

Abstract
Idiosyncratic drug-induced liver injury (IDILI) typically occurs in a small fraction of patients and has resulted in removal of otherwise efficacious drugs from the market. Current preclinical testing methods are ineffective in predicting which drug candidates have IDILI liability. Recent results suggest that immune mediators such as tumor necrosis factor-α (TNF) and interferon-γ (IFN) interact with drugs that cause IDILI to kill hepatocytes. This proof-of-concept study was designed to test the hypothesis that drugs can be classified according to their ability to cause IDILI in humans using classification modeling with covariates derived from concentration-response relationships that describe cytotoxic interaction with cytokines. Human hepatoma (HepG2) cells were treated with drugs associated with IDILI or with drugs lacking IDILI liability and cotreated with TNF and/or IFN. Detailed concentration-response relationships were determined for calculation of parameters such as the maximal cytotoxic effect, slope, and EC50 for use as covariates for classification modeling using logistic regression. These parameters were incorporated into multiple classification models to identify combinations of covariates that most accurately classified the drugs according to their association with human IDILI. Of 14 drugs associated with IDILI, almost all synergized with TNF to kill HepG2 cells and were successfully classified by statistical modeling. IFN enhanced the toxicity mediated by some IDILI-associated drugs in the presence of TNF. In contrast, of 10 drugs with little or no IDILI liability, none synergized with inflammatory cytokines to kill HepG2 cells and were classified accordingly. The resulting optimal model classified the drugs with extraordinary selectivity and specificity.
Introduction
Idiosyncratic drug-induced liver injury (IDILI) is a typically rare reaction that occurs at drug doses that are safe in the majority of patients. Cases of IDILI can be severe, leading to liver transplantation or death (Ostapowicz et al., 2002). In addition to public health concerns, IDILI is a common cause of removal of drugs from the pharmaceutical market due to the occurrence and severity of these reactions and to the poor ability of standard toxicity tests to identify drug candidates with IDILI liability before they reach the market (Watkins, 2005; Aithal et al., 2011). The causes of IDILI are unknown, but it is thought that genetic and/or environmental factors predispose patients to toxicity from an otherwise safe dose of a drug (Roth and Ganey, 2011). Because these reactions are usually rare, drugs with IDILI potential are often not identified during clinical trials that employ limited numbers of human subjects. More effective preclinical strategies to identify drug candidates with IDILI potential could inform decisions about whether to allow a drug candidate to proceed through the development process. An in vitro approach that uses cells that are readily available and easily grown in culture, requires little compound, employs a single relevant endpoint, and is amenable to high-throughput format would be highly desirable.
Development of such an approach has been challenging due to limited knowledge about mechanisms underlying IDILI. Several attempts have been made based on direct effects of drugs on hepatocellular biochemistry and/or on physical-chemical characteristics of drugs (Usui et al., 2009; Low et al., 2011; Sakatis et al., 2012; Thompson et al., 2012; Chen et al., 2013, 2014; Khetani et al., 2013; Morgan et al., 2013; Aleo et al., 2014; Zhu et al., 2014; Choi et al., 2015; Schadt et al., 2015; Susukida et al., 2015; Ware et al., 2015; Zhang et al., 2016a,b). For the most part, the performance of these (selectivity and/or specificity) has not been ideal. Although it is commonly believed that activation of the immune system underlies IDILI, the results of such activation are typically not considered in the design. Such consideration could lead to an improved approach.
Activation of immune cells culminates in the release of immune mediators such as cytokines. Some recently developed animal models as well as human genetic association studies suggest that adaptive immunity plays a role in the precipitation of IDILI responses to some drugs (Lucena et al., 2011; Chakraborty et al., 2015). Mice that have impaired immune tolerance developed liver injury after several administrations of IDILI-associated drugs such as halothane and amodiaquine (Metushi et al., 2015; Chakraborty et al., 2015). Although these models involving activation of the adaptive immune system resulted in only mild liver injury, they could represent an advance in understanding IDILI pathogenesis. Thus far, very few animal models of IDILI have been developed that recapitulate the severity of hepatocellular injury observed in humans. Most of these are based on the interaction of drugs with an activated innate immune system (Roth and Ganey, 2011). Among the models based on the interaction between drugs and the innate immune system, the inflammatory mediators tumor necrosis factor-α (TNF) and interferon-γ (IFN) were critical to the pathogenesis of liver injury (Hassan et al., 2008; Shaw et al., 2009a,b; Zou et al., 2009; Dugan et al., 2011; Lu et al., 2012).
Both innate and adaptive immune responses culminate in the release of these potentially cytotoxic, proinflammatory cytokines. Findings from the animal studies raised the possibility that IDILI-associated drugs sensitize hepatocytes to cell death signaling from cytokines such as TNF and IFN (Roth and Ganey, 2011). Indeed, using a series of drugs, Cosgrove et al. (2009) found a correlation between IDILI liability and the ability of drugs to synergize with cytokines to kill primary human hepatocytes in vitro. Using a smaller subset of drugs, Cosgrove et al. (2009) also found that their results in primary human hepatocytes could be reproduced using HepG2 cells, suggesting that the latter cells hold promise in classifying drugs according to IDILI liability. These and other studies suggest that IDILI-associated drugs act in part by causing stress to hepatocytes, such that they become susceptible to killing mediated by cytokines (Cosgrove et al., 2009; Zou et al., 2009; Fredriksson et al., 2011, 2014; Beggs et al., 2014, 2015; Maiuri et al., 2015; for review, see Roth et al., 2017).
Using HepG2 cells, we recently studied the cytotoxic interaction of TNF/IFN with a series of nonsteroidal anti-inflammatory drugs (NSAIDs) with various IDILI liabilities and also with an antibiotic, trovafloxacin (Beggs et al., 2014, 2015; Maiuri et al., 2015). In studies presented here, we expand on those findings with a larger set of drugs. Importantly, elucidation of detailed concentration-response relationships permitted calculation of various parameters (e.g., EC50, maximal response, slope, etc.) that we then incorporated into statistical models to evaluate the ability of this approach to classify drugs according to their association with IDILI. The results suggest a highly promising, in vitro approach to predict IDILI liability.
Materials and Methods
Materials
All drugs were purchased from Sigma-Aldrich (St. Louis, MO) or Santa Cruz Biotechnology (Dallas, TX) unless otherwise noted. Recombinant human TNF and IFN were purchased from R&D Systems (Minneapolis, MN) or Millipore (Billerica, MA). Phosphate-buffered saline, Dulbecco’s modified Eagle’s medium, fetal bovine serum, antibiotic-antimycotic, and 0.25% trypsin-EDTA were purchased from Life Technologies (Carlsbad, CA).
Cell Culture
Human hepatoma HepG2 cells (American Type Culture Collection, Manassas, VA) were grown in 25-cm2 tissue culture flasks and maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic in a humidified incubator at 37°C under 95% air and 5% CO2. Cells were passed or used for experiments when they reached approximately 80% confluence. Cells were used at passages 6–16; at higher passage numbers, the responses became less consistent and, in some cases, less robust.
IDILI Classification
The 24 drugs evaluated in this study were classified as being associated with (IDILI+) or not associated with (IDILI—) IDILI. Known classification was based on a set of criteria established by Xu et al. (2008). Table 1 lists the drugs evaluated in this study, their maximal plasma concentration (Cmax) after pharmacologic dosing in human patients, and their known IDILI classification. The drugs that were used span several therapeutic classes (i.e., antibiotics, NSAIDs, anticancer, antiepileptic, antidiabetic, and anxiolytic/neuroleptic/antidepressant/antipsychotic drugs). Most of the IDILI+ drugs we chose have caused hepatocellular injury in people, although some (flucloxacillin, chlorpromazine, flutamide, and clavulanate) have been associated with mixed or cholestatic liver damage. Some of the IDILI+ drugs (chlorpromazine, diclofenac, and trovafloxacin) produced liver injury in experimental animals when coupled with an inflammatory stimulus (Buchweitz et al., 2002; Deng et al., 2006; Shaw et al., 2009b).
TABLE 1.
