

Abstract
Background
In the yeast Saccharomyces cerevisiae, as in every eukaryotic organism, the mRNA 5′-untranslated region (UTR) is important for translation initiation. However, the patterns and mechanisms that determine the efficiency with which ribozomes bind mRNA, the elongation of ribosomes through the 5′-UTR, and the formation of a stable translation initiation complex are not clear. Genes that are highly expressed in S. cerevisiae seem to prefer a 5′-UTR rich in adenine and poor in guanine, particularly in the Kozak sequence, which occupies roughly the first six nucleotides upstream of the START codon.
Results
We measured the fluorescence produced by 58 synthetic versions of the S. cerevisiae minimal CYC1 promoter (pCYC1min), each containing a different 5′-UTR. First, we replaced with adenine the last 15 nucleotides of the original pCYC1min 5′-UTR—a theoretically optimal configuration for high gene expression. Next, we carried out single and multiple point mutations on it. Protein synthesis was highly affected by both single and multiple point mutations upstream of the Kozak sequence. RNAfold simulations revealed that significant changes in the mRNA secondary structures occur by mutating more than three adenines into guanines between positions −15 and −9. Furthermore, the effect of point mutations turned out to be strongly context-dependent, indicating that adenines placed just upstream of the START codon do not per se guarantee an increase in gene expression, as previously suggested.
Conclusions
New synthetic eukaryotic promoters, which differ for their translation initiation rate, can be built by acting on the nucleotides upstream of the Kozak sequence. Translation efficiency could, potentially, be influenced by another portion of the 5′-UTR further upstream of the START codon. A deeper understanding of the role of the 5′-UTR in gene expression would improve criteria for choosing and using promoters inside yeast synthetic gene circuits.
Electronic supplementary material
The online version of this article (doi:10.1186/s13036-017-0068-1) contains supplementary material, which is available to authorized users.
Keywords: Synthetic biology, S. cerevisiae, 5′-UTR, Kozak sequence
Background
Rational design of synthetic gene circuits demands proper characterization and categorization of their basic components. These basic components are DNA sequences associated with precise transcription or translation functions and are known as standard biological parts [1]. Bacterial parts are divided into four main groups: promoters, ribosome binding sites (RBSs), coding sequences (CDSs), and terminators. Promoters and terminators are, respectively, the start and stop signals for DNA transcription into mRNA. The RBS is the entry point for the ribosomes into the mRNA, i.e. the place where translation initiation takes place. Protein synthesis terminates when the ribosomes meet the STOP codon at the end of the CDS.
The main feature of the RBS is the Shine-Dalgarno sequence, located approximately 10 nucleotides upstream of the START codon. Its complementarity to a ribosomal region allows it to be recognized and bound by ribosomes. The RBS has been widely studied and, remarkably, software has been developed to design regulated RBSs with the aim of tuning gene expression [2].
Eukaryotic cells do not have a counterpart to the Shine-Dalgarno sequence and mature mRNA is recognized by ribosomes because of the presence of the 5′ cap. Although the mRNA 5′ untranslated region (UTR) plays an important role in determining protein expression [3–5], a eukaryotic standard biological part corresponding to it has never been defined and it is regarded simply as the end of the promoter sequence.
A feature specific to eukaryotic mRNA is the Kozak sequence [6], which extends from approximately position −6 to position +6, where +1 is assigned to the adenine of the START codon (throughout the present paper, all positions are given respective to the START codon). The consensus Kozak sequence varies across organisms in length and nucleotide composition (see for instance [6–8]). However, point mutations in the Kozak sequence affect transcription initiation both in higher [6] and lower [9] eukaryotes.
The consensus Kozak sequence in yeast S. cerevisiae was identified by Hamilton et al. [8], who analyzed the translation initiation site (defined as the region between positions −47 and +50) of 99 genes and calculated the frequency at which each of the four nucleotides occupies any of these positions. Position occupancies were also calculated on the subset of highly expressed genes, a main reference point for our work in constructing synthetic 5′-UTRs that enhance gene expression.
The portion of 5′-UTRs made of 47 nucleotides, taken into account in [8], was rich in adenine and poor in guanine. The abundance of a single nucleotide (adenine) was explained as a means to avoid secondary structures in the leader sequence, which could prevent efficient gene expression. However, this explanation is somewhat challenged by recent in vitro analysis showing a positive correlation between secondary structure in the S. cerevisiae 5′-UTR and protein abundance [10–12]. The optimal S. cerevisiae consensus Kozak sequence (i.e. arising from the group of highly expressed genes) was determined as
where adenine and thymine at position −6 and adenine and cytosine at position −4 showed the same frequency, whereas at position −2 the frequency of adenine was higher than that of cytosine (50 % versus 33 %). Interestingly, adenine was always present at position −3. The triplet at positions −2 to −4 exhibits some analogies with the consensus Kozak sequence in mammalian cells. Here, a cytosine can be found at positions −4 and −2 and a purine (A or G) is highly conserved at position −3.
Following the analysis by Hamilton and co-authors [8], we can define those nucleotides whose frequency at a given position is >50 % as strongly conserved. Accordingly, adenine is strongly conserved at positions −1, −3, −5, −7, and −8. From position −9 to −15, no nucleotide is strongly conserved. In all these positions the most frequent nucleotide is adenine, apart from position −14 where thymine prevails slightly (39 % thymine versus 33 % adenine). At positions −16 to −20 adenine again becomes strongly conserved. More upstream, short traits where adenine is strongly conserved interrupt longer regions where the frequency of no nucleotide reaches 51 %.
A different study on the effect of the 5′-UTR on gene expression in S. cerevisiae was carried out by Dvir et al. [9]. They generated a pool of 2041 distinct leader sequences by performing random mutations in the −10…−1 region of the RPL8A gene (the whole RPL8A 5′-UTR is only 17 nucleotides long). The RPL8A promoter was placed in front of a reporter protein and the influence of diverse 5′-UTRs on gene expression was quantified by fluorescence measurements. The data collected in that study showed that protein synthesis is highly influenced by the nucleotide at position −3: a purine (principally adenine) increases protein expression, whereas a pyrimidine lowers it. A further enhancement in gene expression arises when the purine at position −3 is accompanied by other adenines at positions −1 to −4. Moreover, an adenine at position −1 is sufficient to increase protein expression no matter which nucleotide is present at position −3. In contrast, a guanine at position −2 and a cytosine at position −1 have a negative impact on translation initiation. Cytosine is also moderately under-represented at position −2 among the highly expressed RPL8A variants.
Taking our cue from the works by Hamilton et al. [8] and Dvir et al. [9], we carried out a detailed study on how synthetic 5′-UTRs can alter gene expression in S. cerevisiae. By following an approach similar to that in [9], we focused on a unique gene (CYC1) and built 58 synthetic variants of the minimal CYC1 promoter (pCYC1min) via single and multiple point mutations between positions −15 and −1. The starting point of our work was, however, not the original CYC1 leader sequence but a synthetic one optimized for high gene expression, according to the results in [8]. We fused each synthetic 5′-UTR to the CDS of a green fluorescent protein and quantified gene expression via FACS experiments. Our results indicate clearly that nucleotides upstream of the Kozak sequence (positions −9 to −15) play an important role in protein synthesis. Above all, single point mutations, on an adenine background, tend to increase protein expression, whereas multiple mutations to guanine are negatively correlated to gene expression, even when followed by a Kozak sequence containing only adenines. The latter result is due to changes in the mRNA secondary structure as indicated by the results of simulations with the software RNAfold [13, 14]. Our work paves the way for the characterization of eukaryotic promoters through their translational strength.
