

Abstract
Netrin-1 receptors UNC5H (UNC5H1–4) were originally proposed to mediate the chemorepulsive activity of netrin-1 during axonal guidance processes. However, UNC5H receptors were more recently described as dependence receptors and, as such, able to trigger apoptosis in the absence of netrin-1. They were also proposed as putative tumor suppressors. Here, we show that UNC5H2 physically interacts with the serine/threonine kinase death-associated protein kinase (DAP-kinase) both in cell culture and in embryonic mouse brains. This interaction occurs in part through the respective death domains of UNC5H2 and DAP-kinase. Moreover, part of UNC5H2 proapoptotic activity occurs through this interaction because UNC5H2-induced cell death is partly impaired in the presence of dominant-negative mutants of DAP-kinase or in DAP-kinase mutant murine embryonic fibroblast cells. In the absence of netrin-1, UNC5H2 reduces DAP-kinase autophosphorylation on Ser308 and increases the catalytic activity of the kinase while netrin-1 blocks UNC5H2-dependent DAP-kinase activation. Thus, the pair netrin-1/UNC5H2 may regulate cell fate by controlling the proapoptotic kinase activity of DAP-kinase.
Keywords: apoptosis, DAPK, dependence receptor, netrin-1, UNC5H2
Introduction
Netrin-1, a diffusible laminin-related protein, is a bifunctional molecule (Serafini et al, 1994). It plays a major role during nervous system development by mediating chemoattraction and chemorepulsion of axons/neurons by interacting with its main receptors DCC (Deleted in Colorectal Cancer) and UNC5H (Keino-Masu et al, 1996; Serafini et al, 1996; Leonardo et al, 1997). However, netrin-1 was also more recently described as a survival factor. Indeed, both DCC and UNC5H receptors belong to the so-called dependence receptors family (Mehlen et al, 1998; Llambi et al, 2001). Such receptors, which also include p75NTR, RET, Patched, β-integrin and neogenin, share the functional property of inducing cell death when unbound by ligands (for a review, see Mehlen and Bredesen, 2004). Such receptors induce a cellular state of cell dependence toward ligand availability. Thus, DCC or UNC5H expression leads to apoptosis unless netrin-1 is present.
While DCC was intensively studied because it was initially proposed as a tumor suppressor (Fearon et al, 1990), the interest in UNC5H receptors emerged only recently (Ackerman et al, 1997; Leonardo et al, 1997). UNC5H receptors, which include four members in mammals (UNC5H1–4), are type I transmembrane receptors. While very little is known about UNC5H4, UNC5H1–3 were shown to induce apoptosis via (i) a cleavage of these receptors by caspase (Llambi et al, 2001; Tanikawa et al, 2003) and (ii) intracellular domains required for cell death, that is, a ZU domain (Williams et al, 2003) and a death domain (Llambi et al, 2001). Interestingly, expression of UNC5H1–3 was shown to be downregulated in various cancers, suggesting that UNC5H receptors may be tumor suppressors that would limit tumor growth by inducing apoptosis outside of ligand availability (Thiebault et al, 2003). Moreover, it was shown that UNC5H2 gene is a direct transcriptional target for the tumor suppressor p53 and mediates, in the conditions tested, p53-induced cell death unless netrin-1 is present (Tanikawa et al, 2003).
It is however unclear how UNC5H2 receptor mediates apoptosis. Interestingly, UNC5H proteins display a death domain in their C-terminal extremities related to the death domain of receptors like Fas, TNFr or p75NTR, and the deletion of the death domain of UNC5H2 has been shown to abrogate UNC5H2 proapoptotic activity (Llambi et al, 2001). Sequence analysis reveals that the protein displaying the closest death domain to UNC5H2 death domain is the death-associated protein kinase (DAP-kinase; Deiss et al, 1995; Feinstein et al, 1995) (Figure 1A), that is, 49.4% homology between the two death domains. DAP-kinase is a crucial intracellular protein that mediates cell death induction through a wide spectrum of apoptotic signals via its serine threonine kinase activity (Deiss et al, 1995; Cohen et al, 1999; Jang et al, 2002; Wang et al, 2002). However, the only known signals directly upstream of DAP-kinase that would drive its kinase activity are the calmodulin (CaM) binding through its CaM regulatory region triggered by a calcium spike (Cohen et al, 1997) and an autoinhibitory phosphorylation in this CaM regulatory region, which controls the affinity binding to CaM (Shohat et al, 2001). Because the death domain of DAP-kinase is known to be required for DAP-kinase-induced cell death (Cohen et al, 1999), we investigated the possible link between the transmembrane receptor UNC5H2 that triggers cell death and this cytoplasmic effector for cell death induction. We show here that DAP-kinase is required for a large part of UNC5H2-induced cell death. We also show that the UNC5H2 death domain interacts with the DAP-kinase and regulates the kinase activity of DAP-kinase. We also provide evidence that netrin-1 by interacting with UNC5H2 inhibits DAP-kinase activity, thus suggesting that the dependence receptor UNC5H2 triggers apoptosis by activating DAP-kinase.