IDILI classification, daily dose, Cmax, and references from which Cmax values were taken
| Drug | IDILI Liability | Cmax | Daily Dose | Cmax Reference |
|---|---|---|---|---|
| μM | mg | |||
| Aspirin | IDILI− | 47 | 1300 | Brandon et al., 1986 |
| Azithromycin | IDILI− | 0.5 | 500 | Xu et al., 2008 |
| Buspirone | IDILI− | 0.005 | 15 | Xu et al., 2008 |
| Idarubicin | IDILI− | 0.02 | 1 | Xu et al., 2008 |
| Levofloxacin | IDILI− | 15.7 | 500 | Xu et al., 2008 |
| Moxifloxacin | IDILI− | 6.2 | 400 | Stass et al., 1998 |
| Pioglitazone | IDILI− | 2.67 | 15 | Xu et al., 2008 |
| Promethazine | IDILI− | 0.06 | 25 | Xu et al., 2008 |
| Rofecoxib | IDILI− | 1 | 12.5 | Gottesdiener et al., 2003 |
| Sertraline | IDILI− | 0.06 | 50 | Xu et al., 2008 |
| Bromfenac | IDILI+ | 13.5 | 50 | Gumbhir-Shah et al., 1997 |
| Chlorpromazine | IDILI+ | 0.84 | 200 | Xu et al., 2008 |
| Clavulanate | IDILI+ | 12 | 125 | Hu et al., 2002 |
| Diclofenac | IDILI+ | 7.44 | 100 | Xu et al., 2008 |
| Doxorubicin | IDILI+ | 1 | 1 | Barpe et al., 2010 |
| Flucloxacillin | IDILI+ | 72.6 | 250 | Røder et al., 1995 |
| Flutamide | IDILI+ | 0.36 | 750 | Xu et al., 2008 |
| Ibuprofen | IDILI+ | 164 | 800 | Bramlage and Goldis, 2008 |
| Isoniazid | IDILI+ | 77 | 300 | Xu et al., 2008 |
| Naproxen | IDILI+ | 300 | 500 | Setiawati et al., 2009 |
| Nimesulide | IDILI+ | 21.08 | 200 | Xu et al., 2008 |
| Telithromycin | IDILI+ | 2.77 | 800 | Xu et al., 2008 |
| Trovafloxacin | IDILI+ | 5 | 300 | Xu et al., 2008 |
| Valproic acid | IDILI+ | 175 | 60 | Rha et al., 1993 |
IDILI classification was determined by a set of criteria described in Xu et al. (2008). IDILI− indicates that the drug is not associated with IDILI, whereas IDILI+ indicates that the drug is associated with IDILI.
Cytotoxicity Assessment
HepG2 cells were plated at a density of 4 × 104 cells per well in black-walled, 96-well tissue culture plates and were allowed to attach overnight before being treated with compounds. Drugs were reconstituted in vehicles (VEHs) consisting of sterile water or dimethylsulfoxide (concentration <0.5%). Cells were treated with various concentrations of the drug or its VEH (control) alone or in combination with the cytokines TNF (10 ng/ml) and/or IFN (10 ng/ml) or their phosphate-buffered saline VEH. Cytotoxicity was evaluated as lactate dehydrogenase (LDH) activity released from the cells into culture medium using the Homogeneous Membrane Integrity Assay kit from Promega (Madison, WI). A spectrophotometric method was used to measure the percentage of LDH release in cases in which the drug interfered with the fluorescence-based assay (Vanderlinde, 1985).
Concentration-response curves were generated for 24 drugs, 14 of which are associated with human IDILI and 10 of which are not. Cells were treated with drug concentrations generally ranging from 0 to 100 times the Cmax observed in human patients. This range of concentrations is based on scaling factors described in Xu et al. (2008) and accounts for variability in Cmax as well as exposure of the liver to greater concentrations. The cytokine concentrations used in this study are within 10-fold of the concentrations found in the serum of human patients undergoing an inflammatory response (Pinsky et al., 1993; Taudorf et al., 2007). If a cytotoxic response was observed but did not reach a plateau by the 100× Cmax concentration, further testing was performed with larger concentrations of drug to generate a complete (sigmoidal) concentration-response curve. Typically, the range of drug concentrations included at least two that were without effect, two defining the maximal effect, and two surrounding the EC50. This was necessary because four-parameter logistic modeling used in the statistical analysis requires well defined, sigmoidal concentration-response curves. Cells were exposed to drug/cytokine combinations for 24 hours. This time was chosen based on previous results demonstrating that cytotoxicity from drug/cytokine exposure begins within 24 hours (Beggs et al., 2014; Maiuri et al., 2015). For analysis of drug concentration-response data, concentrations of each drug were expressed as a fraction of its Cmax.
Statistical Analysis
The statistical approach used in this study can be divided into three phases: 1) drug concentration-response modeling and covariate development using four-parameter logistic models, 2) classification model development using logistic regression models, and 3) analysis of classification accuracy with receiver operating characteristic (ROC) curves.
Defining Covariates: Four-Parameter Logistic Concentration-Response Model.
In the first phase, variables (potential covariates) for use in the classification analysis were defined from the analysis of drug concentration-response data. Initially, a one-way analysis of variance was used as an omnibus test to determine whether a particular treatment (e.g., drug alone or in combination with TNF and/or IFN) caused a significant change in LDH release relative to baseline (i.e., LDH release in the absence of drug, hereafter designated “min”). The criterion for significance for the analysis of variance was set at α = 0.01. A 1% level of significance was used to rule out more vigorously marginal relationships between concentration and response. For treatments that did not result in a significant change in LDH above min (P > 0.01), the following was assumed for the purpose of concentration-response modeling: the minimum LDH response (min) = the maximum LDH response (max). For drug/cytokine treatment combinations that did result in a statistically significant LDH response, the concentration-response data were modeled using the following four-parameter logistic function:
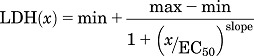 |
where LDH(x) is the percentage of LDH released at a given concentration x, x is [drug]/Cmax, min is the percentage of LDH release at 0 drug concentration (i.e., baseline), and max is the maximal LDH response (i.e., maximum percentage of LDH release). From this equation, the drug concentration associated with the 50% maximal response (EC50) and the slope of the concentration-response curve were calculated. The four-parameter logistic models were generated using R statistical software (R package “drc”; R Foundation for Statistical Computing, Vienna, Austria) (R Core Team, 2015; Ritz and Streibig, 2005).
In addition to slope and EC50, several other “base covariates” were calculated from the concentration-response curves for use in further analyses. These were calculated for each of the 96 drug/cytokine treatment combinations evaluated in this study (24 drugs × 4 cytokine combinations) (Supplemental Tables 1–9). Delta was defined as max minus min. In addition to the covariates determined from concentration-response curves, Cmax was considered as another base covariate.
Similar to EC50, the covariate EC10 represents the [drug]/Cmax value associated with a 10% increase above min relative to max and was determined by the following equation:
where D10 is a categorical variable related to reaching a threshold LDH response above which a drug is classified as positively associated with IDILI. D10 is defined as 0 if delta is < 10% LDH release and as 1 if delta is ≥ 10% LDH release.
The base covariate R10 represents the [drug]/Cmax value associated with an increase in 10 LDH percentage points above min for a particular treatment condition and was determined by the following equation:
R10 is considered to be 0 when delta is < 10% LDH (i.e., when D10 = 0).
From the base covariates defined above, several other covariates were derived. These included the EC50 quotient, EC10 quotient, R10 quotient, deltadiff, and TNF change. Each of these “derived” covariates is explained in more detail below.
The EC50 quotient, EC10 quotient, and R10 quotient represent the ratio between the EC50, EC10, or R10 of the drug/cytokine concentration-response curve and the respective value for the drug/VEH concentration-response curve. In some instances, the value derived from this calculation is indeterminate (i.e., when the denominator = 0). To incorporate the quotient values into the classification models described below, the categorical variable “Q” was used to eliminate the possibility of the quotient being indeterminate. Q is defined as 0 if delta VEH and/or delta cytokine is < 10% LDH and 1 if both delta VEH and delta cytokine are ≥ 10% LDH. For the purposes of calculating the EC50 quotient, the following condition was applied: if Q = 0, then the EC50 quotient = 0; if Q = 1, then the EC50 quotient = EC50 VEH/EC50 cytokine. The same condition was applied for calculation of the EC10 quotient and R10 quotient.
Deltadiff represents the difference between the delta of the drug/cytokine concentration-response curve and the delta of the drug/VEH curve. In other words, deltadiff = (delta cytokine) – (delta VEH). Figure 1 graphically illustrates several of the covariates defined above. Supplemental Tables 1–9 list the values of all of the covariates computed in this study. In addition to the covariates derived from the concentration-response curves, the maximal therapeutic plasma drug concentration (Cmax) in human patients was used in some models (Table 1).
Fig. 1.

Depiction of base covariates considered for evaluation. The blue curve represents hypothetical response to drug alone; the red curve represents response to drug in the presence of cytokine. Min is the LDH release in the absence of drug, max is the maximal LDH release. Delta = max(LDH) – min(LDH) for each curve. Although not depicted in the figure, deltadiff = delta cytokine − deltaVEH. EC50 is the [drug]/Cmax value at 50% delta. R10 is not depicted but it represents the [drug]/Cmax value associated with a 10% increase in LDH release above min(LDH).
TNF change is a categorical variable related to the alteration in the drug-induced cytotoxic response in the presence and absence of TNF, determined as follows:
Recall that D10 is defined as 0 if delta is  10% change in LDH release, and D10 is defined as 1 if delta is > 10% change in LDH release. Accordingly, TNF change = 1 if the TNF curve has a delta > 10% LDH release and the VEH curve has a delta ≤ 10% LDH release; TNF change = 0 in all other situations.