Results and discussion
We chose the 5′-UTR of the well-studied S. cerevisiae CYC1 promoter [15, 16]. We fused pCYC1min (starting at position −143) to a yeast-enhanced green fluorescent protein (yEGFP) [17] and the CYC1 terminator. Compared to the complete CYC1 promoter, pCYC1min contains two of the three TATA boxes and no upstream activating sequences. pCYC1min is a moderately weak promoter and, for this reason, appears to be an ideal candidate for detecting both positive and negative effects of point mutations in the leader sequence on the expression of the downstream reporter protein. The CYC1 promoter 5′-UTR is 71 nucleotides long.
In the following analysis, we refer to the portion of CYC1 5′-UTR at position −1 to −8 as the extended Kozak sequence and that at −9 to −15 as the upstream region. In the extended Kozak sequence adenine is strongly conserved in five positions, whereas in the upstream region no nucleotide is strongly conserved. However, adenine is the most frequent at almost every site (see Background).
The extended Kozak sequence
The original CYC1 sequence from positions −15 to −1 is CACACTAAATTAATA (hereafter referred to as k 0). According to Dvir et al. [9], the presence of an adenine at positions −1, −3, and −4, together with the absence of guanine at position −2, should make this leader sequence almost optimal for high expression. However, thymine at position −2 and cytosine at position −13 have a frequency lower than 20 % and 10 %, respectively, among highly expressed S. cerevisiae genes [8]. We built our first synthetic CYC1 leader sequence (k 1) by placing an adenine at each position from −1 to −15.
The fluorescence level associated with k 1 was 6.5 % higher than that measured with k 0. However, no statistically significant difference arose from the data gathered on these two leader sequences (p-value =0.13). We kept k 1 (the optimized leader sequence) as a template for our next synthetic constructs and built 57 more synthetic 5′-UTRs by mutating single or multiple nucleotides in k 1.
The first group of synthetic leader sequences was made by a single point mutation from position −1 to position −8 (see Table 1). Hence, we modified the extended Kozak sequence only, whereas the upstream region was kept in an optimized configuration for high gene expression with adenines at positions −9 to −15.
Table 1.
Synthetic CYC1 5′-UTR terminal sequences from k 1 to k 25
| ID | Mutation at | Sequence |
|---|---|---|
| k 0 | - | CACACTAAATTAATA |
| k 1 | - | AAAAAAAAAAAAAAA |
| k 2 | AAAAAAAAAAAAAAC | |
| k 3 | −1 | AAAAAAAAAAAAAAT |
| k 4 | AAAAAAAAAAAAAAG | |
| k 5 | AAAAAAAAAAAAACA | |
| k 6 | −2 | AAAAAAAAAAAAATA |
| k 7 | AAAAAAAAAAAAAGA | |
| k 8 | AAAAAAAAAAAACAA | |
| k 9 | −3 | AAAAAAAAAAAATAA |
| k 10 | AAAAAAAAAAAAGAA | |
| k 11 | AAAAAAAAAAACAAA | |
| k 12 | −4 | AAAAAAAAAAATAAA |
| k 13 | AAAAAAAAAAAGAAA | |
| k 14 | AAAAAAAAAACAAAA | |
| k 15 | −5 | AAAAAAAAAATAAAA |
| k 16 | AAAAAAAAAAGAAAA | |
| k 17 | AAAAAAAAACAAAAA | |
| k 18 | −6 | AAAAAAAAATAAAAA |
| k 19 | AAAAAAAAAGAAAAA | |
| k 20 | AAAAAAAACAAAAAA | |
| k 21 | −7 | AAAAAAAATAAAAAA |
| k 22 | AAAAAAAAGAAAAAA | |
| k 23 | AAAAAAACAAAAAAA | |
| k 24 | −8 | AAAAAAATAAAAAAA |
| k 25 | AAAAAAAGAAAAAAA |
First column, 5′-UTR identifiers. Second column, position of single point mutations with respect to k 1. Third column, bold font represents mutated nucleotides. k 0 represents the original CYC1 5′-UTR terminal sequence
The highest fluorescence was recorded for k 16 (where a guanine substituted the adenine at position −5) and the lowest by k 9 (where a thymine replaced the adenine at position −3). Moreover, the fluorescence level of k 16 was statistically significantly different from that of k 0 and k 1. An enhancement in fluorescence due to a guanine at position −5 was a surprising result because guanine is the least frequent nucleotide in yeast S. cerevisiae leader sequences. Moreover, no guanine was ever detected at this position among highly expressed genes [8] or provoked any fluorescence enhancement in the work by Dvir et al. [9].
Despite the absence of a statistically significant difference from k 1, the only constructs other than k 16 that resulted in an increase of >5 % on the fluorescence level of k 1 were k 3, k 10, and k 24. In particular, in k 3, a thymine replaced an adenine at position −1, and in k 10 the adenine at position −3 was mutated into a guanine. As reported above, adenine at positions −1 and −3 should guarantee high gene expression. Nevertheless, on such an adenine background, less frequent nucleotides at positions −1 or −3 seem to be required to further enhance gene expression. In contrast, a thymine instead of an adenine at position −3 (k 9) was the only mutation that induced a >5 % reduction in k 1 fluorescence level. This result is consistent with the observation in [9] that a thymine at position −3 is abundant in poorly expressed genes (Fig. 1 a).
Fig. 1.