Figure 1.
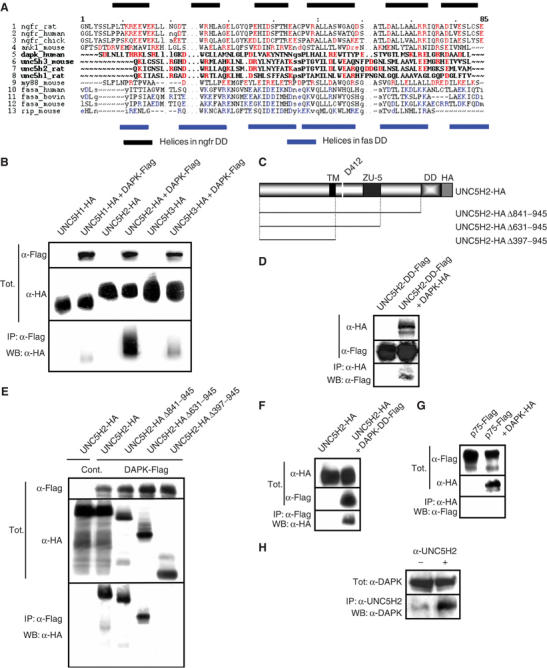
UNC5H2 interacts with DAP-kinase. (A) Sequence alignment of the death domain of different death domain-containing proteins p75NTR (ngfr), Ankyrin-1 (ank1), DAPK, UNC5H, MyD88 (my88), Fas and RIP. Helices in the death domain of p75NTR (ngfr DD) and of Fas (fas DD) are shown. (B) UNC5H2 interacts with DAP-kinase in HEK 293T. Lysates of HEK 293T transiently transfected with HA-tagged UNC5H1-H2 or H3, together with Flag-tagged DAP-kinase, were subjected to Flag (DAP-kinase) pull-down. Tot: Western blot on lysate before pull-down; IP: α-Flag; WB: α-HA—Western blot with anti-HA antibody after pull-down). (C) Schematic representation of UNC5H2 and the various mutants used in this study. TM: transmembrane domain; DD: death domain; D412: caspase cleavage site. (D) UNC5H2 death domain interacts with DAP-kinase in HEK 293T. Similar experiment as in (B) but HA-tagged DAP-kinase was expressed together with the Flag-tagged UNC5H2-DD. Pull-down here was performed with anti-HA and UNC5H2-DD was analyzed by Western blot using an anti-Flag antibody. (E) UNC5H2 interacts with DAP-kinase through multiple domains in HEK 293T. Similar experiment as in (B) using deletion mutants of UNC5H2. (F) Similar as in (D), but HA-tagged UNC5H2 was expressed together with Flag-tagged DAP-kinase DD and pull-down was performed using anti-FlagM2 antibody. (G) Similar as in (D), but Flag-tagged p75ntr was used instead of UNC5H2. (H) Brains from E13 mice embryos were dissected out and resuspended in the lysis buffer described in Materials and methods supplemented with 1% NP-40 as described before (Forcet et al, 2002). Immunoprecipitation was performed using either a UNC5H2 antibody (α-UNC5H2) or a control antibody (−), and DAP-kinase was detected by immunoblot.
Results
The death domain of UNC5H2 interacts with DAP-kinase
We first analyzed whether UNC5H2 interacts with DAP-kinase. Co-immunoprecipitation experiments were performed from human embryonic kidney (HEK) 293T cells coexpressing Flag-tagged DAP-kinase and HA-tagged UNC5H2. As shown in Figure 1B, HA-tagged UNC5H2 is found immunoprecipitated with Flag-tagged DAP-kinase when anti-FlagM2 antibody was used for pull-down. Interestingly, such interaction can barely be detected when UNC5H3 or UNC5H1, the other members of UNC5H family, is used. To analyze whether the death domain of UNC5H2 is sufficient by itself for the interaction with DAP-kinase, we assessed whether the death domain module of UNC5H2—that is, UNC5H2-DD: UNC5H2 death domain only—was able per se to co-immunoprecipitate with DAP-kinase. As shown in Figure 1D, UNC5H2-DD was pulled down by DAP-kinase when a Flag-tagged UNC5H2-DD-expressing construct was coexpressed in HEK 293T cells together with HA-tagged DAP-kinase-expressing construct and when the immunoprecipitation was performed using anti-HA antibody.