10% change in LDH release, and D10 is defined as 1 if delta is > 10% change in LDH release. Accordingly, TNF change = 1 if the TNF curve has a delta > 10% LDH release and the VEH curve has a delta ≤ 10% LDH release; TNF change = 0 in all other situations.
Classification Modeling Using Defined Covariates.
In the second phase of analysis, classification models were developed using logistic regression with covariates as independent variables in the analysis and known IDILI classification as the dependent variable. The ability to classify drugs accurately was evaluated using the known IDILI classifications shown in Table 1. A model selection process was used to determine whether a covariate or set of covariates is associated with IDILI liability. Covariates were first evaluated individually to determine how well a particular covariate classified drugs according to IDILI liability, and then covariates were evaluated in combination. Combinations of covariates were selected to maximize the ability of the model to distinguish between drugs associated or not with IDILI. Specifically, covariates that describe changes in efficacy (delta, deltadiff, etc.) were paired with covariates that describe changes in potency (EC50, EC10, R10, etc.) to find covariate combinations that led to models that most accurately discriminated between drugs that are and are not associated with IDILI. The best-fit logistic regression models were used as classification models to compute a probability that a given drug is associated with IDILI. The logistic regression models use the following equation:
where 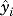 is the calculated (predicted) probability that drug i with a vector of covariates xi is associated with IDILI. The β coefficients (β0, the regression intercept; and βi, the regression slopes for model covariates, xi) were derived from the logistic regression models using 1) combinations of the covariates (xi) generated by concentration-response modeling for each of the 24 drugs evaluated in this study plus daily dose and Cmax, and 2) the known IDILI classification for the dependent variable yi (i.e., 1 for IDILI+ drugs and 0 for IDILI− drugs). For treatments that did not result in a significant change in LDH above min (P > 0.01), a value of 0 was assigned for covariates derived from EC50 for purposes of calculating βi. The regression coefficients (βi) were calculated using Firth’s method, which eliminates bias when estimating the value βi (Firth, 1993). Firth’s method was necessary since many of the covariates used in this study exhibited quasi-complete separation. This occurs when a covariate almost perfectly separates observations into the appropriate categories. In this study, several covariates almost completely separated drugs according to their IDILI liability. When separation or quasi-complete separation occurs, use of the standard method (i.e., maximum likelihood estimation) provides biased, unreliable estimates of βi. Firth’s method uses a penalized likelihood regression to rectify this and is an appropriate method to use for estimating βi when quasi-complete separation of data occurs (Firth, 1993). All logistic regression models were computed using R statistical software (R package “logistf”) (Heinze et al., 2013; R Core Team, 2015).
is the calculated (predicted) probability that drug i with a vector of covariates xi is associated with IDILI. The β coefficients (β0, the regression intercept; and βi, the regression slopes for model covariates, xi) were derived from the logistic regression models using 1) combinations of the covariates (xi) generated by concentration-response modeling for each of the 24 drugs evaluated in this study plus daily dose and Cmax, and 2) the known IDILI classification for the dependent variable yi (i.e., 1 for IDILI+ drugs and 0 for IDILI− drugs). For treatments that did not result in a significant change in LDH above min (P > 0.01), a value of 0 was assigned for covariates derived from EC50 for purposes of calculating βi. The regression coefficients (βi) were calculated using Firth’s method, which eliminates bias when estimating the value βi (Firth, 1993). Firth’s method was necessary since many of the covariates used in this study exhibited quasi-complete separation. This occurs when a covariate almost perfectly separates observations into the appropriate categories. In this study, several covariates almost completely separated drugs according to their IDILI liability. When separation or quasi-complete separation occurs, use of the standard method (i.e., maximum likelihood estimation) provides biased, unreliable estimates of βi. Firth’s method uses a penalized likelihood regression to rectify this and is an appropriate method to use for estimating βi when quasi-complete separation of data occurs (Firth, 1993). All logistic regression models were computed using R statistical software (R package “logistf”) (Heinze et al., 2013; R Core Team, 2015).
ROC Analysis.
In the third phase of the statistical approach, the classification models, generated as logistic regression models using single covariates or combinations of covariates, were evaluated by ROC analysis to determine which model and corresponding sets of covariates led to the most accurate classification of drugs according to their potential to cause IDILI. An ROC curve was created for each model by graphing the true positive rate (sensitivity; i.e., proportion of drugs correctly classified as associated with IDILI) against the true negative rate specificity (1 − false positive rate; i.e., proportion of drugs correctly classified as not associated with IDILI) at various probability cutoff thresholds (k). ROC curves were generated using R package pROC (Robin et al., 2011; R Core Team, 2015). An area under the curve (AUC) and confidence interval were computed for each ROC curve (where each logistic regression model has one ROC curve). Plots depicting the AUCs and 95% confidence intervals of the ROC curves were generated for the purpose of comparing multiple logistic regression models using R package Metafor (Viechtbauer, 2010). Corresponding to each ROC curve is an optimal threshold value (k*), the threshold yielding the highest point of accuracy on the curve [i.e., the point nearest the point (1,1) on the curve]. Thus, each ROC curve has a corresponding AUC and an optimal cutoff (k*) that corresponds to the highest point of accuracy on that ROC curve.
Combinations of covariates were strategically selected for evaluation based on what was deemed to lead to the most accurate classification of drugs. ROC curves and corresponding whisker plots were generated to illustrate graphically the ability of each classification model to classify drugs accurately. This allowed for selection of optimal set(s) of covariates for accurate drug classification according to IDILI liability. Our goal was to achieve a classification model and a corresponding set of covariates with an AUC as close to 1 as possible with the narrowest 95% confidence interval. A model that is able to classify drugs perfectly according to their potential to cause IDILI would have an ROC curve with an AUC = 1. DeLong’s method was used to determine whether there were statistically significant differences among ROC curves (DeLong et al., 1988).
A separate classification analysis was also performed to evaluate the ability of Cmax to classify a larger set of drugs with known IDILI potential. Cmax values were obtained for 272 drugs from a study conducted by Xu et al. (2008) and evaluated using ROC analysis.
Results
Drug/Cytokine Cytotoxicity: Concentration Response In Vitro
HepG2 cells were treated with various concentrations of a drug alone or in combination with TNF and/or IFN, and cytotoxicity was assessed 24 hours later as increased LDH activity in the culture medium. Detailed cytotoxicity concentration-response curves were generated for 24 drugs (Table 1): 14 drugs that are associated with IDILI and 10 that are not (negative comparators) (Fig. 2; Supplemental Fig. 4). Of the 14 drugs associated with IDILI, almost all synergized with cytokines in causing cell death (Fig. 2). Four of these (diclofenac, bromfenac, nimesulide and clavulanate) caused no cytotoxicity on their own but synergized with TNF to cause cytotoxicity. Nine IDILI-associated drugs led to a statistically significant increase in LDH release (relative to no drug) in the absence of cytokines (valproic acid, doxorubicin, telithromycin, ibuprofen, naproxen, chlorpromazine, flutamide, trovafloxacin, and isoniazid). Interestingly TNF significantly enhanced the cytotoxicity mediated by eight of these drugs (valproic acid, doxorubicin, telithromycin, ibuprofen, naproxen, chlorpromazine, trovafloxacin, and isoniazid). In contrast to TNF, IFN did not interact with any drug to cause cytotoxicity. However, coexposure to IFN enhanced the cytotoxic interaction between TNF and several of the drugs (diclofenac, bromfenac, trovafloxacin, valproic acid, chlorpromazine, telithromycin, and isoniazid). Two of the 14 IDILI-associated drugs (flutamide and flucloxacillin) did not synergize with cytokines to kill HepG2 cells. Of the 10 negative comparators, pioglitazone was the only drug that caused cytotoxicity on its own; however, this effect was not enhanced by the addition of cytokines. With the exception of azithromycin, which was modestly cytotoxic in the presence of TNF/IFN, none of the remaining negative comparator drugs synergized with cytokines to kill HepG2 cells (Fig. 2).
Fig. 2.




Drug/cytokine-induced cytotoxicity: concentration-response. HepG2 cells were treated with 14 drugs associated with IDILI and 10 drugs not associated with IDILI, alone (VEH) and in combination with TNF and/or IFN. Cytotoxicity (% LDH release) was evaluated 24 hours after treatment. The numbers listed on the x-axis represent the concentration of drug relative to Cmax (fold Cmax). Refer to Table 1 for the Cmax information and known IDILI classification for each drug and to the Materials and Methods for the rationale concerning the range of drug concentrations evaluated. Each data point represents the mean ± standard error of the mean (SEM) of at least three separate experiments. The dotted curves indicate that the treatment condition (i.e., VEH, TNF, IFN, or TNF/IFN) resulted in a statistically significant change in LDH from baseline (no drug) at one or more drug concentrations (ANOVA P < 0.01). Solid lines indicate that the treatment condition did not result in a statistically significant change in LDH relative to baseline at any drug concentration (ANOVA P ≥ 0.01). The dotted curves were modeled using a four-parameter logistic function as described in the Materials and Methods to compute parameters describing curve characteristics (minimum cytotoxic effect, maximum cytotoxic effect, EC50, etc.).