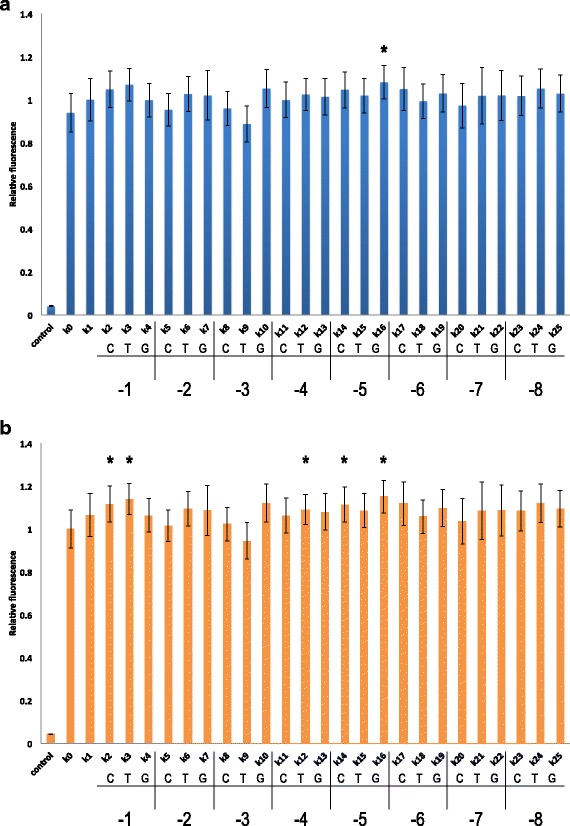
Effect of point mutations in the extended Kozak sequence on fluorescence expression. Fluorescence levels are plotted relative to k 1 (a) and k 0 (b). Control corresponds to a yeast strain without the yEGFP gene. The nucleotide that replaced an adenine in k 1 and the position at which the mutation took place are given below the name of each synthetic leader sequence. Asterisks, p-value <0.05 vs. k 1 (a) or k 0 (b)
With respect to k 0, all 25 new synthetic leader sequences contained between six and eight mutations. Apart from k 9, all synthetic 5′-UTRs showed a fluorescence level higher than that of k 0, five of which were significantly higher. These included positions −1, −4, and −5. As already noted in the comparison with k 1, an adenine just upstream of the START codon seemed to be of no particular advantage for gene expression. Here, a cytosine and a thymine (k 2 and k 3, respectively) performed much better than an adenine. However, with respect to k 0, there were seven more point mutations upstream. At position −4 a thymine (k 12) resulted in the highest fluorescence increment, whereas at position −5, both a cytosine (k 14) and a guanine (k 16) enhanced fluorescence to >10 % above that of k 0. Since k 0 has a thymine at positions −2, −5, and −6, each of the five synthetic 5′-UTRs that showed statistically significant differences from k 0 were affected by a point mutation at two or more adjacent sites. Three more synthetic leader sequences (k 10,k 17, and k 24) caused a >10 % increase in fluorescence compared to k 0, though these differences were not significant (p-value >0.05). k 10 and k 17 also had double point mutations at adjacent sites (Fig. 1 b).
Multiple mutations to guanine
The analysis of our first 25 synthetic 5′-UTR sequences gave the surprising result that a single point mutation to guanine—which is essentially absent from the extended Kozak sequence of highly expressed S. cerevisiae genes—can enhance the fluorescence level of k 1, a leader sequence optimized for gene expression. Moreover, five of our synthetic 5′-UTRs unambiguously (>9 %) increased the fluorescence level associated with pCYC1min.
According to our data, a single mutation to guanine can enhance gene expression. However, two previous papers [18, 19] reported that multiple guanines placed in front of a START codon would considerably reduce protein synthesis. Therefore, we assessed how multiple point mutations to guanine affected the translation efficiency of pCYC1min, to determine if they could be used to modulate gene expression.
According to [8], among highly expressed S. cerevisiae genes, guanine is the least frequent nucleotide between positions −1 and −15, with the exception of position −7, in which the least frequent nucleotide is cytosine. We constructed a synthetic 5′-UTR that reflects this sequence (k 26; Table 2). This shut down gene expression, as shown by the corresponding fluorescence level not being significantly different (p-value =0.21) from our negative control (an S. cerevisiae strain that did not contain the yEGFP gene).
Table 2.
Synthetic CYC1 5′-UTR terminal sequences from k 26 to k 38
| ID | MFE (kcal/mol) | Sequence |
|---|---|---|
| k 0 | -241.21 | CACACTAAATTAATA |
| k 1 | -241.21 | AAAAAAAAAAAAAAA |
| k 26 | −261.39 | GGGGGGGGCGGGGGG |
| k 27 | −247.04 | AAAAAAAGCGGGGGG |
| k 28 | −246.41 | GGGGGGGAAAAAAAA |
| k 29 | −245.97 | AAAAAAAGCGGGGGA |
| k 30 | −244.28 | AAAAAAAGCGGGGAG |
| k 31 | −247.46 | AAAAAAAGCGGGAGG |
| k 32 | -241.21 | GGGGAAAAAAAAAAA |
| k 33 | −241.93 | GAGAGAGAAAAAAAA |
| k 34 | −246.59 | GGGGGGAAAAAAAAA |
| k 35 | −242.5 | GGGGGAAAAAAAAAA |
| k 36 | -241.21 | GGGAAAAAAAAAAAA |
| k 37 | -241.21 | GGAAAAAAAAAAAAA |
| k 38 | -241.21 | GAAAAAAAAAAAAAA |
Nucleotides in bold are mutations with respect to k 1 sequence. k 0 is shown for comparison. MFE, minimum free energy, computed in RNAfold
We tested whether multiple mutations to guanine (cytosine at position −7) would affect gene expression in a different way when they covered either the whole extended Kozak sequence (k 27) or the upstream region (k 28). Since mutations were made with respect to k 1, all the non-mutated sites contained an adenine. Surprisingly, we found that the two configurations were equivalent for gene expression (p-value >0.40) and reduced k 1 fluorescence level by about half.
Starting from k 27, we replaced the guanine at positions −1 (k 29), −2 (k 30), and −3 (k 31) with an adenine to determine whether a single adenine at the three positions just upstream of the START codon would enhance fluorescence expression when the other sites of the extended Kozak sequence were occupied either by a guanine or a cytosine. At position −1 an adenine showed no improvement on the fluorescence of k 27. Interestingly, at positions −2 and −3, an adenine caused a drop in gene expression to approximately 7 % of the k 1 fluorescence level. These results demonstrate that an adenine per se cannot improve gene expression even when it occupies position −3 or −1. More generally, we can conclude that the effect on gene expression of a single point mutation in the leader sequence is strongly context-dependent.
Finally, to understand better how important the upstream region is for gene expression, we progressively reduced the number of guanines from seven (k 28) to one (k 38). Starting from position −9, we replaced a guanine with an adenine at each step and saw that the fluorescence level increased almost linearly with the number of adenines (Fig. 2 and Additional file 1). The last sequence in which the fluorescence level was statistically significantly different from that of k 1 was k 36, in which guanines were present at positions −13 to −15. A guanine alone at position −15 or accompanied by another at position −14 did not result in a significant difference in fluorescence level from that of k 1. Therefore, even in the presence of an extended Kozak sequence optimized for high gene expression, multiple mutations in the upstream region have evident repercussions on protein synthesis and can be used as a means of tuning protein abundance. An explanation for this result is presented in the Computational Analysis section, below. Interestingly, four guanines intermixed with adenines (k 33) in the upstream region reduced k 1 fluorescence to a smaller extent than four guanines in a row (k 32), providing further confirmation that the effect on gene expression of point mutations inside the 5′-UTR is highly dependent on the nucleotidic context (Fig. 2; see Additional file 1 for a comparison with k 0 fluorescence).
Fig. 2.