To check more precisely whether the death domain of UNC5H2 was the only domain required for interaction with DAP-kinase, we then expressed in HEK 293T cells a series of HA-tagged UNC5H2 mutants truncated at various points within the intracellular domain together with Flag-tagged DAP-kinase. As opposed to our initial expectations, the deletion of the death domain of UNC5H2 was not sufficient to inhibit the interaction between UNC5H2 and DAP-kinase observed after a pull-down performed with anti-FlagM2 antibody (Figure 1E). However, the deletion of the complete intracellular domain of UNC5H2 totally inhibited its interaction with DAP-kinase (Figure 1E). Thus, in addition to the association of DAP-kinase with the death domain of UNC5H2, DAP-kinase indirectly or directly interacts with other domain(s) of UNC5H2. On the other hand, we wondered whether DAP-kinase interacts with UNC5H2 via the death domain. Similarly, while a Flag-tagged DAP-kinase death domain only, that is, DAP-kinase DD, could pull down the HA-tagged UNC5H2 when anti-FlagM2 was used for immunoprecipitation (Figure 1F and not shown), the deletion of the death domain of DAP-kinase was not sufficient to inhibit DAP-kinase/UNC5H2 interactions (data not shown). To discard a nonspecific interaction of DAP-kinase DD with death domain-containing proteins, we assessed whether the closest death domain-containing receptor after UNC5H proteins, that is, p75ntr, interacts with DAP-kinase. HEK 293T cells were then cotransfected with Flag-tagged p75ntr-expressing construct and HA-tagged DAP-kinase-expressing plasmid and HA pull-down was performed. As shown in Figure 1G, p75ntr failed to be pulled down by DAP-kinase, thereby supporting a specific interaction between UNC5H2 and DAP-kinase. Thus, taken together, these data suggest that the UNC5H2/DAP-kinase interaction occurs via their respective death domains and also through other(s) region(s) of both UNC5H2 and DAP-kinase proteins.
To assay whether the interaction between DAP-kinase and UNC5H2 may be detected in vivo with endogenous proteins, we performed immunoprecipitations on brains of mice embryo (E13) in which both UNC5H2 and DAP-kinase are known to be expressed (Leonardo et al, 1997; Llambi et al, 2001) (Figure 1H). As shown in Figure 1H, DAP-kinase was pulled down with an antibody raised against UNC5H2, while a nonrelevant antibody, that is, goat polyclonal antibody, was not able to specifically immunoprecipitate DAP-kinase above the low background levels. Thus, either when ectopically expressed in HEK 293T cells or when endogenously expressed in mice embryos, UNC5H2 interacts with DAP-kinase.
The UNC5H2 proapoptotic activity is mediated by DAP-kinase
We then investigated whether UNC5H2-induced cell death may be mediated by DAP-kinase. In a first attempt to implicate DAP-kinase in UNC5H2-induced cell death, UNC5H2 and DAP-kinase were cotransfected in either HeLa or HEK 293T cells. The presence of DAP-kinase potentiated cell death morphology (i.e., cell detachment, nuclear condensation) in UNC5H2-transfected population, while in the same assay DAP-kinase had no cumulative effect on DCC-induced cell death (Figure 2A and not shown). To further demonstrate the role of DAP-kinase in UNC5H2-mediated cell death, we analyzed whether inhibition of endogenous DAP-kinase activity by dominant-negative mutants may modulate UNC5H2-induced cell death. As previously reported, HEK 293T cells express DAP-kinase endogenously (Cohen et al, 1999). Moreover, while comparing parental HEK 293T and UNC5H2-transfected population, UNC5H2 was detected only in transfected cells (Figure 2B). Along the same lines, the expression levels of UNC5H2 mRNA were estimated by quantitative RT–PCR to be at least 10 times lower than in a normal tissue, for example, in adult human colorectal tissue (not shown), in agreement with the observed loss of UNC5H2 expression in immortalized/transformed cells (Thiebault et al, 2003). In this setting, two different dominant-negative mutants of DAP-kinase, that is, kinase dead mutant (K42A) and the death domain only, were individually coexpressed in HEK 293T cells together with UNC5H2. Cell death was analyzed either by Trypan blue exclusion assay (Figure 2B) or by measurement of caspase activity (not shown). Various plasmid ratios of the UNC5H2-expressing construct and the dominant-negative mutants were used. In most cases, UNC5H2-mediated cell death was inhibited by the presence of these dominant-negative mutants of DAP-kinase (Figure 2B and not shown), while as a negative control, a dominant negative for caspase-8 had no effect (Figure 2B). The 1:2 ratio was found to have the most important inhibitory effect on UNC5H2-induced cell death (Figure 2B). Finally, UNC5H2-induced cell death was analyzed in immortalized murine embryonic fibroblasts (MEFs) derived from either wild-type mouse or DAP-kinase null mouse. UNC5H2- or DCC-expressing construct was transiently transfected in these cells and the UNC5H2- or DCC-transfected cells were concentrated using a microbeads system. While DCC triggered cell death to a similar extent in both parental and DAP-kinase null MEF cells, UNC5H2-induced cell death was reduced in DAP-kinase null MEF cells (Figure 2C).
Figure 2.