Classification Models and ROC Analysis
Data used in the classification models consisted of covariate data generated as described above (i.e., parameters of the concentration-response curves for each of the 24 drugs and additional variables derived from these parameters) in addition to the daily dose and Cmax for each drug. Daily dose and Cmax values are shown in Table 1. Supplemental Tables 1–9 summarize parameters of the concentration-response curves for each drug and all derived covariates. Numerous logistic regression models were constructed. We began with models employing each covariate alone, then moved onto models populated with combinations of covariates (Supplemental Table 10). All classification models were evaluated for their ability to classify the 24 drugs using ROC analysis. Findings are described below.
Cmax Is Moderately Associated with IDILI Potential
IDILI reactions were once thought not to be dose related; however, the observation that most drugs that have been withdrawn from the market or have received a black box warning due to IDILI were prescribed at doses >50 mg/d suggested that daily dose plays some role in the propensity of a drug to cause IDILI (Uetrecht, 1999). Based on this observation, we evaluated how accurately the daily dose or the Cmax of a drug classifies drugs listed in Table 1 according to their potential to cause IDILI. We used logistic regression modeling and ROC analysis as described in the Materials and Methods. The AUC of the ROC curve generated for the model incorporating daily dose was 0.64 (95% confidence interval, 0.37, 0.9) (Fig. 3). Since the 95% confidence interval for the ROC curve from the daily dose model contained the value 0.5, representing no better than random classification, it cannot be concluded that the magnitude of the daily dose is predictive of IDILI for this drug set. A larger set of drugs may be needed to determine whether daily dose can predict IDILI.
Fig. 3.

Comparison of the model incorporating daily dose to that incorporating Cmax. (A) AUCs and 95% confidence intervals are depicted for the ROC curves derived from the models incorporating either daily dose or Cmax. The covariates are listed on the left, and the AUC for each one is shown on the right next to the 95% confidence interval (shown in brackets). (B) The ROC curves for the model incorporating daily dose and Cmax are indicated by red and blue lines, respectively. The 95% confidence intervals for the model incorporating daily dose and Cmax are shaded red or blue, respectively. Overlap between the confidence intervals for the two ROC curves appears in violet.
The AUC of the ROC curve generated for the model employing Cmax for our set of 24 drugs was 0.80 (95% confidence interval, 0.61, 0.98) (Fig. 3). Similar results were obtained by Shah et al. (2015) for a set of 125 drugs. These results suggest that Cmax is associated with IDILI.
To determine whether our set of 24 drugs is representative of a larger set of drugs and to further evaluate the ability of Cmax to predict IDILI liability, Cmax values were obtained for 272 drugs from a study conducted by Xu et al. (2008) and converted to micromolar units. Cmax was significantly associated with IDILI for this larger data set (βCmax= 0.044, P < 0.001). The AUC of the ROC curve generated from this larger set of drugs was 0.70 (95% confidence interval, 0.64, 0.76). The ROC curves derived from the set of 24 drugs and from the set of 272 drugs are depicted along with their 95% confidence intervals in Supplemental Fig. 2. The confidence interval corresponding to the ROC curve derived from the set of 272 drugs (shaded red) is contained within the confidence interval for the ROC curve derived from the set of 24 drugs (shaded gray). This suggests that the smaller set of drugs adequately represents the relationship between Cmax and IDILI potential seen in a much larger set of drugs.
ROC Analysis of Models Incorporating the Base Covariates
Almost all of the 14 IDILI-associated drugs synergized with TNF to cause death of HepG2 cells, and some of them were cytotoxic by themselves (Fig. 2). These results suggested that cytotoxic synergy with TNF might be associated with IDILI liability. Accordingly, classification models were constructed using base covariates from the concentration-response curves to determine whether the presence of TNF improved a model’s ability to classify drugs according to IDILI liability. The base covariates were modeled for each drug individually; base covariates that were at least moderately associated with IDILI liability included delta VEH, delta TNF, EC50 VEH, EC50 TNF, EC10 VEH, EC10 TNF, R10 VEH, and R10 TNF. AUCs and 95% confidence intervals are shown in Fig. 4A for each of these covariates. It can be seen that the confidence interval for each of these covariates does not contain the value 0.5, indicating a significantly better than random ability to classify the 24 drugs according to IDILI potential. The model incorporating delta TNF produced the ROC curve with the greatest AUC (0.93) and the narrowest 95% confidence interval (0.83, 1.00); this suggesting that of these models, it provided the most accurate classification of the drugs (Fig. 4A). Furthermore, the base covariates that described the response to drug/TNF (labeled “TNF”) led to models that produced ROC curves that had improved AUCs with narrower confidence intervals than those that described the response to drug alone (i.e., labeled “VEH”) (Fig. 4).
Fig. 4.

Evaluation of models incorporating the base covariates. (A) AUCs and 95% confidence intervals are illustrated for the ROC curves derived from the models incorporating the base covariates delta VEH, delta TNF, EC50 VEH, EC50 TNF, EC10 VEH, EC10 TNF, R10 VEH, or R10 TNF. The covariates are listed on the left, and the AUC for each is shown on the right next to the 95% confidence interval (shown in brackets). *P < 0.05 (statistically significant difference between ROC curves as determined by DeLong’s test) (DeLong et al., 1988). (B) ROC curves were generated and indicate for each model the 95% confidence interval shaded in gray. The covariates incorporated in the model are listed on the bottom right corner of each ROC curve.
ROC Analysis of Models Incorporating Derived Covariates
Probability models were also generated using the individual covariates that were derived from the base covariates: EC50 quotient, EC10 quotient, R10 quotient, and deltadiff. In Fig. 5, the EC50, EC10, and R10 quotient covariates represent the ratio of the drug/TNF concentration-response curve to the drug/VEH curve, and deltadiff represents the difference between the deltas from the drug/TNF concentration-response curve and the drug/VEH curve. Each of these covariates (except deltadiff) was moderately associated with IDILI liability (Fig. 5); however, the ROC curves generated based on these models did not have greater AUCs or narrower confidence intervals than the models produced by incorporating the base covariates (compare Figs. 4 and 5).
Fig. 5.

Evaluation of models incorporating the derived covariates. (A) AUCs and 95% confidence intervals for the ROC curves are depicted for the models incorporating the derived covariates individually. The covariates are listed on the left, and the AUC for each is shown on the right next to the 95% confidence interval (shown in brackets). (B) ROC curves were generated and indicate for each model the 95% confidence interval shaded in gray. The covariate incorporated in the model is listed on the bottom-right corner of each ROC curve.
Addition of IFN Data Did Not Improve the Classification of Drugs
None of the drugs synergized with IFN in the absence of TNF to cause cytotoxicity, but several IDILI-associated drugs synergized with IFN in the presence of TNF (Fig. 2). Accordingly, we examined whether incorporation of the TNF/IFN responses would improve the performance of models that employed only TNF responses. The drug/TNF/IFN models tended to have smaller AUCs and larger confidence intervals than the drug/TNF models (Supplemental Fig. 3), indicating that the addition of data describing the IFN response did not enhance the ability of models to classify drugs.
ROC Analysis of Models Incorporating Combinations of the Base and Derived Covariates
Although it was illustrative to evaluate the base and derived covariates individually, we hypothesized that incorporation of several covariates into a model would lead to more accurate classification of drugs than incorporation of a single covariate. Accordingly, various combinations of the base and derived covariates were evaluated to identify a set of covariates that led to the most accurate drug classification. Combining base and derived covariates led to several models with greater AUCs and narrower confidence intervals than the models incorporating only a single covariate. A representative set including the best-performing models is presented in Fig. 6. Furthermore, when Cmax was added as a covariate, it improved the performance (AUC and confidence interval) of some models but not others (Fig. 6). Supplemental Table 10 shows the coefficients (β values) and their P values for the models shown in Fig. 6.
Fig. 6.

Evaluation of models incorporating combinations of the base and derived covariates with and without Cmax. AUCs and 95% confidence intervals for the ROC curves are depicted for the models incorporating various combinations of base and derived covariates in the absence (upper half) and presence (lower half) of Cmax. The covariates are listed on the left, and the AUC for each is shown on the right next to the 95% confidence interval (shown in brackets).
Some of the combination models were associated with remarkably high AUCs, and some of these were associated with small confidence intervals. There were no statistically significant differences among the models with an AUC > 0.95 as determined by DeLong’s method for comparing ROC curves (P > 0.05) (DeLong et al., 1988). The ROC curves that met this criterion (AUC > 0.95) are shown in Fig. 7.
Fig. 7.