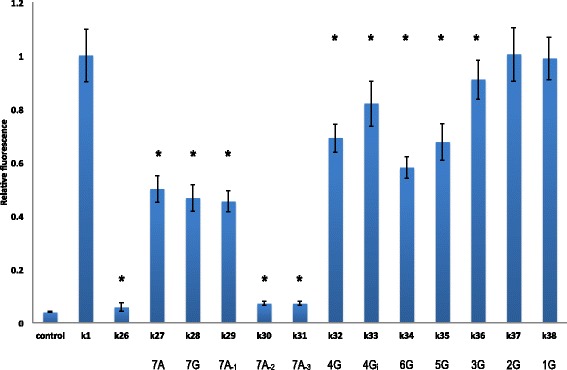
Multiple point mutations to guanine. The ratio between the fluorescence level of the synthetic 5′-UTRs from k 26 to k 38 and that of k 1 are reported. The number of adenines or guanines in the upstream region is given below the leader sequence name (from k 27 to k 38). The subscripts −1, −2, and −3 indicate that an adenine is present in the extended Kozak sequence only at the corresponding position. Subscript i represents intermixed (see main text). Asterisks, p-value <0.05 vs. k 1
The upstream region
The previous analysis confirmed that the effect on gene expression due to both single and multiple mutations within the 5′-UTR is strongly context-dependent. Moreover, our data clearly showed that changes not only in the Kozak sequence but also inside the upstream region markedly affect gene expression. We therefore performed point mutations on k 1 between positions −9 and −15 (Table 3) to assess whether a single nucleotide different from adenine can change the translation rate when placed into the upstream region.
Table 3.
Synthetic CYC1 5′-UTR terminal sequences from k 39 to k 58
| ID | Mutation at | Sequence |
|---|---|---|
| k 0 | - | CACACTAAATTAATA |
| k 1 | - | AAAAAAAAAAAAAAA |
| k 39 | AAAAAACAAAAAAAA | |
| k 40 | −9 | AAAAAATAAAAAAAA |
| k 41 | AAAAAAGAAAAAAAA | |
| k 42 | AAAAACAAAAAAAAA | |
| k 43 | −10 | AAAAATAAAAAAAAA |
| k 44 | AAAAAGAAAAAAAAA | |
| k 45 | AAAACAAAAAAAAAA | |
| k 46 | −11 | AAAATAAAAAAAAAA |
| k 47 | AAAAGAAAAAAAAAA | |
| k 48 | AAACAAAAAAAAAAA | |
| k 49 | −12 | AAATAAAAAAAAAAA |
| k 50 | AAAGAAAAAAAAAAA | |
| k 51 | AACAAAAAAAAAAAA | |
| k 52 | −13 | AATAAAAAAAAAAAA |
| k 53 | AAGAAAAAAAAAAAA | |
| k 54 | ACAAAAAAAAAAAAA | |
| k 55 | −14 | ATAAAAAAAAAAAAA |
| k 56 | AGAAAAAAAAAAAAA | |
| k 57 | CAAAAAAAAAAAAAA | |
| k 58 | −15 | TAAAAAAAAAAAAAA |
| k 38 | GAAAAAAAAAAAAAA |
As in Table 1, the position of single point mutations with respect to k 1 is given in the second column, whereas the mutated nucleotides are written in bold in the third column. Also reported are k 0 and k 38, the latter initially used to study the effect of guanines in the upstream region
All point mutations (except the one in k 38) resulted in a fluorescence level higher than that associated with k 1. Notably, in eight cases, the increase in fluorescence was statistically significant (>10 % higher than k 1 fluorescence). These eight mutations included four contiguous positions, from −11 to −14. None of these were taken into account in the reference work by Dvir et al. [9].
At position −11, a guanine instead of an adenine (k 47) enhanced fluorescence expression by >15 %, whereas cytosine and thymine had no significant effects. Every mutation at position −12 increased the fluorescence of k 1. The greatest change (>15 %) was due to a guanine (k 50). Mutations at position −13 also strongly enhanced k 1 fluorescence level. Two point mutations—cytosine (k 51) and guanine (k 53)—resulted in statistically significant differences in fluorescence from k 1, whereas a thymine (k 52) augmented k 1 fluorescence by about 14 % but this did not reach statistical significance. It should be noted that among all our 58 synthetic 5′-UTRs, k 51 had the highest fluorescence level—almost 17 % higher than that of k 1.
Finally, two different point mutations at position −14 led to an increase in fluorescence: a cytosine (k 54) and a thymine (k 55) (Fig. 3; see Additional file 1 for a comparison with k 0).
Fig. 3.
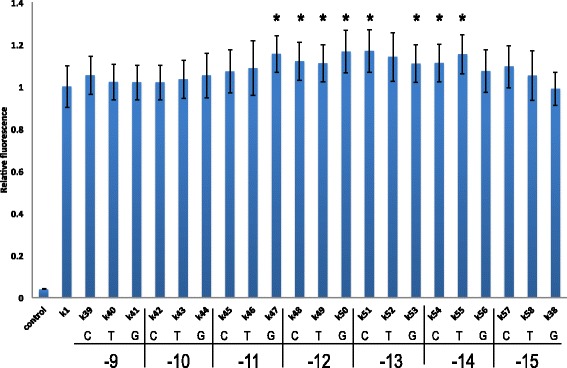
Effect of point mutations in the upstream region on fluorescence relative to k 1. The nucleotide that replaced an adenine in k 1 and the position at which the mutation took place are given below the name of each synthetic leader sequence. Asterisks, p-value <0.05 vs. k 1
Together, the results of this last analysis of the upstream region underline another surprising result: single point mutations upstream of the Kozak sequence, in particular at positions −12 and −13, were those that most enhanced gene expression from a context rich in adenines.
Computational analysis
We carried out simulations with RNAfold to investigate possible correlations between computed mRNA secondary structures, together with their corresponding minimum free energies (MFEs), and measured fluorescence levels. Our analysis provides an explanation for the drop in fluorescence due to multiple mutations from adenine to guanine (and cytosine) in the −15…−1 region. In contrast, no plausible justification for the effects of single point mutations on translational efficiency emerged from simulations with RNAfold.
As an input for RNAfold, we used mRNA sequences starting at the transcription start site of pCYC1min [16] and ending at the poly-A site of the CYC1 terminator [20]. Each sequence was 937 nucleotides long. From preliminary simulations, we observed that a poly-A chain with a variable length of 150–200 nucleotides had no significant effect on mRNA folding. All mRNA secondary structures were calculated at 30 °C (the temperature at which we grew S. cerevisiae cells for the FACS experiments).
k 0 and k 1 have the same MFE: −241.21 kcal/mol. This is the highest—and the most common—within the collection of 59 sequences analyzed in this work (see Additional file 1). The mRNA secondary structure corresponding to this MFE is characterized by the presence of a giant hairpin between positions −40 and +10. The hairpin loop goes from position −31 to position +1 and contains the whole 5′-UTR portion we have targeted here. The hairpin stem is made of nine base-pairs, of which only one gave a “mismatch” because of an adenine at position −38 and +8 (see Fig. 4 a).
Fig. 4.