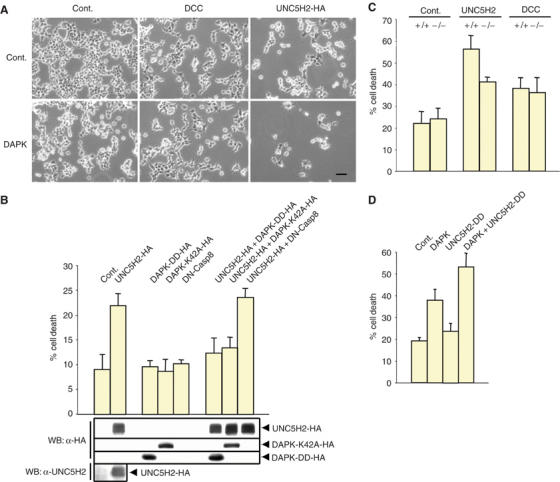
UNC5H2 mediates apoptosis through DAP-kinase. (A) Overexpression of DAP-kinase synergizes UNC5H2-induced cell death. HEK 293T cells were transfected with either UNC5H2- or DCC-expressing construct together or not with DAP-kinase. 48 h after transfection, cell death morphology was examined. Scale bar: 20 μm. (B) Cell death was measured by Trypan blue exclusion assay on HEK 293T cells transfected with UNC5H2 together with dominant-negative mutant of DAP-kinase, that is, the DAP-kinase death domain only, or DAP-kinase K42A, or as a control a dominant-negative mutant of caspase-8. In the assay presented, 0.33 μg of UNC5H2-encoding plasmid versus 0.66 μg of dominant-negative mutant-expressing construct was used for Lipofectamine transfection of 1.5 × 105 cells and the viability of about 900 cells was estimated. Western blots on endogenous UNC5H2 using anti-human UNC5H2 antibody (provided by H Arakawa), HA-tagged UNC5H2 and HA-tagged DAP-kinase mutant using anti-HA antibody were performed. (C) Cell death triggered by DCC or UNC5H2 expression was measured by Trypan blue exclusion assay in MEFs from wild-type mice (+/+) or in MEFs from DAP-kinase knockout mice (−/−). Standard deviations are indicated (n=4). **Student's test, P<0.001. (D) Cell death was analyzed by Trypan blue exclusion assay from HEK 293T cells transfected with DAP-kinase in the presence or not of UNC5H2-DD. Standard deviations are indicated (n=3).
It is however interesting to note that neither the dominant-negative mutants nor the use of DAP-kinase null MEF cells completely abrogated the UNC5H2 proapoptotic activity, hence suggesting that besides DAP-kinase, UNC5H2 may also recruit other effector(s) for cell death induction. However, together, these data support the fact that DAP-kinase is involved specifically in UNC5H2-mediated cell death. To investigate whether the UNC5H2 death domain interaction with DAP-kinase was required for this process, we transiently transfected UNC5H2-DD together with DAP-kinase in HEK 293T cells. The death domain of UNC5H2 was not able to trigger cell death by itself. The expression of DAP-kinase alone triggered cell death to a limited extent (37%), while coexpression of DAP-kinase and the UNC5H2 death domain significantly enhanced 293T cell death (52%) (Figure 2D). Thus, UNC5H2 interacts with DAP-kinase and requires, at least in part, DAP-kinase to behave as a proapoptotic receptor probably because of the interaction between UNC5H2 death domain and DAP-kinase.
UNC5H2 triggers the DAP-kinase catalytic activity
In light of previous studies documenting that DAP-kinase mediates apoptosis through its catalytic activity (Cohen et al, 1997), we further investigated whether its interaction with UNC5H2 may lead to activation of the kinase catalytic activity. DAP-kinase activity has been shown to be tightly controlled by an inhibitory type of autophosphorylation. As previously shown, in the absence of a calcium spike, DAP-kinase is autophosphorylated on Ser308 located in the CaM-binding domain, a process that turns off the kinase activity (Cohen et al, 1997; Shohat et al, 2001). Monoclonal antibodies directed against phospho-Ser308 or immunoprecipitation with a phosphoserine antibody detected this phosphorylation event. HEK 293T cells were then transiently transfected with DAP-kinase- and/or HA-tagged UNC5H2-expressing constructs in the presence or absence of netrin-1. In a first assay, autophosphorylation of DAP-kinase was analyzed by performing an immunoprecipitation with a phosphoserine antibody followed by a DAP-kinase immunoblot (Figure 3A). In a second assay, an immunoblot was directly performed using a monoclonal antibody directed against phospho-Ser308 of DAP-kinase (Figure 3B). In a third assay, this phospho-antibody was used to perform immunostaining (Figure 3C and D). In both cases, it was observed that the expression of UNC5H2 in HEK 293T cells leads to the inhibition of DAP-kinase autophosphorylation on Ser308 (Figure 3A–D). Interestingly, when UNC5H2 is expressed, the presence of netrin-1 completely restored DAP-kinase autophosphorylation. Thus, the pair UNC5H2/netrin-1 regulates autophosphorylation of DAP-kinase.
Figure 3.
