiodt M, Andersen L, Shear M, Smith CJ. Leukoplakia-like lesions developing in patients with oral discoid lupus erythematosus. Acta Odontol Scand. 1981;39 (4):209–16. doi: 10.3109/00016358109162282. [DOI] [PubMed] [Google Scholar]
- 106.Nelson ND, Bertuch AA. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat Res. 2012;730(1-2):43–51. doi: 10.1016/j.mrfmmm.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–6. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 108.Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413(6854):432–5. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 109.Holman JD, Dyer JA. Genodermatoses with malignant potential. Curr Opin Pediatr. 2007;19(4):446–54. doi: 10.1097/MOP.0b013e3282495939. [DOI] [PubMed] [Google Scholar]
- 110.Ogden GR, Lane DP, Chisholm DM. p53 expression in dyskeratosis congenita: a marker for oral premalignancy? J Clin Pathol. 1993;46(2):169–70. doi: 10.1136/jcp.46.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cavalcante AS, Anbinder AL, Carvalho YR. Actinic cheilitis: clinical and histological features. Journal of oral and maxillofacial surgery : official journal of the American Association of Oral and Maxillofacial Surgeons. 2008;66(3):498–503. doi: 10.1016/j.joms.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 112.Vieira RA, Minicucci EM, Marques ME, Marques SA. Actinic cheilitis and squamous cell carcinoma of the lip: clinical, histopathological and immunogenetic aspects. An Bras Dermatol. 2012;87(1):105–14. doi: 10.1590/s0365-05962012000100013. [DOI] [PubMed] [Google Scholar]
- 113.Moy RL. Clinical presentation of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42(1 Pt 2):8–10. doi: 10.1067/mjd.2000.103343. [DOI] [PubMed] [Google Scholar]
- 114.Souza Freitas V, de Andrade Santos PP, de Almeida Freitas R, Pereira Pinto L, de Souza LB. Mast cells and matrix metalloproteinase 9 expression in actinic cheilitis and lip squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112(3):342–8. doi: 10.1016/j.tripleo.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 115.Grimbaldeston MA, Skov L, Finlay-Jones JJ, Hart PH. Increased dermal mast cell prevalence and susceptibility to development of basal cell carcinoma in humans. Methods. 2002;28(1):90–6. doi: 10.1016/s1046-2023(02)00213-x. [DOI] [PubMed] [Google Scholar]
- 116.Gulubova MV. Structural examination of tryptase- and chymase-positive mast cells in livers, containing metastases from gastrointestinal cancers. Clin Exp Metastasis. 2003;20(7):611–20. doi: 10.1023/a:1027310827655. [DOI] [PubMed] [Google Scholar]
- 117.Araujo CP, Gurgel CA, Ramos EA, Freitas VS, Barbosa Ade A Jr, Ramalho LM, et al. Accumulation of CD1a-positive Langerhans cells and mast cells in actinic cheilitis. J Mol Histol. 2010;41(6):357–65. doi: 10.1007/s10735-010-9297-z. [DOI] [PubMed] [Google Scholar]
- 118.Rigel DS, Stein Gold LF. The importance of early diagnosis and treatment of actinic keratosis. J Am Acad Dermatol. 2013;68(1 Suppl 1):S20–7. doi: 10.1016/j.jaad.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 119.Rossi R, Mori M, Lotti T. Actinic keratosis. Int J Dermatol. 2007;46(9):895–904. doi: 10.1111/j.1365-4632.2007.03166.x. [DOI] [PubMed] [Google Scholar]
- 120.Lalji A KN, Lalji M. Actinic keratosis and Squamous Cell Carcinoma. Clin Res Dermatol Open. 2014;1(1):1–3. [Google Scholar]