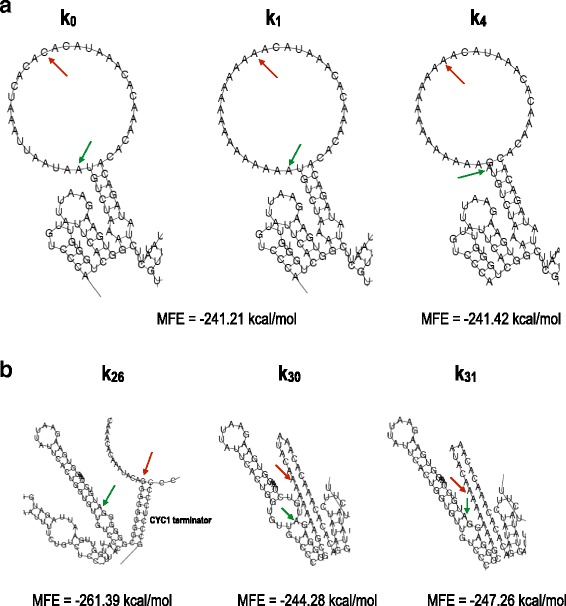
mRNA secondary structures. a A giant hairpin is present in the mRNA secondary structure corresponding to the MFE of both k 0 and k 1. The hairpin loop contains the −15…−1 region. The portion of the 5′-UTR in our analysis is free from any pairing interactions in its wild-type configuration (k 0) and in that theoretically optimized for high protein expression (k 1). The loop of the giant hairpin is reduced in k 4 owing to the base-pairing interaction between the guanine at position −1 and the cytosine at position −31. In every mRNA structure presented, a green arrow indicates position +1, and a red arrow indicates position −15. b The disruption of the giant hairpin induces a decrease in the MFE of the mRNA secondary structure. k 26 and k 31 are associated with the lowest MFEs computed in our analysis. The two sequences contain multiple guanines in the extended Kozak sequence involved in pairing interactions with the CDS. A similar pattern is also present in k 30. Here, however, a second mini-loop around the START codon provokes an increase in MFE. The MFE of k 26 is substantially lower than those of k 30 and k 31 because of the presence of another stem due to pairing interactions between the upstream region and the CYC1 terminator. Nevertheless, the fluorescence levels of k 30 and k 31 are only approximately 1.2-fold higher than that of k 26
Multiple mutations to guanines either in the upstream region or the extended Kozak sequence originate base-pairing interactions between, at least, a portion of the −15…−1 region and the CDS (yEGFP) or the CYC1 terminator. As a consequence, the giant hairpin is destroyed and replaced by one or two stems that lower the MFE of the mRNA secondary structure (Table 2). Most of the MFE values smaller than −241.21 kcal/mol were associated with fluorescence levels lower than that of k 1 (Fig. 5). This result is in agreement with the notion, supported also by [8, 9], that stable mRNA secondary structures in the 5′-UTR reduce protein expression. However, the fluorescence levels we measured did not increase proportionally to increments in the MFE. Moreover, in two cases (k 32 and k 36) RNAfold predicted a giant hairpin in the mRNA structure, whereas the fluorescence levels from our experiments were significantly lower than that of k 1 (Fig. 5 and Additional file 1).
Fig. 5.
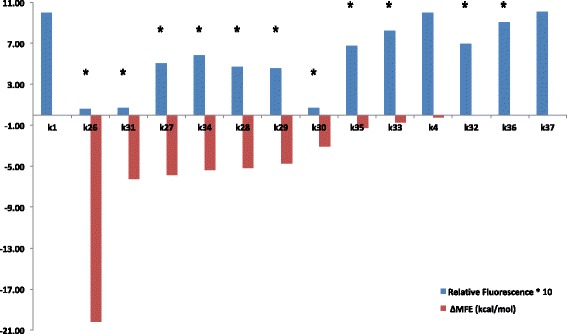
Low MFE values are associated with reduced fluorescence expression. Red bars, difference between MFEs of the corresponding 5′-UTR and k 1 (ΔMFE). Blue bars, 10-fold magnified ratio between the fluorescence level of the indicated 5′-UTR and that of k 1. Apart from k 1, sequences are sorted by increasing ΔMFE. All sequences except k 4 contain multiple point mutations with respect to k 1. Asterisks above blue bars, p-value <0.05 vs. k 1
k 26 was designed by choosing the least frequent nucleotides between positions −15 and −1 among a set of highly expressed S. cerevisiae genes. The corresponding MFE (−261.39 kcal/mol) was the lowest within the ensemble of transcription units considered in this work. No giant hairpin was present in the MFE mRNA secondary structure as the −15…−1 region was sequestered into two different stems. The guanines between positions −1 and −6 were part of a long stem and paired with a hexamer at the beginning of the yEGFP sequence (positions +33 to +38). In contrast, positions −9 to −15 paired with a region of the CYC1 terminator, at positions +750 to +758 (Fig. 4 b).
A fluorescence level just above that of k 26 was registered for k 30 and k 31. Both differed from k 26 for the upstream region (made of seven adenines) and the presence of an adenine in the extended Kozak region (at positions −2 and −3, respectively). Similarly to k 26, the first five nucleotides of the extended Kozak region of k 30 and the first six of k 31 were sequestered into a stem with the CDS. However, differently from k 26, the upstream regions of k 30 and k 31 were entirely free from any pairing interactions (see Fig. 4 b). Their MFEs (−244.28 and −247.26 kcal/mol, respectively) were also significantly higher than that of k 26. These three sequences suggest that a condition for markedly lowering protein expression is to enclose the nucleotides at positions −1 to −5 in an mRNA secondary structure. Moreover, not all of these nucleotides have to participate in base-pairing interactions. Indeed, a guanine at position −1 (k 30) or −2 (k 26 and k 31) is “free” and responsible for the presence of a mini-loop in the mRNA structure.
However, this hypothesis is contradicted by k 29. The MFE of this sequence (−245.97 kcal/mol) is comparable to that of k 30 and k 31, and the corresponding mRNA secondary structure is very similar to that of k 31 (Fig. 6 a). Nevertheless, the fluorescence level associated with k 29 was more than 6-fold higher than that of k 31 and amounted to 45% of that of k 1.
Fig. 6.
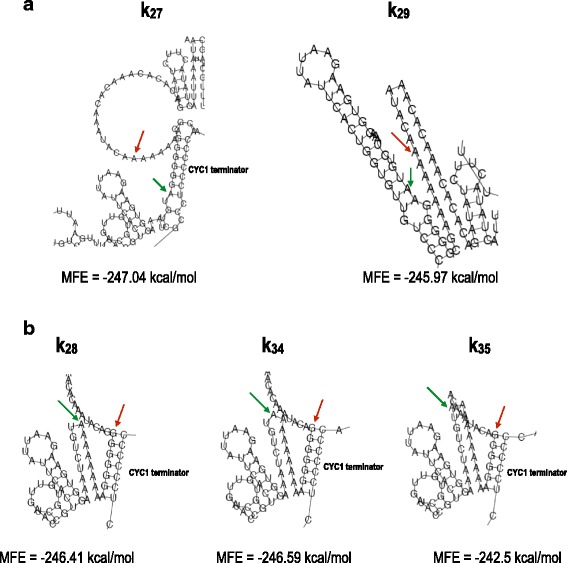
mRNA secondary structures. a k 27 differs from k 29 only by a guanine instead of an adenine at position −1. However, their mRNA secondary structures are dissimilar. In k 27, the extended Kozak sequence is involved in base-pairing interactions with the CYC1 terminator, whereas in k 29 the extended Kozak sequence is locked into a stem with the CDS. The MFE associated with k 27 is lower than that of k 29, but there is no difference between the fluorescence levels of the two sequences (p-value =0.20). b Multiple guanines in the upstream region give rise to mRNA structures characterized by base-pairing interactions between the 5′-UTR and the CYC1 terminator. k 28 and k 34 have six guanines in a stem with the CYC1 terminator, whereas k 35 has only 5 guanines in an analogous structure. This causes an increase in MFE and consequently a higher fluorescence
k 27 shared with k 29– k 31 an upstream region made only of adenines. However, unlike in these three sequences, the extended Kozak sequence of k 27 did not contain any adenine. The MFE of k 27 (−247.04 kcal/mol) was comparable to that of k 29– k 31, but its corresponding mRNA secondary structure had a different configuration. Indeed, all nucleotides of the extended Kozak sequence (with the exception of the cytosine at position −7) were involved in base-pairing interaction not with the CDS but with the CYC1 terminator (positions +755 to +762; Fig. 6 a). The fluorescence level of k 27 was slightly higher than that of k 29, i.e. almost 7-fold greater than that of k 31.