ROC curves with an AUC ≥ 0.95. ROC curves for which AUC ≥ 0.95 are depicted. The 95% confidence interval is shaded gray. The covariates incorporated into the model are listed at the bottom-right corner of each ROC curve. The ROC curves shown were not significantly different from each other as determined by DeLong’s test (P > 0.05).
Discussion
The purpose of this proof-of-concept study was to develop and evaluate an in vitro approach combined with statistical modeling to classify drugs according to their potential to cause IDILI. The overall hypothesis tested was that the ability of a drug to synergize with the cytokines TNF and/or IFN to kill HepG2 cells is associated with the drug’s propensity to cause IDILI in humans. Detailed concentration-response curves were generated, and this proved to be critical for development of a statistical classification model with the capacity to classify drugs correctly.
Since it has been suggested that the daily dose of a drug might be associated with its potential to cause IDILI and since dose is often related to Cmax, we evaluated how well daily dose or Cmax classifies drugs according to their IDILI liability. Daily dose was not effective at classifying the set of 24 drugs according to their potential to cause IDILI (Fig. 3). Plasma drug concentration is used as a surrogate for tissue or intracellular concentration, which is difficult to obtain. The relationship between Cmax and the concentration of free drug in liver cells can differ among drugs. The intrahepatocellular concentration of drugs can be affected not only by physicochemical properties of the drug, but also by uptake and efflux mechanisms and metabolism as well as binding to cellular or acellular components (Chu et al., 2013). Accordingly, for some drugs, the intrahepatocellular drug concentration can be many times the plasma concentration. Despite this potential disconnect, Cmax was somewhat effective at classifying the set of 24 drugs, suggesting that the magnitude of plasma drug concentration is a contributor to IDILI. However, it is clear that the use of Cmax as the sole covariate did not lead to a model with great classification ability (Supplemental Fig. 2).
We then determined whether cytotoxicity induced by treatment with drugs in the absence of cytokines could produce a high-performing model. Models employing only individual base covariates describing cytotoxicity in the absence of TNF performed no better than Cmax (compare Figs. 3 and 4). In contrast, the models incorporating TNF performed significantly better in classifying the drugs.
The derived covariates, when evaluated individually, did not produce more desirable ROC curves than the base covariates (compare Fig. 5 with Fig. 4). However, when covariates that account for TNF-induced changes in potency and/or efficacy were combined with those derived from drug alone, much better models resulted (Figs. 6 and 7). Furthermore, incorporating Cmax into these models led to the ROC curve with the greatest AUC (0.99) and narrowest confidence interval (0.97, 1) (Figs. 6 and 7). The coefficients and test statistic values for this best-performing classification model, which incorporated deltadiff, EC50 VEH, EC50 TNF, delta VEH, and Cmax as covariates, are listed in Table 2. A cutoff value is an estimated probability above which a drug would be classified as associated with IDILI (1 = associated with IDILI) and below which a drug would be classified as not associated with IDILI (0 = not associated with IDILI). The optimal cutoff threshold is the probability cutoff that permits the most accurate classification of drugs according to IDILI liability for a given model [i.e., the point on the ROC curve closest to the coordinate (1, 1)]. Table 3 shows the sensitivity and specificity of the best-performing model when the optimal cutoff threshold was applied. Based on this model, the estimated probability that a specific drug from the set of 24 drugs is associated with IDILI is shown in Table 4. As can be seen in Table 4, this classification model led to almost complete separation between IDILI-associated drugs and drugs that are not associated with IDILI. If this model were to be used in a preclinical safety evaluation setting to predict IDILI potential of a set of drugs, the user could either select the optimal cutoff threshold or choose a cutoff threshold that is either more or less sensitive depending on what false positive rate is deemed acceptable.
TABLE 2.
Logistic regression coefficients and P values for the optimal classification model incorporating the covariates deltadiff, EC50 VEH, EC50 TNF, delta VEH, and Cmax
| Covariate | β | χ2 | P Value |
|---|---|---|---|
| Intercept | −1.924 | 7.091 | 0.008 |
| Deltadiff | 0.108 | 2.471 | 0.116 |
| EC50 VEH | −0.066 | 6.215 | 0.013 |
| EC50 TNF | 0.050 | 4.480 | 0.034 |
| DeltaVEH | 0.081 | 11.038 | 0.001 |
| Cmax | 0.0031 | 0.129 | 0.720 |
The coefficients (β values) were computed using Firth’s approach as described in the Materials and Methods (Firth, 1993). A P value < 0.05 indicates that the covariate contributes significantly to the prediction of outcome (IDILI liability).
TABLE 3.
Sensitivity and specificity for the optimal classification model incorporating the covariates deltadiff, EC50 VEH, EC50 TNF, delta VEH, and Cmax
| Measure | Value | 95% Confidence Interval |
|---|---|---|
| Optimal cutoff threshold (k*) | 0.46 | |
| True negative rate (specificity) using threshold k* | 1 | (0.7, 1) |
| True positive rate (sensitivity) using threshold k* | 0.93 | (0.79, 1) |
| AUC | 0.99 | (0.97, 1) |
The optimal cutoff threshold (k*) is shown as are the specificity and sensitivity of the model at k*. Also indicated are the area under the ROC curve (AUC) and the 95% confidence intervals for the specificity, sensitivity, and AUC.
TABLE 4.
Estimated probabilities that a drug is associated with IDILI computed from the best-performing logistic regression model employing deltadiff, EC50 VEH, EC50 TNF, delta VEH, and Cmax as covariates
| Drug | Estimated Probability | True Classification |
|---|---|---|
| Buspirone | 0.1275 | IDILI− |
| Idarubicin | 0.1275 | IDILI− |
| Promethazine | 0.1275 | IDILI− |
| Sertraline | 0.1275 | IDILI− |
| Azithromycin | 0.1276 | IDILI− |
| Rofecoxib | 0.1278 | IDILI− |
| Moxifloxacin | 0.1296 | IDILI− |
| Levofloxacin | 0.1329 | IDILI− |
| Aspirin | 0.1445 | IDILI− |
| Flucloxacillin | 0.1545 | IDILI+ |
| Pioglitazone | 0.2935 | IDILI− |
| Telithromycin | 0.6685 | IDILI+ |
| Flutamide | 0.7036 | IDILI+ |
| Trovafloxacin | 0.7448 | IDILI+ |
| Isoniazid | 0.7562 | IDILI+ |
| Diclofenac | 0.8578 | IDILI+ |
| Naproxen | 0.8589 | IDILI+ |
| Doxorubicin | 0.8839 | IDILI+ |
| Bromfenac | 0.9397 | IDILI+ |
| Clavulanate | 0.9509 | IDILI+ |
| Chlorpromazine | 0.961 | IDILI+ |
| Ibuprofen | 0.9733 | IDILI+ |
| Valproic acid | 0.9909 | IDILI+ |
| Nimesulide | 1 | IDILI+ |
With regard to the true IDILI classification of drugs, IDILI− indicates that the drug is not associated with IDILI and IDILI + indicates that the drug is associated with IDILI in human patients.
IFN contributed to hepatotoxicity in several animal models of IDILI and was therefore of interest to include in our examination (Hassan et al., 2008; Shaw et al., 2009a; Dugan et al., 2011; Roth et al., 2017). Interestingly, IFN did not synergize with any of the drugs in our study to cause cell death (Fig. 2); however, as we showed previously for diclofenac (Maiuri et al., 2015), IFN enhanced the cytotoxic interaction between several IDILI-associated drugs and TNF. What underlies this pattern of response to IFN is not known; however, IFN can increase expression of TNF receptors (Wang et al., 2006) and, conversely, TNF can increase IFN signaling (Han et al., 1999; Robinson et al., 2003). These actions could explain why IFN had an effect only in the presence of TNF.
We evaluated whether a change in the concentration-response curves due to exposure to IFN could improve the classification of drugs. The classification model developed from the covariates that described the response to drug/TNF/IFN produced ROC curves that were not improved from those incorporating covariates that describe the response to drug/TNF (Supplemental Fig. 3). These results indicate that cytotoxic synergy between IDILI-associated drugs and TNF is sufficient to produce a statistical model that accurately classifies drugs irrespective of the presence of IFN. They also suggest that the cell killing activity of IFN depends on the presence of TNF.
It is worth considering the possibility that other cytokines play a role in the pathogenesis of IDILI, and it would be interesting to examine whether other cytokines could interact with drugs to cause cytotoxicity in vitro and/or synergize with TNF to enhance cytotoxicity. Cosgrove et al. (2009) performed a study examining drug/cytokine interactions in vitro and found that interleukin (IL)-1β in combination with TNF and IFN interacted with some drugs to cause cytotoxicity. It is unclear to what extent IL-1β contributed to this interaction. However, Shaw et al. (2009b) demonstrated that IL-1β levels are elevated in mice cotreated with trovafloxacin and lipopolysaccharide, raising the possibility that IL-β plays a role in the hepatotoxicity observed in these mice. Whether the presence of IL-1β or other immune mediators would improve the ability of the models presented herein to classify drugs is unknown but worth considering in future studies.