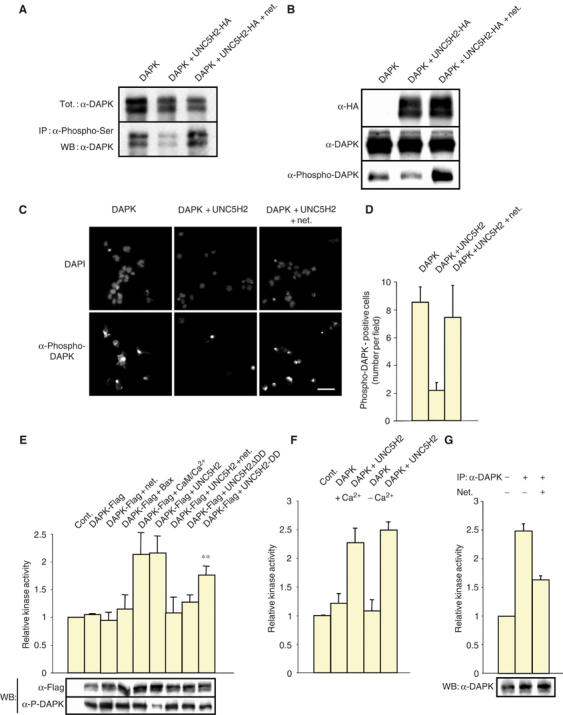
The pair netrin-1/UNC5H2 modulates DAP-kinase activity. (A) DAP-kinase phosphorylation is regulated by the pair UNC5H2/netrin-1. HEK 293T cells were transiently transfected with a control vector or with HA-tagged UNC5H2-expressing construct together with DAP-kinase-expressing construct. Netrin-1 was added 24 h after transfection. Pull-down with a phospho-serine antibody followed by Western blot using anti-DAP-kinase was performed. (B) Similar transfection assay and netrin-1 incubation as in (A) but a series of Western blot was performed using either anti-DAP-kinase, anti-HA (UNC5H2) or anti-phospho-DAP-kinase. (C) Phospho-DAP-kinase immunostaining on HEK 293T transfected with HA-tagged UNC5H2-expressing construct together with DAP-kinase-expressing construct. Representative images are shown. DAPI staining was performed to monitor the total number of cells. Scale bar: 20 μm. (D) Quantitative analysis from (C) was performed by counting the number of cells stained by the phospho-DAP-kinase antibody. About 1000 total cells were counted in an average of 50 fields from five different experiments. (E) DAP-kinase activity is regulated by the pair UNC5H2/netrin-1. Lysates from HEK 293T transfected with Flag-tagged DAP-kinase together with UNC5H2 or Bax, incubated or not with netrin-1 (0.3 μg/ml, 24 h), were subjected to anti-Flag (DAP-kinase) pull-down and assessed for DAP-kinase phosphorylating activity in vitro as described in Materials and methods. To induce calcium-mediated DAP-kinase activation, CaM (1 μM)/Ca2+ (0.5 mM) was added to the kinase assay reaction medium during 10 min. Standard deviations are indicated (n=5). **Student's test, P<0.005. Insets: Control Western blots showing DAP-kinase expression level (α-Flag) and DAP-kinase autophosphorylation (α-P-DAPK). (F) Similar experiment as in (E) but HEK 293T cells were cultured in Ca2+-free medium 24 h after transfection. (G) Semidissociated P5 rat hippocampi were incubated for 1 h with purified netrin and subjected to lysis and DAP-kinase pull-down using the anti-DAP-kinase antibody described in Shani et al (2004). DAP-kinase catalytic activity was then measured as in (E). Standard deviations are indicated (n=4). Inset: Control Western blot showing DAP-kinase expression level.
To demonstrate that the regulation of autophosphorylation by this pair consequently affects DAP-kinase activity, we performed in vitro kinase assays using the DAP-kinase substrate peptide KKRPQRRYSNVF. HEK 293T cells were then transfected with Flag-tagged DAP-kinase and/or UNC5H2-expressing constructs in the presence or absence of netrin-1. Immunoprecipitations were then performed using anti-FlagM2 antibody and the resulting pull-downs were tested for their ability to phosphorylate in vitro the DAP-kinase substrate. It was found that while UNC5H2 increases DAP-kinase activity to a similar extent as a treatment with CaM/calcium, the presence of netrin-1 is sufficient to block this effect (Figure 3E). To further confirm that these general changes observed in DAP-kinase activity were related to a specific effect of UNC5H2, bound or unbound to netrin-1, rather than to an effect of UNC5H2 on cell death that consequently affects DAP-kinase activity, we assessed whether Bax, a general apoptosis inducer, also triggered DAP-kinase activation. As shown in Figure 3E, Bax transfection while affecting caspase activity (data not shown) had no effect on DAP-kinase activity. Thus, the pair UNC5H2/netrin-1 regulates DAP-kinase activity. Moreover, the UNC5H2 death domain that was previously shown to interact with DAP-kinase is required for this activation since transfection of a mutant of UNC5H2 deleted in its death domain instead of full-length UNC5H2 impaired DAP-kinase catalytic activity. To further confirm the role of the UNC5H2 death domain, HEK 293T cells were transfected with UNC5H2-DD instead of full-length UNC5H2. This resulted in a significant increase in DAP-kinase activity, demonstrating that the death domain of UNC5H2 per se activates DAP-kinase.