The five sequences considered so far (k 26, k 27, k 29– k 31) have in common an extended Kozak region rich in guanine that was sequestered into a stem in the MFE mRNA secondary structure. In four cases, the extended Kozak sequence paired (partially) with the CDS, and in one case (k 27) with the CYC1 terminator. The MFE of k 26 was the lowest, as its upstream region was also sequestered into a stem. The other four sequences showed very similar MFE values but rather different fluorescence levels.
The other group of sequences affected by multiple mutations with respect to k 1 had only adenines in the extended Kozak sequence and a variable number of guanines in the upstream region.
k 28, k 34, and k 35 had, respectively, 7, 6, and 5 guanines in a row from position −15 downstream. Although the MFE of k 35 was clearly higher than that of k 28 and k 34 (Table 2), the three sequences gave rise to similar mRNA structures where at least five guanines of the upstream region (plus the first adenine downstream) were locked into a stem due to base-pairing interactions with the CYC1 terminator (see Fig. 6 b).
Interestingly, both the MFE and fluorescence level of k 28 were comparable to those of k 27 and k 29. Hence, even if the Kozak sequence was free of pairing interactions, the sequestering of the upstream region into a stem was enough to guarantee a clear drop in protein expression. This is further confirmation of the role played by the nucleotides upstream of the Kozak sequence in tuning protein expression.
A different MFE mRNA secondary structure was obtained for k 33 (four guanines, intermixed with adenines), in which half of the extended Kozak sequence and almost the whole upstream region were involved in base-pairing interactions with the CDS, giving rise to a long stem. However, compared to k 35, where only five nucleotides of the upstream region were locked into a stem with the CYC1 terminator, k 33 showed a higher MFE as well as a higher fluorescence level (Fig. 5 and Additional file 1).
Finally, for k 32, k 36, and k 37 (with four, three, and two guanines in the upstream region, respectively) RNAfold returned the same MFE as for k 1. The corresponding mRNA secondary structures were all characterized by the presence of the the giant hairpin (see Additional file 1). Compared to our experimental data, this result was plausible only for k 37 but in apparent disagreement with the measurements for k 32 and k 36, whose fluorescence levels were significantly lower than that of k 1 (Fig. 5). In particular, the fluorescence of k 32 only corresponded to about 69% of that of k 1. Therefore, it can be argued that in vivo k 32 and k 1 share the same MFE and mRNA secondary structure, as suggested by the in silico simulations.
In contrast to the multiple point mutations, of the single point mutations on k 1, only k 4 caused a modification in the structure of the giant hairpin and a consequent decrease in the MFE. k 4 carries a guanine at position −1 that pairs with the cytosine at position −31 such that the length of the loop is reduced from 32 to 29 nucleotides and the MFE is lowered to −241.42 kcal/mol (Fig. 4 a). According to our data, this minimal change has no effect on fluorescence expression. All the other point mutations that induced a fluorescence level significantly higher than that of k 1 (namely, k 16, k 47– k 51, and k 53– k 55) were characterized by the same MFE and corresponding mRNA secondary structure as k 1, according to the RNAfold simulations.
Conclusions
To date, the role played by 5′-UTR in eukaryotic cell transcription initiation has not been studied deeply nor clearly understood. As for S. cerevisiae, two main works in the literature [8, 9] showed that the 5′-UTR is rich in adenine and poor in guanine, mainly in the proximity of the START codon. The consensus Kozak sequence, determined on highly expressed genes, shows a strongly conserved adenine (frequency >50 %) at positions −1, −3, and −5, whereas cytosines at positions −2 and −4 and a thymine at position −6 are also strongly present. Adenines at positions −3 and −1 are suggested to be necessary for enhancing gene expression. In contrast, two other works [18, 19] showed that several adjacent guanines placed just upstream of the START codon induced a significant drop in protein synthesis.
We analyzed the 5′-UTR of the constitutive S. cerevisiae CYC1 promoter. We fused pCYC1min to a reporter protein and quantified the strength of 58 synthetic leader sequences using fluorescence measurements. We took into account only the 15 nucleotides just upstream of the START codon. In a previous report, Dvir et al. [9] examined positions −1 to −10 of a different S. cerevisiae gene, RPL8A.
Our starting point was the construction of a synthetic 5′-UTR where each position from −1 to −15 was taken by an adenine—the nucleotide that seems the most favorable for high gene expression. We called this sequence k 1. With respect to the original CYC1 5′-UTR (here termed k 0), k 1 did not represent a significant improvement. In contrast, a statistically significant enhancement in fluorescence was achieved by single point mutations in k 1. Surprisingly, a cytosine and a thymine in position −1 proved to be more efficient than an adenine, which disagrees with previous claims that an adenine at position −1 is on its own sufficient for enhancing gene expression. Furthermore, point mutations at positions −4 and −5 also showed a statistically significant improvement with respect to the fluorescence of k 0. In particular, a guanine (the least frequent nucleotide in S. cerevisiae leader sequences) at position −5 resulted in a statistically significant increase in fluorescence, even compared to k 1. Hence, single point mutations on an adenine background seem to constitute a novel technique for improving translational strength.
We also studied the effects that multiple mutations to guanine can have on gene expression. Here, the starting point was a leader sequence containing the least frequent nucleotides in highly expressed S. cerevisiae genes [8] at positions −1 to −15. They were all guanines apart from a cytosine at position −7. This sequence, here termed k 26, switched off fluorescence expression. By mutating only the extended Kozak sequence (positions −1 to −8) or the upstream region (positions −9 to −15), we obtained almost identical results, namely about half of the fluorescence expressed by k 1. This was the first hint that mutations in the upstream region (or, more generally, outside the Kozak sequence) can markedly affect gene expression. Furthermore, we showed that gene expression can be tuned just by varying the number of adjacent guanines in the upstream region while keeping the extended Kozak sequence made of adenines only. This represents another possible approach to engineering synthetic promoters that differ in their translational strength. We also noticed that four guanines intermixed with adenines reduce translation initiation less than four guanines in a row. This confirms that the effect of point mutations (multiple or single) on gene expression is highly context-dependent.