We reported recently that IFN-mediated enhancement of NSAID/TNF-induced cytotoxicity occurs with some IDILI-associated NSAIDs but not others, and this effect was related to chemical structure and to the magnitude of clinical concern about IDILI for specific NSAIDs (Maiuri et al., 2015). Specifically, several acetic acid derivatives that are associated with IDILI of greatest clinical concern synergized with TNF to cause HepG2 cell death, and IFN enhanced this effect. In contrast, two propionic acid derivatives, which are associated with IDILI that is of less clinical concern, also synergized with TNF, but IFN was without effect. In the analysis presented here, we were seeking a binary answer (IDILI potential yes or no) and inclusion of IFN did not affect the outcome. It would be interesting if the ability of drugs to sensitize cells to the harmful effects of IFN could distinguish drugs of greater concern clinically for IDILI from those of less concern. Clearly, a larger number of drugs would need to be analyzed to evaluate this.
A potential challenge that might be faced when employing this assay during preclinical safety evaluation is the inability to generate complete concentration-response relationships due to solubility limitations of the drug or other factors. Computing covariates using the four-parameter logistic model requires complete concentration-response curves; however, we defined several covariates that can be computed without the need to generate a complete concentration-response curve. One of these is R10, or the drug concentration at which there is an increase of 10% LDH activity above min. Another covariate that could be computed without the need to generate a complete concentration-response curve we defined as “TNF change.” TNF change identifies those drugs that are only cytotoxic in the presence of TNF without the need for complete concentration-response curves. Interestingly, combining the covariates R10 quotient (i.e., R10 TNF/R10 VEH) and TNF change resulted in an ROC curve with an AUC of 0.88 (95% confidence interval, 0.75, 1) (Supplemental Fig. 1). This suggests that a model that leads to good classification of drugs according to their potential to cause IDILI can be generated without the need to delineate complete concentration-response relationships. This model might be useful for predicting the IDILI potential of drug candidates when availability of compound is limited or when solubility limitation prevents generation of a complete concentration-response curve.
Although HepG2 cells are human derived, their use for drug toxicity evaluation has been criticized because they have limited capacity to bioactivate drugs to toxic metabolites via cytochrome P450–mediated pathways. Despite this potential limitation, Cosgrove et al. (2009) found that HepG2 cells behave similarly to primary human hepatocytes in their cytotoxic responses to drug-cytokine combinations. We have also observed comparable responses in primary murine hepatocytes (Zou et al., 2009; Beggs et al., 2014; Maiuri et al., 2015). These findings suggest either that 1) metabolic activation of drugs by HepG2 cells, although limited, is sufficient to stress cells so that they respond to cytokine exposure by dying or 2) metabolism is not generally needed for the cytotoxic interaction of drugs with cytokines.
Visual inspection of Fig. 2 revealed three IDILI+ drugs for which there was modest (flutamide, clavulanate) or no (flucloxacillin) response. Despite this, statistical modeling classified flutamide and clavulanate as IDILI+. In the case of flucloxacillin, analysis of variance of the concentration-response data determined that there was no statistically significant increase in LDH alone or in the presence of TNF. As a result, covariates were set to zero with the exception of Cmax, and the calculated probability for flucloxacillin was low (0.154; see the formula in the Materials and Methods). In contrast, analysis of variance applied to the concentration-response data for flutamide alone and in combination with TNF detected very small but statistically significant increases in LDH release relative to baseline (Fig. 2; Supplemental Fig. 1). Consequently, nonzero covariates were generated from the concentration-response data. Similarly for clavulanate, there was an interaction with TNF that was small but statistically significant, resulting in nonzero covariates. These nonzero covariates led to calculated probabilities that, at the optimal cutoff, identified flutamide and clavulanate correctly as IDILI+. The pronounced reproducibility of even small changes using HepG2 cells and the apparent sensitivity of the model in these two cases may be a strength of this approach to classification.
Flutamide is metabolized in vivo to 2-hydroxyflutamide (Cmax, 5.74 μM), which is more potent pharmacologically as an antiandrogen (Brogden and Clissold, 1989) and is thought to contribute to IDILI responses (Ball et al., 2016). To strengthen this proof-of-concept study and to further evaluate the modest cytotoxic effect of flutamide, we investigated the performance of 2-hydroxyflutamide in the assay. Concentration-response curves were generated (Supplemental Fig. 5), and covariates derived from the curves were used in the best-performing classification model. A high probability for association with IDILI was calculated (0.999) for 2-hydroxyflutamide. Furthermore, we replaced the flutamide covariate data in the best-performing model with the 2-hydroxyflutamide covariates. Recalculation of the best-performing classification model yielded coefficients similar to those presented in Table 2 (Supplemental Table 11), the same AUC for the ROC curve, and no change in classification of drugs as IDILI+ or IDILI−. These results strengthen the conclusions based on the modest cytotoxic response to flutamide and support the approach to classifying IDILI-associated drugs.
The observation that flucloxacillin was incorrectly classified as not associated with IDILI suggests that the best-performing model, although seemingly promising, has limitations. It is worth noting that flucloxacillin typically produces liver injury in humans that is classified as cholestatic rather than hepatocellular (Enat et al., 1980; Williams and Malatjalian, 1981; Bengtsson et al., 1985; Moseley, 2013). Clavulanate is also associated predominately with a cholestatic pattern of injury in human patients (Sánchez-Ruiz-Granados et al., 2012; Beraldo et al., 2013) and interacted only weakly with TNF. Accordingly, it is possible that the approach described herein is more robust in classifying drugs that cause hepatocellular rather than cholestatic patterns of injury.
In summary, these results add to evidence that drug-induced stress can sensitize hepatocytes to the killing actions of cytokines such as TNF and IFN (reviewed in Roth et al., 2017). Moreover, this could be requisite for the pathogenesis of IDILI, since numerous IDILI-associated drugs show cytotoxic synergy with cytokines in vitro at drug concentrations near those that occur in patients. Currently, effective assays to screen preclinically for IDILI potential are lacking. A method that accurately identifies drug candidates with the potential to cause IDILI could revolutionize preclinical testing strategies. Our results suggest an in vitro assay that could do just that (i.e., by delineating drug concentration-response curves in the absence and presence of TNF and employing resulting covariates in an appropriate statistical model for classification). One of the strengths of this approach is that the user would have discretion to choose a level of risk tolerance guided by the results of ROC analysis. That choice could depend on a variety of factors, including risk tolerance in the context of the therapeutic use, other drug candidates that are in contention for going forward into development, and so forth. For example, if several potentially effective compounds with no apparent toxicity were identified in early preclinical screens but some returned an “IDILI+” result in a drug-cytokine assay, this might prompt a decision to pursue other candidates for development. The magnitude of interaction with TNF assessed by direct inspection of concentration-response curves (Fig. 2) might also be useful to inform such decisions. Overall, this classification approach is attractive because it 1) uses a cell type that is easily obtained and maintained in culture and yields consistent results, 2) requires minimal amounts of test compound, 3) employs a single, easily and inexpensively measured phenotypic endpoint that is directly relevant to IDILI (hepatocellular death), 4) is based on interaction between drug and a product of immune system activation likely to be relevant to IDILI pathogenesis, and 5) is adaptable to high-throughput technology. Validation of this approach as a screening tool will require the evaluation of additional drugs, but the results presented herein are quite promising.
Acknowledgments
The authors thank Holly Mollon for technical assistance.
Abbreviations
- AUC
area under the curve
- IDILI
idiosyncratic drug-induced liver injury
- IFN
interferon-γ
- IL
interleukin
- LDH
lactate dehydrogenase
- NSAID
nonsteroidal anti-inflammatory drug
- ROC
receiver operating characteristic
- TNF
tumor necrosis factor-α
- VEH
vehicle
Authorship Contributions
Participated in research design: Maiuri, Wassink, Hession, Ganey, Roth.
Conducted experiments: Maiuri, Turkus, Breier, Lansdell, Kaur.
Performed data analysis: Maiuri, Wassink, Hession, Ganey, Roth.
Wrote or contributed to the writing of the manuscript: Maiuri, Hession, Ganey, Roth.