- 121.Marks R, Ponsford MW, Selwood TS, Goodman G, Mason G. Non-melanotic skin cancer and solar keratoses in Victoria. Med J Aust. 1983;2(12):619–22. doi: 10.5694/j.1326-5377.1983.tb122724.x. [DOI] [PubMed] [Google Scholar]
- 122.Bagan JV, Jimenez-Soriano Y, Diaz-Fernandez JM, Murillo-Cortes J, Sanchis-Bielsa JM, Poveda-Roda R, et al. Malignant transformation of proliferative verrucous leukoplakia to oral squamous cell carcinoma: a series of 55 cases. Oral oncology. 2011;47(8):732–5. doi: 10.1016/j.oraloncology.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 123.Fuchs A, Marmur E. The kinetics of skin cancer: progression of actinic keratosis to squamous cell carcinoma. Dermatol Surg. 2007;33(9):1099–101. doi: 10.1111/j.1524-4725.2007.33224.x. [DOI] [PubMed] [Google Scholar]
- 124.Smoller BR. Squamous cell carcinoma: from precursor lesions to high-risk variants. Mod Pathol. 2006;19 (Suppl 2):S88–92. doi: 10.1038/modpathol.3800509. [DOI] [PubMed] [Google Scholar]
- 125.Choi KH, Kim GM, Kim SY. The keratin-14 expression in actinic keratosis and squamous cell carcinoma: is this a prognostic factor for tumor progression? Cancer Res Treat. 2010;42(2):107–14. doi: 10.4143/crt.2010.42.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grimbaldeston MA, Finlay-Jones JJ, Hart PH. Mast cells in photodamaged skin: what is their role in skin cancer? Photoch Photobiol Sci. 2006;5(2):177–83. doi: 10.1039/b504344a. [DOI] [PubMed] [Google Scholar]
- 127.Kim MS, Kim YK, Lee DH, Seo JE, Cho KH, Eun HC, et al. Acute exposure of human skin to ultraviolet or infrared radiation or heat stimuli increases mast cell numbers and tryptase expression in human skin in vivo. Br J Dermatol. 2009;160(2):393–402. doi: 10.1111/j.1365-2133.2008.08838.x. [DOI] [PubMed] [Google Scholar]
- 128.Berhane T, Halliday GM, Cooke B, Barnetson RS. Inflammation is associated with progression of actinic keratoses to squamous cell carcinomas in humans. Br J Dermatol. 2002;146(5):810–5. doi: 10.1046/j.1365-2133.2002.04720.x. [DOI] [PubMed] [Google Scholar]
- 129.Chopra V, Tyring SK, Johnson L, Fine JD. Patients with severe forms of inherited epidermolysis bullosa exhibit decreased lymphokine and monokine production. J Clin Immunol. 1990;10(6):321–9. doi: 10.1007/BF00917477. [DOI] [PubMed] [Google Scholar]
- 130.Arbiser JL, Fan CY, Su X, Van Emburgh BO, Cerimele F, Miller MS, et al. Involvement of p53 and p16 tumor suppressor genes in recessive dystrophic epidermolysis bullosa-associated squamous cell carcinoma. J Invest Dermatol. 2004;123(4):788–90. doi: 10.1111/j.0022-202X.2004.23418.x. [DOI] [PubMed] [Google Scholar]
- 131.Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17(7):553–68. doi: 10.1111/j.1600-0625.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- 132.Bauer JW, Schaeppi H, Kaserer C, Hantich B, Hintner H. Large melanocytic nevi in hereditary epidermolysis bullosa. J Am Acad Dermatol. 2001;44(4):577–84. doi: 10.1067/mjd.2001.112217. [DOI] [PubMed] [Google Scholar]
- 133.Pellicano R, Fabrizi G, Cerimele D. Multiple keratoacanthomas and junctional epidermolysis bullosa A therapeutic conundrum. Arch Dermatol. 1990;126(3):305–6. [PubMed] [Google Scholar]