Simulations with RNAfold led to an explanation for the changes in fluorescence expression observed in the presence of multiple mutations from adenine to guanine (and cytosine at position −7). The k 1 mRNA sequence folds into an MFE secondary structure characterized by a giant hairpin whose loop contains the entire −15…−1 region. Therefore, in this configuration, both the extended Kozak sequence and the upstream region are free from base-pairing interactions. Moreover, this mRNA secondary structure returns the highest MFE among the 59 sequences analyzed in this work and seems to foster protein synthesis. Multiple mutations to guanine (and cytosine) cause either the extended Kozak sequence or the upstream region to be locked into a stem due to base-pairing interactions with the CDS or the CYC1 terminator. As a result, the giant hairpin is destroyed, the MFE is lowered, and fluorescence expression is decreased to different extents. Although it was not possible to identify the precise relationship between MFEs and fluorescence levels, our results mostly agree with the notion that stable mRNA structures in the 5′-UTR hinder protein expression.
Finally, we carried out single point mutations on the sole upstream region of k 1 and determined whether positions “far” from the START codon played an important role in protein synthesis. We found that eight sequences, with a single point mutation between positions −11 and −14, produced significant increases in k 1 fluorescence level. Position −12 turned out to be the most critical, because each mutation led to significantly greater fluorescence. Moreover, the highest increase in fluorescence—from all 58 synthetic 5′-UTRs—was obtained by turning the adenine at position −13 into a cytosine. Therefore, the leader configuration upstream of the Kozak sequence has a marked influence on translation initiation.
As in the reference work by Dvir et al. [9], the main limit in our analysis is that it was carried out on a single gene. Hence, the results shown here might be not completely valid for other S. cerevisiae genes. On the whole, our work shows that 5′-UTRs can be exploited to generate new libraries of synthetic S. cerevisiae promoters. In particular, mutations on the 5′-UTR should not be limited to the Kozak sequence but they should also involve upstream non-conserved regions. As in the CYC1 gene, these modifications might lead to very high increases (or decreases) in translation initiation. However, further studies investigating entire 5′-UTRs are advisable. Our simulations with RNAfold showed how, at the mRNA level, interactions between the promoter (5′-UTR) and either the CDS or the terminator (3’-UTR) sequence influence gene expression considerably. This notion should be taken into account to improve the modelling of basic modules for eukaryotic gene circuits (as we previously described in [21]). Our hope is that this work will emphasize the fact that part characterization—a fundamental concept in Synthetic Biology—is still far from being achieved and more basic experiments on standard biological parts (and subparts) are required for a comprehensive description of the basic components of synthetic gene circuits. Only with such accurate knowledge could Synthetic Biology be regarded as a proper engineering discipline.
Methods
Plasmid construction
Backbones for all the plasmids used in this work were either the yeast integrative shuttle-vector pRSII406 (Addgene-35442, a gift from Steven Haase) [22] or the modified version pMM125, where the BsaI site in the ampicillin resistance gene and the BpiI site in the URA3 marker were removed via silent mutations. The minimal CYC1 promoter (pCYC1min) was extracted from the yeast S. cerevisiae genome (strain FY1679-08A, see below) following the procedure described in [23].
Every transcription unit expresses the yeast enhanced green fluorescent protein (yEGFP) obtained from pRS31-glag [24] (courtesy of the Hasty lab, University of California, San Diego, USA). A slightly different version termed yEGFPgg (where the BsaI site was removed through a silent mutation) was used in the plasmids assembled using the Golden Gate method [25]. The CYC1 terminator (CYC1t) [26] placed at the end of every transcription unit was obtained from pRS403-pGAL1-strongSC_GFP (Addgene-22316, a gift from David Bartel).
Plasmids containing 5′-UTR from k 0 to k 38 were constructed via isothermal assembly [27]. Those containing 5′-UTR from k 39 to k 58 were assembled using the Golden Gate method [25]. For this purpose, we built an acceptor vector (pMM247) where 15-nucleotide-long sequences could be inserted between two BsaI cutting sites, namely TACA (i.e. position −19…−16 of pCYC1min) and ATGT (the first four nucleotides of yEGFPgg). Short DNA sequences containing k 39 to k 58 extended with the above BsaI sites were prepared by Comate Bioscience Co., Ltd. (Harbin, China).
To extract DNA sequences from plasmids, we used touchdown PCR. DNA elution from agarose gel was carried out with the QIAGEN-28604 DNA Elution Kit. Isothermal assembly required 1 h at 50 °C. For Golden Gate assembly, the insert (k i sequences, i:39,…,58) and the pMM247 acceptor vector were combined in an equimolar ratio and mixed with a master mix (1 μ l BsaI 20 units /μ l, NEB-R0535S; 2 μ l Cutsmart buffer NEB; 1 μ l T4 ligase 400 units /μ l, NEB-M0202S; 2 μ l10 mM ATP, Sigma-Aldrich-A7699) to a final 15 μ l volume. The thermocycler program was set to three cycles of 10 min at 40 °C and another 10 min at 16 °C. These cycles were followed by 10 min at 50 °C, 20 min at at 80 °C, and the final temperature was set to 16 °C.
E. coli competent cells (strain DH5 α, Life Technology, 18263-012) transformed with our plasmids (30-s heatshock at 42 °C) were grown overnight at 37 °C in LB (Luria-Bertani) broth or plates (Bacto tryptone 10%, yeast extract 5%, NaCl 10%, agar 15% for the plates) supplied with ampicillin. Plasmid extraction from bacterial cells was carried out using standard methods [28]. All plasmids were sequenced (Sanger method) to check the correctness of the new synthetic constructs.
Yeast strain construction
Our synthetic plasmids were integrated into the genome of the yeast S. cerevisiae strain FY1679-08A (MATa; ura3-52; leu2 Δ1; trp1 Δ63; his3 Δ200; GAL2), Euroscarf (Johann Wolfgang Goethe University, Frankfurt, Germany). Genomic integration was carried out as described in [29]. About 5 μ g of plasmidic DNA was linearized at the URA3 marker with the restriction enzyme StuI (NEB-R0187S). Transformed cells were grown on plates containing synthetic selective medium (SD-URA; 2% glucose, 2% agar) for about 36 h at 30 °C.
Flow cytometry
Yeast cells were grown overnight in synthetic complete medium (SDC) at 30 °C. They were diluted in the morning to approximately 1:100 and allowed to grow in SDC for up to 5 h longer, so that their optical density at 600 nm wavelength (OD 600) was always between 0.2 and 2.0 (exponential phase). Fluorescence measurements were performed with a BD Accuri C6 cytometer (488 nm laser, 533/30 filter). The FACS machine set-up was checked at the beginning and end of each experiment, using fluorescent beads (AlignFlow, Life Technologies-A16500). Reliable measurements were considered as only those where the relative difference between the initial and final values of the peaks of the beads was lower than 5%. Data were analyzed with the flowcore R-Bioconductor package [30]. Fluorescence levels were compared using Welch’s two-sided t-test (p-value <0.05 was considered significant) and were estimated as the mean values of at least three independent experiments (carried out on different days and each with 30000 recorded samples). Standard deviations were calculated on these mean values. The error on relative fluorescence values (ratios) was computed using the error propagation formula.