Footnotes
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant R01DK112695 (to R.A.R. and P.E.G.)], the National Institutes of Health National Institute of General Medical Sciences [Grant T32GM092715 (to A.R.M.)], and a Colgate-Palmolive Award for Research in Alternative Methods.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, Hunt CM, Wilke RA, Avigan M, Kaplowicz N, et al. (2011) Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther 89(6):806–15. [DOI] [PubMed] [Google Scholar]
- Aleo MD, Luo Y, Swiss R, Bonin PD, Potter DM, Will Y. (2014) Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 60:1015–1022. [DOI] [PubMed] [Google Scholar]
- Ball AL, Kamalian L, Alfirevic A, Lyon JJ, Chadwick AE. (2016) Identification of the additional mitochondrial liabilities of 2-hydroxyflutamide when compared with its parent compound, flutamide in HepG2 cells. Toxicol Sci 153:341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barpe DR, Rosa DD, Froehlich PE. (2010) Pharmacokinetic evaluation of doxorubicin plasma levels in normal and overweight patients with breast cancer and simulation of dose adjustment by different indexes of body mass. Eur J Pharm Sci 41:458–463. [DOI] [PubMed] [Google Scholar]
- Beggs KM, Fullerton AM, Miyakawa K, Ganey PE, Roth RA. (2014) Molecular mechanisms of hepatocellular apoptosis induced by trovafloxacin-tumor necrosis factor-alpha interaction. Toxicol Sci 137:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs KM, Maiuri AR, Fullerton AM, Poulsen KL, Breier AB, Ganey PE, Roth RA. (2015) Trovafloxacin-induced replication stress sensitizes HepG2 cells to tumor necrosis factor-alpha-induced cytotoxicity mediated by extracellular signal-regulated kinase and ataxia telangiectasia and Rad3-related. Toxicology 331:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson F, Florén CH, Hägerstrand I, Söderström C, Aberg T. (1985) Flucloxacillin-induced cholestatic liver damage. Scand J Infect Dis 17:125–128. [DOI] [PubMed] [Google Scholar]
- Beraldo DO, Melo JF, Bonfim AV, Teixeira AA, Teixeira RA, Duarte AL. (2013) Acute cholestatic hepatitis caused by amoxicillin/clavulanate. World J Gastroenterol 19:8789–8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramlage P, Goldis A. (2008) Bioequivalence study of three ibuprofen formulations after single dose administration in healthy volunteers. BMC Pharmacol 8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon RA, Eadie MJ, Curran AC, Nolan PC, Presneill JJ, Patterson MC. (1986) A new formulation of aspirin: bioavailability and analgesic efficacy in migraine attacks. Cephalalgia 6:19–27. [DOI] [PubMed] [Google Scholar]
- Brogden RN, Clissold SP. (1989) Flutamide. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in advanced prostatic cancer. Drugs 38:185–203. [DOI] [PubMed] [Google Scholar]
- Buchweitz JP, Ganey PE, Bursian SJ, Roth RA. (2002) Underlying endotoxemia augments toxic responses to chlorpromazine: is there a relationship to drug idiosyncrasy? J Pharmacol Exp Ther 300:460–467. [DOI] [PubMed] [Google Scholar]
- Chakraborty M, Fullerton AM, Semple K, Chea LS, Proctor WR, Bourdi M, Kleiner DE, Zeng X, Ryan PM, Dagur PK, et al. (2015) Drug-induced allergic hepatitis develops in mice when myeloid-derived suppressor cells are depleted prior to halothane treatment. Hepatology 62:546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Borlak J, Tong W. (2013) High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 58:388–396. [DOI] [PubMed] [Google Scholar]
- Chen M, Tung CW, Shi Q, Guo L, Shi L, Fang H, Borlak J, Tong W. (2014) A testing strategy to predict risk for drug-induced liver injury in humans using high-content screen assays and the ‘rule-of-two’ model. Arch Toxicol 88:1439–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JM, Oh SJ, Lee JY, Jeon JS, Ryu CS, Kim YM, Lee K, Kim SK. (2015) Prediction of drug-induced liver injury in HepG2 cells cultured with human liver microsomes. Chem Res Toxicol 28:872–885. [DOI] [PubMed] [Google Scholar]
- Chu X, Korzekwa K, Elsby R, Fenner K, Galetin A, Lai Y, Matsson P, Moss A, Nagar S, Rosania GR, et al. International Transporter Consortium (2013) Intracellular drug concentrations and transporters: measurement, modeling, and implications for the liver. Clin Pharmacol Ther 94:126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove BD, King BM, Hasan MA, Alexopoulos LG, Farazi PA, Hendriks BS, Griffith LG, Sorger PK, Tidor B, Xu JJ, et al. (2009) Synergistic drug-cytokine induction of hepatocellular death as an in vitro approach for the study of inflammation-associated idiosyncratic drug hepatotoxicity. Toxicol Appl Pharmacol 237:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong ER, DeLong DM, Clarke-Pearson DL. (1988) Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44:837–845. [PubMed] [Google Scholar]
- Deng X, Stachlewitz RF, Liguori MJ, Blomme EA, Waring JF, Luyendyk JP, Maddox JF, Ganey PE, Roth RA. (2006) Modest inflammation enhances diclofenac hepatotoxicity in rats: role of neutrophils and bacterial translocation. J Pharmacol Exp Ther 319:1191–1199. [DOI] [PubMed] [Google Scholar]
- Dugan CM, Fullerton AM, Roth RA, Ganey PE. (2011) Natural killer cells mediate severe liver injury in a murine model of halothane hepatitis. Toxicol Sci 120:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enat R, Pollack S, Ben-Arieh Y, Livni E, Barzilai D. (1980) Cholestatic jaundice caused by cloxacillin: macrophage inhibition factor test in preventing rechallenge with hepatotoxic drugs. BMJ 280:982–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth D. (1993) Bias reduction of maximum likelihood estimates. Biometrika 80:27–38. [Google Scholar]
- Fredriksson L, Herpers B, Benedetti G, Matadin Q, Puigvert JC, de Bont H, Dragovic S, Vermeulen NP, Commandeur JN, Danen E, et al. (2011) Diclofenac inhibits tumor necrosis factor-α-induced nuclear factor-κB activation causing synergistic hepatocyte apoptosis. Hepatology 53:2027–2041. [DOI] [PubMed] [Google Scholar]
- Fredriksson L, Wink S, Herpers B, Benedetti G, Hadi M, de Bont H, Groothuis G, Luijten M, Danen E, de Graauw M, et al. (2014) Drug-induced endoplasmic reticulum and oxidative stress responses independently sensitize toward TNFα-mediated hepatotoxicity. Toxicol Sci 140:144–159. [DOI] [PubMed] [Google Scholar]
- Gottesdiener K, Agrawal N, Porras A, Wong P, Rogers JD, Gertz BJ, Redfern JS, Marbury T. (2003) Effects of renal insufficiency and hemodialysis on the pharmacokinetics of rofecoxib. Am J Ther 10:252–258. [DOI] [PubMed] [Google Scholar]
- Gumbhir-Shah K, Cevallos WH, DeCleene SA, Halstenson CE, Korth-Bradley JM. (1997) Absolute bioavailability of bromfenac in humans. Ann Pharmacother 31:395–399. [DOI] [PubMed] [Google Scholar]
- Han Y, Rogers N, Ransohoff RM. (1999) Tumor necrosis factor-alpha signals to the IFN-gamma receptor complex to increase Stat1alpha activation. J Interferon Cytokine Res 19:731–740. [DOI] [PubMed] [Google Scholar]
- Hassan F, Morikawa A, Islam S, Tumurkhuu G, Dagvadorj J, Koide N, Naiki Y, Mori I, Yoshida T, Yokochi T. (2008) Lipopolysaccharide augments the in vivo lethal action of doxorubicin against mice via hepatic damage. Clin Exp Immunol 151:334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinze G, Ploner M, Dunkler D, and Southworth H (2013) Logistf: Firth’s Bias Reduced Logistic Regression, R Package Version 1.21. R Foundation for Statistical Computing, Vienna, Austria.