- 134.Weber F, Bauer JW, Sepp N, Hogler W, Salmhofer W, Hintner H, et al. Squamous cell carcinoma in junctional and dystrophic epidermolysis bullosa. Acta Derm Venereol. 2001;81(3):189–92. doi: 10.1080/000155501750376285. [DOI] [PubMed] [Google Scholar]
- 135.Millard LG, Barker DJ. Development of squamous cell carcinoma in chronic discoid lupus erythematosus. Clin Exp Dermatol. 1978;3(2):161–6. doi: 10.1111/j.1365-2230.1978.tb01480.x. [DOI] [PubMed] [Google Scholar]
- 136.Harper JG, Pilcher MF, Szlam S, Lind DS. Squamous cell carcinoma in an African American with discoid lupus erythematosus: a case report and review of the literature. South Med J. 2010;103(3):256–9. doi: 10.1097/SMJ.0b013e3181c98ba9. [DOI] [PubMed] [Google Scholar]
- 137.Greenberg MS GM. Burket’s oral medicine. Diagnosis and Treatment. 10th ed. Philadelphia: Lippincott Company; 2003. [Google Scholar]
- 138.Bhat MR, Hulmani M, Dandakeri S, Kambil SM, Gatti R. Disseminated discoid lupus erythematosus leading to squamous cell carcinoma. Indian J Dermatol. 2012;57(2):158–61. doi: 10.4103/0019-5154.94298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Copcu E, Aktas A, Sisman N, Oztan Y. Thirty-one cases of Marjolin's ulcer. Clin Exp Dermatol. 2003;28(2):138–41. doi: 10.1046/j.1365-2230.2003.01210.x. [DOI] [PubMed] [Google Scholar]
- 140.Mallipeddi R, Keane FM, McGrath JA, Mayou BJ, Eady RA. Increased risk of squamous cell carcinoma in junctional epidermolysis bullosa. J Eur Acad Dermatol Venerol. 2004;18(5):521–6. doi: 10.1111/j.1468-3083.2004.00968.x. [DOI] [PubMed] [Google Scholar]
- 141.Schiodt M. Oral discoid lupus erythematosus III A histopathologic study of sixty-six patients. Oral Surg Oral Med Oral Pathol. 1984;57(3):281–93. doi: 10.1016/0030-4220(84)90184-1. [DOI] [PubMed] [Google Scholar]
- 142.Atkinson JC, Harvey KE, Domingo DL, Trujillo MI, Guadagnini JP, Gollins S, et al. Oral and dental phenotype of dyskeratosis congenita. Oral Dis. 2008;14(5):419–27. doi: 10.1111/j.1601-0825.2007.01394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Salopek TG, Kopf AW, Stefanato CM, Vossaert K, Silverman M, Yadav S. Differentiation of atypical moles (dysplastic nevi) from early melanomas by dermoscopy. Dermatol Clin. 2001;19(2):337–45. doi: 10.1016/s0733-8635(05)70271-0. [DOI] [PubMed] [Google Scholar]
- 144.Goldstein AM, Struewing JP, Chidambaram A, Fraser MC, Tucker MA. Genotype-phenotype relationships in U.S. melanoma-prone families with CDKN2A and CDK4 mutations. J Nati Cancer Inst. 2000;92(12):1006–10. doi: 10.1093/jnci/92.12.1006. [DOI] [PubMed] [Google Scholar]
- 145.Farber MJ, Heilman ER, Friedman RJ. Dysplastic nevi. Dermatol Clin. 2012;30(3):389–404. doi: 10.1016/j.det.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 146.Elder DE. Dysplastic naevi: an update. Histopathology. 2010;56(1):112–20. doi: 10.1111/j.1365-2559.2009.03450.x. [DOI] [PubMed] [Google Scholar]
- 147.de Wit PE, van't Hof-Grootenboer B, Ruiter DJ, Bondi R, Brocker EB, Cesarini JP, et al. Validity of the histopathological criteria used for diagnosing dysplastic naevi An interobserver study by the pathology subgroup of the EORTC Malignant Melanoma Cooperative Group. Eur J Cancer. 1993;29a(6):831–9. doi: 10.1016/s0959-8049(05)80419-8. [DOI] [PubMed] [Google Scholar]
- 148.Lyon VB. Congenital melanocytic nevi. Pediatr Clin North Am. 2010;57(5):1155–76. doi: 10.1016/j.pcl.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 149.Boente Mdel C, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN) A distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15(6):451–3. [PubMed] [Google Scholar]