Because of the calcium-dependent DAP-kinase activity (Cohen et al, 1997), we next wondered whether UNC5H2 modulates DAP-kinase activity through calcium modulation. Similar DAP-kinase catalytic activity assays were then performed from transfected HEK 293T cells cultured in medium devoid of calcium/magnesium. As shown in Figure 3F, no change in UNC5H2-mediated DAP-kinase catalytic activity was observed, hence supporting a direct effect of UNC5H2-DD on DAP-kinase activity.
To further analyze in vivo relevance of this UNC5H2-mediated DAP-kinase activation, we next explored whether in vivo netrin-1 level may affect DAP-kinase catalytic activity. The hippocampus or cerebellum of P5 rat was dissected out, semidissociated and further incubated or not with netrin-1. To first determine whether DAP-kinase catalytic activity can be detected in the hippocampus, pull-downs were performed using either an anti-DAP-kinase antibody or an unrelated antibody, that is, goat polyclonal antibody, and the DAP-kinase catalytic activity within the pull-downs was assessed by measuring KKRPQRRYSNVF phosphorylation. Specific DAP-kinase catalytic activity can be detected in the hippocampus (Figure 3G). A similar experiment was then performed after incubation of semidissociated hippocampus with netrin-1. As shown in Figure 3G, DAP-kinase catalytic activity was significantly reduced, hence strengthening the in vivo relevance of the UNC5H2/DAP-kinase interaction. Interestingly, when a similar experiment was performed in semidissociated cerebellum, DAP-kinase catalytic activity was not modulated by the presence of netrin-1 (not shown), hence suggesting that the effect observed in the hippocampus is a tissue-specific effect.
UNC5H2 cleavage is a prerequisite for its death signaling through DAP-kinase
One common feature of dependence receptors is the fact that most of them have been shown to be cleaved by caspase, a cleavage required, at least in part, for cell death induction by these receptors (Mehlen and Bredesen, 2004). Hence, UNC5H1–3 were shown to be cleaved in vitro, and mutation of the caspase cleavage site of UNC5H2 reduced or turned off UNC5H2 proapoptotic activity (Llambi et al, 2001; Tanikawa et al, 2003). We then investigated whether DAP-kinase autophosphorylation and activity were dependent on UNC5H2 cleavage by measuring DAP-kinase Ser308 phosphorylation and catalytic activity in the presence of UNC5H2 D412N, a mutant of UNC5H2 unable to be cleaved (Llambi et al, 2001), or in the presence of a mutant of UNC5H2 lacking the whole C-terminal domain lying below the caspase cleavage site. HEK 293T cells were then transiently transfected with either wild-type UNC5H2-, UNC5H2 D412N- or UNC5H2 1–412-expressing construct. Autophosphorylation was then monitored by immunoblot using the phospho-specific antibody directed against DAP-kinase Ser308, while DAP-kinase catalytic activity was determined by measuring the ability of DAP-kinase pull-down to phosphorylate the DAP-kinase substrate peptide. As shown in Figure 4, the D412N mutant form was less efficient than the wild-type UNC5H2 in reducing DAP-kinase autophosphorylation (Figure 4A) and in inducing the DAP-kinase phosphorylating activity (Figure 4B). The mutant of UNC5H2 lacking the cleaved fragment was unable to either inhibit DAP-kinase autophosphorylation or to enhance DAP-kinase catalytic activity (Figure 4B and data not shown). It is also interesting to note that the amplitude of reduction in DAP-kinase activity from wild-type UNC5H2 to UNC5H2 D412N was comparable to the reduction of the UNC5H2 proapoptotic activity observed after D412N mutation (Llambi et al, 2001). Thus, UNC5H2-mediated DAP-kinase activation probably requires the preliminary caspase cleavage of UNC5H2.
Figure 4.
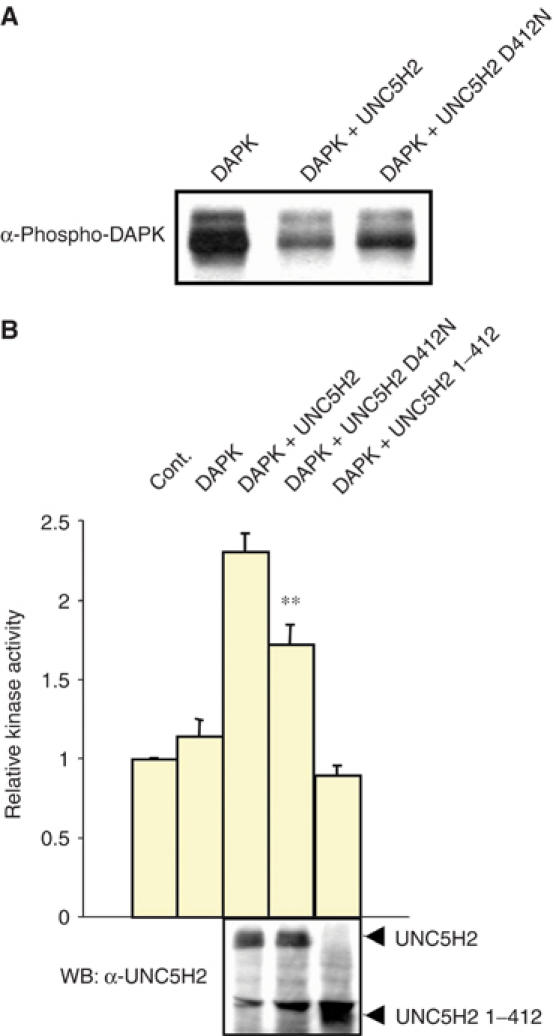
DAP-kinase autophosphorylation and activation by UNC5H2 is dependent on UNC5H2 caspase cleavage. (A) Similar experiment as in Figure 3A but HEK 293T cells were transfected with either full-length UNC5H2 or UNC5H2 mutated in the caspase site (D412N). (B) Similar experiment as in Figure 3E but UNC5H2 D412N or UNC5H2 1–412, that is, UNC5H2 deleted below the caspase cleavage site, was used. Standard deviations are indicated (n=5). **Student's test, P<0.005. Inset: Control Western blot showing expression of UNC5H2 and its related mutants.