- Hu G, Dai Z, Long L, Han Y, Hou S, Wu L. (2002) Bioequivalence of clavulanate potassium and amoxicillin (1:7) dispersible tablets in healthy volunteers. J Huazhong Univ Sci Technolog Med Sci 22:224–227. [DOI] [PubMed] [Google Scholar]
- Khetani SR, Kanchagar C, Ukairo O, Krzyzewski S, Moore A, Shi J, Aoyama S, Aleo M, Will Y. (2013) Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol Sci 132:107–117. [DOI] [PubMed] [Google Scholar]
- Low Y, Uehara T, Minowa Y, Yamada H, Ohno Y, Urushidani T, Sedykh A, Muratov E, Kuz’min V, Fourches D, et al. (2011) Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem Res Toxicol 24:1251–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Jones AD, Harkema JR, Roth RA, Ganey PE. (2012) Amiodarone exposure during modest inflammation induces idiosyncrasy-like liver injury in rats: role of tumor necrosis factor-alpha. Toxicol Sci 125:126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, Day CP, Ruiz-Cabello F, Donaldson PT, Stephens C, et al. Spanish DILI Registry. EUDRAGENE. DILIN. DILIGEN. International SAEC (2011) Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 141:338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri AR, Breier AB, Gora LF, Parkins RV, Ganey PE, Roth RA. (2015) Cytotoxic synergy between cytokines and NSAIDs associated with idiosyncratic hepatotoxicity is driven by mitogen-activated protein kinases. Toxicol Sci 146:265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metushi IG, Hayes MA, Uetrecht J. (2015) Treatment of PD-1(−/−) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 61(4):1332–42. [DOI] [PubMed] [Google Scholar]
- Morgan RE, van Staden CJ, Chen Y, Kalyanaraman N, Kalanzi J, Dunn RT, 2nd, Afshari CA, Hamadeh HK. (2013) A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci 136:216–241. [DOI] [PubMed] [Google Scholar]
- Moseley R. (2013) Hepatotoxicity of antimicrobials and antifuncal agents, in Drug-Induced Liver Disease, 3rd ed (Kaplowitz N, Deleve L. eds) pp 463–482, Elsevier, Amsterdam. [Google Scholar]
- Ostapowicz G, Fontana RJ, Schiødt FV, Larson A, Davern TJ, Han SH, McCashland TM, Shakil AO, Hay JE, Hynan L, et al. U.S. Acute Liver Failure Study Group (2002) Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 137:947–954. [DOI] [PubMed] [Google Scholar]
- Pinsky MR, Vincent JL, Deviere J, Alegre M, Kahn RJ, Dupont E. (1993) Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest 103:565–575. [DOI] [PubMed] [Google Scholar]
- R Core Team (2015) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Rha JH, Jang IJ, Lee KH, Chong WS, Shin SG, Lee N, Myung HJ. (1993) Pharmacokinetic comparison of two valproic acid formulations--a plain and a controlled release enteric-coated tablets. J Korean Med Sci 8:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C, Streibig J. (2005) Bioassay analysis using R. J Stat Softw 12:1–22. [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J, and Müller M (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CM, Shirey KA, Carlin JM. (2003) Synergistic transcriptional activation of indoleamine dioxygenase by IFN-gamma and tumor necrosis factor-alpha. J Interferon Cytokine Res 23:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Røder BL, Frimodt-Møller N, Espersen F, Rasmussen SN. (1995) Dicloxacillin and flucloxacillin: pharmacokinetics, protein binding and serum bactericidal titers in healthy subjects after oral administration. Infection 23:107–112. [DOI] [PubMed] [Google Scholar]
- Roth RA, Ganey PE. (2011) Animal models of idiosyncratic drug-induced liver injury--current status. Crit Rev Toxicol 41:723–739. [DOI] [PubMed] [Google Scholar]
- Roth RA, Maiuri AR, Ganey PE. (2017) Idiosyncratic drug-induced liver injury: is drug-cytokine interaction the linchpin? J Pharmacol Exp Ther 360:461–470. [DOI] [PubMed] [Google Scholar]
- Sakatis MZ, Reese MJ, Harrell AW, Taylor MA, Baines IA, Chen L, Bloomer JC, Yang EY, Ellens HM, Ambroso JL, et al. (2012) Preclinical strategy to reduce clinical hepatotoxicity using in vitro bioactivation data for >200 compounds. Chem Res Toxicol 25:2067–2082. [DOI] [PubMed] [Google Scholar]
- Sánchez-Ruiz-Granados E, Bejarano-García A, Uceda-Torres E. (2012) Recurrent cholestasis by amoxicillin-clavulanic acid: the importance of a correct diagnosis of hepatotoxicity. Rev Esp Enferm Dig 104:616–617. [DOI] [PubMed] [Google Scholar]
- Schadt S, Simon S, Kustermann S, Boess F, McGinnis C, Brink A, Lieven R, Fowler S, Youdim K, Ullah M, et al. (2015) Minimizing DILI risk in drug discovery - a screening tool for drug candidates. Toxicol In Vitro 30 (Pt B):429–437. [DOI] [PubMed] [Google Scholar]
- Setiawati E, Deniati S, Yunaidi D, Handayani L, Harinanto G, Santosos ID, Purnomo Sari A, and Rimainar A (2009) Bioequivalence study with two naproxen sodium tablet formulations in healthy subjects. J Bioequivalence Bioavailability 1:28–33. [Google Scholar]
- Shah F, Leung L, Barton HA, Will Y, Rodrigues AD, Greene N, Aleo MD. (2015) Setting clinical exposure levels of concern for drug-induced liver injury (DILI) using mechanistic in vitro assays. Toxicol Sci 147:500–514. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Ditewig AC, Waring JF, Liguori MJ, Blomme EA, Ganey PE, Roth RA. (2009a) Coexposure of mice to trovafloxacin and lipopolysaccharide, a model of idiosyncratic hepatotoxicity, results in a unique gene expression profile and interferon gamma-dependent liver injury. Toxicol Sci 107:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Ganey PE, Roth RA. (2009b) Tumor necrosis factor alpha is a proximal mediator of synergistic hepatotoxicity from trovafloxacin/lipopolysaccharide coexposure. J Pharmacol Exp Ther 328:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stass H, Dalhoff A, Kubitza D, Schühly U. (1998) Pharmacokinetics, safety, and tolerability of ascending single doses of moxifloxacin, a new 8-methoxy quinolone, administered to healthy subjects. Antimicrob Agents Chemother 42:2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susukida T, Sekine S, Nozaki M, Tokizono M, Ito K. (2015) Prediction of the clinical risk of drug-induced cholestatic liver injury using an in vitro sandwich cultured hepatocyte assay. Drug Metab Dispos 43:1760–1768. [DOI] [PubMed] [Google Scholar]
- Taudorf S, Krabbe KS, Berg RM, Pedersen BK, Møller K. (2007) Human models of low-grade inflammation: bolus versus continuous infusion of endotoxin. Clin Vaccine Immunol 14:250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RA, Isin EM, Li Y, Weidolf L, Page K, Wilson I, Swallow S, Middleton B, Stahl S, Foster AJ, et al. (2012) In vitro approach to assess the potential for risk of idiosyncratic adverse reactions caused by candidate drugs. Chem Res Toxicol 25:1616–1632. [DOI] [PubMed] [Google Scholar]
- Uetrecht JP. (1999) New concepts in immunology relevant to idiosyncratic drug reactions: the “danger hypothesis” and innate immune system. Chem Res Toxicol 12:387–395. [DOI] [PubMed] [Google Scholar]
- Usui T, Mise M, Hashizume T, Yabuki M, Komuro S. (2009) Evaluation of the potential for drug-induced liver injury based on in vitro covalent binding to human liver proteins. Drug Metab Dispos 37:2383–2392. [DOI] [PubMed] [Google Scholar]
- Vanderlinde RE. (1985) Measurement of total lactate dehydrogenase activity. Ann Clin Lab Sci 15:13–31. [PubMed] [Google Scholar]
- Viechtbauer W. (2010) Conducting meta-analyses in R with the metafor package. J Stat Softw 36:1–48. [Google Scholar]
- Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, Abraham C, Turner JR. (2006) IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology 131:1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware BR, Berger DR, Khetani SR. (2015) Prediction of drug-induced liver injury in micropatterned co-cultures containing iPSC-derived human hepatocytes. Toxicol Sci 145:252–262. [DOI] [PubMed] [Google Scholar]
- Watkins (2005) Idiosyncratic liver injury: challenges and approaches. Toxicol Pathol 33(1):1–5. [DOI] [PubMed]
- Williams CN, Malatjalian DA. (1981) Severe penicillin-induced cholestasis in a 91-year-old woman. Dig Dis Sci 26:470–473. [DOI] [PubMed] [Google Scholar]
- Xu JJ, Henstock PV, Dunn MC, Smith AR, Chabot JR, de Graaf D. (2008) Cellular imaging predictions of clinical drug-induced liver injury. Toxicol Sci 105:97–105. [DOI] [PubMed] [Google Scholar]
- Zhang J, Doshi U, Suzuki A, Chang CW, Borlak J, Li AP, Tong W. (2016a) Evaluation of multiple mechanism-based toxicity endpoints in primary cultured human hepatocytes for the identification of drugs with clinical hepatotoxicity: results from 152 marketed drugs with known liver injury profiles. Chem Biol Interact 255:3–11. [DOI] [PubMed] [Google Scholar]
- Zhang J, Salminen A, Yang X, Luo Y, Wu Q, White M, Greenhaw J, Ren L, Bryant M, Salminen W, et al. (2016b) Effects of 31 FDA approved small-molecule kinase inhibitors on isolated rat liver mitochondria. Arch Toxicol 91:2921–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu XW, Sedykh A, Liu SS. (2014) Hybrid in silico models for drug-induced liver injury using chemical descriptors and in vitro cell-imaging information. J Appl Toxicol 34:281–288. [DOI] [PubMed] [Google Scholar]
- Zou W, Beggs KM, Sparkenbaugh EM, Jones AD, Younis HS, Roth RA, Ganey PE. (2009) Sulindac metabolism and synergy with tumor necrosis factor-alpha in a drug-inflammation interaction model of idiosyncratic liver injury. J Pharmacol Exp Ther 331:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]