Additional file
Supplementary Material. (PDF 474 kb)
Acknowledgements
We thank all the students of the Synthetic Biology laboratory, Harbin Institute of Technology, China, for their kind help.
Funding
This study was supported by the National Natural Science Foundation of China (project number 31571373).
Availability of data and materials
A complete list of the plasmids and yeast strains we constructed, together with the DNA sequences of each genetic part (promoter, CDS, and terminator) used in this work, as well as further experimental data, are provided as Additional file 1.
Authors’ contributions
Study conception: MAM. Plasmid construction and integration into yeast cells: JL, QL, and WS. FACS experiments and data analysis: JL and MAM. Manuscript writing: MAM. All authors read and approved the final manuscript.
Competing interests
The authors declare they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- 5’-UTR
5’untranslated region
- CDS
Coding sequence
- CYC1t
CYC1 terminator
- FACS
Fluorescence activated cell sorting
- LB
Luria-Bertani (medium)
- MFE
Minimum free energy
- pCYC1min
Minimal CYC1 promoter
- RBS
Ribosome binding site
- SDC
Synthetic complete medium
- yEGFP
Yeast-enhanced green fluorescent protein
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13036-017-0068-1) contains supplementary material, which is available to authorized users.
References
- 1.Endy D. Foundations for engineering biology. Nature. 2005;438(7067):449–53. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- 2.Carothers JM, Goler JA, Juminaga D, Keasling JD. Model-driven engineering of RNA devices to quantitatively program gene expression. Science. 2011;334(6063):1716–9. doi: 10.1126/science.1212209. [DOI] [PubMed] [Google Scholar]
- 3.Kozak M. Point mutations close to the AUG initiator codon affect the efficiency of translation of rat preproinsulin in vivo. Nature. 1984;308(5956):241–6. doi: 10.1038/308241a0. [DOI] [PubMed] [Google Scholar]
- 4.Tuller T, Ruppin E, Kupiec M. Properties of untranslated regions of the S. cerevisiae genome. BMC Genomics. 2009;10(1):391. doi: 10.1186/1471-2164-10-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lubliner S, Keren L, Segal E. Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res. 2013;41(11):5569–81. doi: 10.1093/nar/gkt256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44(2):283–92. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 7.Cavener DR. Comparison of the consensus sequence flanking translational start sites in Drosophila and vertebrates. Nucleic Acids Res. 1987;15(4):1353–61. doi: 10.1093/nar/15.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamilton R, Watanabe CK, de Boer HA. Compilation and comparison of the sequence context around the AUG startcodons in Saccharomyces cerevisiae mRNAs. Nucleic Acids Res. 1987;15(8):3581–93. doi: 10.1093/nar/15.8.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dvir S, Velten L, Sharon E, Zeevi D, Carey LB, Weinberger A, Segal E. Deciphering the rules by which 5’-UTR sequences affect protein expression in yeast. Proc Natl Acad Sci U S A. 2013;110(30):2792–801. doi: 10.1073/pnas.1222534110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zur H, Tuller T. Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep. 2012;13(3):272–7. doi: 10.1038/embor.2011.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park C, Chen X, Yang JR, Zhang J. Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2013;110(8):678–86. doi: 10.1073/pnas.1218066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mao Y, Liu H, Liu Y, Tao S. Deciphering the rules by which dynamics of mRNA secondary structure affect translation efficiency in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42(8):4813–22. doi: 10.1093/nar/gku159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol AMB. 2011;6(1):26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9(1):133–48. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guarente L, Lalonde B, Gifford P, Alani E. Distinctly regulated tandem upstream activation sites mediate catabolite repression of the CYC1 gene of S. cerevisiae. Cell. 1984;36(2):503–11. doi: 10.1016/0092-8674(84)90243-5. [DOI] [PubMed] [Google Scholar]
- 16.Hahn S, Hoar ET, Guarente L. Each of three "TATA elements" specifies a subset of the transcription initiation sites at the CYC-1 promoter of Saccharomyces cerevisiae. Proc Natl Acad Sci. 1985;82(24):8562–6. doi: 10.1073/pnas.82.24.8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheff MA, Thorn KS. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast. 2004;21(8):661–70. doi: 10.1002/yea.1130. [DOI] [PubMed] [Google Scholar]
- 18.Valenzuela P, Gray P, Quiroga M, Zaldivar J, Goodman HM, Rutter WJ. Nucleotide sequence of the gene coding for the major protein of hepatitis B virus surface antigen. Nature. 1979;280(5725):815–9. doi: 10.1038/280815a0. [DOI] [PubMed] [Google Scholar]
- 19.Kniskern PJ, Hagopian A, Montgomery DL, Burke P, Dunn NR, Hofmann KJ, Miller WJ, Ellis RW. Unusually high-level expression of a foreign gene (hepatitis B virus core antigen) in Saccharomyces cerevisiae. Gene. 1986;46(1):135–41. doi: 10.1016/0378-1119(86)90177-0. [DOI] [PubMed] [Google Scholar]
- 20.Guo Z, Sherman F. 3’-end-forming signals of yeast mRNA. Trends Biochem Sci. 1996;21(12):477–81. doi: 10.1016/S0968-0004(96)10057-8. [DOI] [PubMed] [Google Scholar]
- 21.Marchisio MA, Colaiacovo M, Whitehead E, Stelling J. Modular, rule-based modeling for the design of eukaryotic synthetic gene circuits. BMC Syst Biol. 2013;7(1):42. doi: 10.1186/1752-0509-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chee MK, Haase SB. New and Redesigned pRS Plasmid Shuttle Vectors for Genetic Manipulation of Saccharomycescerevisiae. G3, Genes|Genomes|Genetics. 2012;2(5):515–26. doi: 10.1534/g3.111.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Looke M, Kristjuhan K, Kristjuhan A. Extraction of genomic DNA from yeasts for PCR-based applications. BioTechniques. 2011;50(5):325–8. doi: 10.2144/000113672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grilly C, Stricker J, Pang WL, Bennett MR, Hasty J. A synthetic gene network for tuning protein degradation in Saccharomyces cerevisiae. Mol Syst Biol. 2007;3:127. doi: 10.1038/msb4100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE. 2008;3(11):3647. doi: 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mumberg D, Müller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156(1):119–22. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 27.Gibson DG. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides. Nucleic Acids Res. 2009;37(20):6984–90. doi: 10.1093/nar/gkp687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green MR, Sambrook J, editors. Molecular Cloning. New York: Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- 29.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/S0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 30.Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, Spidlen J, Strain E, Gentleman R. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinforma. 2009;10(1):106–8. doi: 10.1186/1471-2105-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material. (PDF 474 kb)
Data Availability Statement
A complete list of the plasmids and yeast strains we constructed, together with the DNA sequences of each genetic part (promoter, CDS, and terminator) used in this work, as well as further experimental data, are provided as Additional file 1.