Discussion
Dependence receptors can be viewed as membrane sensors for cell fate decision. In the presence of ligand, these receptors mediate signals that drive the cell toward a positive fate, for example, axon/neuronal guidance for UNC5H. In the absence of ligand, these receptors trigger apoptosis. While the mechanisms by which these receptors induce cell death remain mainly unknown, we show here that UNC5H2 may activate a protein kinase that in turn initiates a cell death program. Interestingly, while it has been shown that DAP-kinase can mediate apoptosis induction upon numerous stimuli, this is the first report of (i) the interaction of DAP-kinase with a death-inducing receptor and (ii) the kinase activation of DAP-kinase independently of a spike of calcium. This would argue for an important pathway for cell death induction and would match with (i) the finding that UNC5H and DAP-kinase were both proposed as tumor suppressors that would eliminate unneeded cells (Inbal et al, 1997; Thiebault et al, 2003) and (ii) the observation that DAP-kinase requires functional p53 for cell death induction (Raveh et al, 2001), while UNC5H2 seems to be transcriptionally regulated by p53 (Tanikawa et al, 2003).
The observation that UNC5H2 recruits and activates DAP-kinase raises several questions. The first one resides in the specificity of UNC5H2 to interact with DAP-kinase. Why does UNC5H3 or UNC5H1 fail to interact with DAP-kinase while UNC5H1–3 appear very similar ? A possible explanation may hold in the fact that the death domain of UNC5H3 is less charged than that of UNC5H2. Thus, this increased hydrophobicity may somehow prevent this protein from interacting with DAP-kinase that is more charged. However, because UNC5H2 interacts with DAP-kinase not only through its death domain but also through other domain(s), this would not be a sufficient explanation for the absence of interaction between UNC5H3 and DAP-kinase. Thus, it has to be hypothesized that other domains in UNC5H2 and UNC5H3 differ. Along these lines, NRAGE, which was shown to interact with the ZU domain of UNC5H1, fails to recruit UNC5H2 (Williams et al, 2003). Thus, even though the different UNC5H receptors are closely related and probably all act as dependence receptors (Llambi et al, 2001), they signal differently to cell death and are consequently not functionally redundant.
A second question relates to the mechanism by which netrin-1 affects the DAP-kinase activation by UNCH2. Because the presence of netrin-1 completely inhibits UNC5H2-mediated induction of DAP-kinase activity, we could have expected that netrin-1 may block or reduce UNC5H2 interaction with DAP-kinase. However, in most cases, the presence of netrin-1 did not change quantitatively the interaction between DAP-kinase and UNC5H2 (not shown). This has to be considered concomitant with the findings that the UNC5H2 death domain is necessary (i.e., its deletion abrogates UNC5H2-mediated induction of DAP-kinase activity) and sufficient (i.e., expression of the UNC5H2 death domain triggers DAP-kinase activity), yet it is not the only domain of UNC5H2 that interacts with DAP-kinase. Thus, a hypothesis may be raised according to which in the presence of netrin-1, DAP-kinase binds to UNC5H2 through the non-‘death domain' interacting region(s) of UNC5H2 and DAP-kinase and under these conditions DAP-kinase is autophosphorylated and inactive. However, when netrin-1 is withdrawn, DAP-kinase still binds to UNC5H2 but now also interacts via the UNC5H2 death domain and this interacting event is necessary and sufficient to trigger DAP-kinase activation. This mechanism may then be either an important safeguard control or, conversely, a more efficient system to speed up cell death induction. From the former point of view, when a cell expresses a ‘dangerous' proapoptotic kinase like DAP-kinase, any accidental activation would trigger cell death induction. Continuous recruitment of DAP-kinase to UNC5H2 when the survival signal netrin-1 is present would be a good way to avoid inappropriate activation. However, this hypothesis would be difficult to conciliate with the fact that only a fraction of DAP-kinase interacts with UNC5H2. From the latter point of view, to have DAP-kinase positioned close to the death domain of UNC5H2 would be an efficient system to activate rapidly DAP-kinase when netrin-1 is withdrawn.