- 150.Gozel S, Donmez M, Akdur NC, Yikilkan H. Development of six tumors in a sebaceus nevus of jadassohn: report of a case. Korean J Pathol. 2013;47(6):569–74. doi: 10.4132/KoreanJPathol.2013.47.6.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lanssens S, Ongenae K. Dermatologic lesions and risk for cancer. Acta Clin Belg. 2011;66(3):177–85. doi: 10.2143/ACB.66.3.2062543. [DOI] [PubMed] [Google Scholar]
- 152.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56(11):2488–92. [PubMed] [Google Scholar]
- 153.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 154.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74(6):363–96. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 155.Zeidler R, Albermann K, Lang S. Nicotine and apoptosis. Apoptosis. 2007;12(11):1927–43. doi: 10.1007/s10495-007-0102-8. [DOI] [PubMed] [Google Scholar]
- 156.Arora S, Aggarwal P, Pathak A, Bhandari R, Duffoo F, Gulati SC. Molecular genetics of head and neck cancer (Review) Mol Med Rep. 2012;6(1):19–22. doi: 10.3892/mmr.2012.889. [DOI] [PubMed] [Google Scholar]
- 157.Gainza-Cirauqui ML, Nieminen MT, Novak Frazer L, Aguirre-Urizar JM, Moragues MD, Rautemaa R. Production of carcinogenic acetaldehyde by Candida albicans from patients with potentially malignant oral mucosal disorders. J Oral Pathol Med. 2013;42(3):243–9. doi: 10.1111/j.1600-0714.2012.01203.x. [DOI] [PubMed] [Google Scholar]
- 158.Johnson NW, Jayasekara P, Amarasinghe AA. Squamous cell carcinoma and precursor lesions of the oral cavity: epidemiology and aetiology. Periodontol. 2000. 2011;57(1):19–37. doi: 10.1111/j.1600-0757.2011.00401.x. [DOI] [PubMed] [Google Scholar]
- 159.Chen AC, Halliday GM, Damian DL. Non-melanoma skin cancer: carcinogenesis and chemoprevention. Pathology. 2013;45(3):331–41. doi: 10.1097/PAT.0b013e32835f515c. [DOI] [PubMed] [Google Scholar]
- 160.Salajegheh A, Dolan-Evans E, Sullivan E, Irani S, Rahman MA, Vosgha H, et al. The expression profiles of the galectin gene family in primary and metastatic papillary thyroid carcinoma with particular emphasis on galectin-1 and galectin-3 expression. Exp Mol Pathol. 2014;96(2):212–8. doi: 10.1016/j.yexmp.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 161.Raica M, Cimpean AM, Ribatti D. Angiogenesis in pre-malignant conditions. Eur J Cancer. 2009;45(11):1924–34. doi: 10.1016/j.ejca.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 162.Degen M, Natarajan E, Barron P, Widlund HR, Rheinwald JG. MAPK/ERK-dependent translation factor hyperactivation and dysregulated laminin gamma2 expression in oral dysplasia and squamous cell carcinoma. Am J Pathol. 2012;180(6):2462–78. doi: 10.1016/j.ajpath.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nordemar S, Hogmo A, Lindholm J, Auer G, Munck-Wikland E. Laminin-5 gamma 2: a marker to identify oral mucosal lesions at risk for tumor development? Anticancer Res. 2003;23(6d):4985–9. [PubMed] [Google Scholar]
- 164.Inoue H, Miyazaki Y, Kikuchi K, Yoshida N, Ide F, Ohmori Y, et al. Podoplanin expression during dysplasia-carcinoma sequence in the oral cavity. Tumour Biol. 2012;33(1):183–94. doi: 10.1007/s13277-011-0261-7. [DOI] [PubMed] [Google Scholar]