A third question deals with the role of the caspase cleavage of UNC5H2 with regard to UNC5H2-mediated induction of DAP-kinase activity. The initial view of the receptor cleavage by caspase suggested that a preliminary cleavage allows exposure of a proapoptotic moiety that in turn induces apoptosis (Llambi et al, 2001; Tanikawa et al, 2003). Consequently, DAP-kinase activation by unbound UNC5H2 should be a consequence of the UNC5H2 caspase cleavage. In agreement with this model, UNC5H2 D412N was less efficient in inducing DAP-kinase activity. However, an important question refers to the initiation of this process: if DAP-kinase activation by UNC5H2 requires the prior caspase cleavage of UNC5H2, then how is the process initiated since caspase activation is usually thought to occur downstream of DAP-kinase activation? One possibility is that the process may be initiated by a non-caspase protease, and then propagated via caspase cleavage. Only few cleavage events by a non-caspase protease would then be sufficient to initiate DAP-kinase and consequently the death program by activating locally enough caspase to generate a caspase amplification loop via UNC5H2. An alternative view could also be that the now old dogma, suggesting that caspases are completely inactive in nonapoptotic cells and are only activated massively upon proapoptotic stimuli, is just wrong. Indeed, recent findings have shown that caspase zymogens display some protease activity (Salvesen and Dixit, 1997; Yang et al, 1998; Salvesen and Duckett, 2002). Along the same lines, it is noteworthy that cells express endogenous inhibitors of active caspases like IAPs, suggesting that cells are equipped with inhibitors that function at the point of the active caspases (Salvesen and Duckett, 2002). Similarly, local caspase activation without cell death induction starts to be documented (Fernando et al, 2002; Campbell and Holt, 2003). Thus, it seems that cell death induction could be the result of caspase activation amplification rather than caspase initiation per se. Here, netrin-1 withdrawal would trigger the shift from low/local caspase activation to high/widespread caspase activation by allowing a caspase amplification loop that includes (i) UNC5H2 cleavage and (ii) DAP-kinase activation.
A fourth point regards the control by UNC5H2 of DAP-kinase autophosphorylation. Indeed, we have proposed that DAP-kinase autophosphorylation inhibits DAP-kinase phosphorylating activity, by inducing a conformational change and by consequently masking the N-terminal catalytic domain of DAP-kinase (Shohat et al, 2001). How does UNC5H2 induce DAP-kinase dephosphorylation and DAP-kinase phosphorylating activity? A tempting view would be that unbound UNC5H2 recruits DAP-kinase and consequently unmasks the catalytic domain of DAP-kinase, this event leading to the induction of kinase activity of DAP-kinase. In this scheme, however, it is not clear how DAP-kinase gets dephosphorylated. We may then speculate that when the catalytic domain is active, the accessibility for the regulatory domain is reduced, leading to the rapid loss of autophosphorylation. An alternative view may be that the interaction of DAP-kinase with UNC5H2 death domain is accompanied by its exposure to dephosphorylation, by specific phosphatases bound directly or indirectly to UNC5H2. In this case, the dephosphorylation would be the triggering event leading to DAP-kinase activation.
Finally, we have shown here the importance of the interaction between UNC5H2 and DAP-kinase in the killing activity of UNC5H2. However, it has to be noted that UNC5H receptors, before their demonstration as dependence receptors, were proposed to mediate the response of growth cones to the chemorepulsive activity of netrin-1 during nervous system development (Ackerman et al, 1997; Leonardo et al, 1997; Hong et al, 1999). Thus, DAP-kinase, which has been extensively described as a proapoptotic molecule, may also be involved in axon guidance processes, but such an implication has now to be investigated.
Materials and methods
Cells, transfection procedures, immunoblotting and netrin-1 production
Transient transfections of HEK 293T and HeLa cells were performed using Lipofectamine and the manufacturer's suggested procedure (Life Technologies). MEFs derived from DAP-kinase null mice as previously described (Raveh et al, 2001) were spontaneously immortalized by prolonged passages in culture and transfected using Lipofectamine. Netrin-1 was purified from netrin-1-producing 293-EBNA cells according to Serafini et al (1994).
Plasmid constructs
Full-length UNC5H2-, UNC5h2-HA Δ841–945- and UNC5H2 1–412-expressing constructs were in pcDNA3.1 (Invitrogen) as described before (Llambi et al, 2001); DCC-expressing construct pDCC-CMV.S, netrin-1-expressing construct pGNET1-myc and dominant-negative mutant of caspase-8-expressing construct have already been described (Forcet et al, 2001); pcDNA3.1-p75ntr and pcDNA3.1-Bax were from DE Bredesen. HA-tagged DAPK- and dominant-negative DAPK-DD-expressing constructs were described previously (Cohen et al, 1999; Raveh et al, 2001). The constructs encoding various deletions in the C-terminal tail of UNC5H2 were generated by a TOPO directional cloning strategy (Invitrogen) using pcDNA3.1-UNC5H2 as template and the following primers: 5′-CACCATGAGGGCCCGGAGCGGG-3′ and 5′-GCTGTAGGAGTGATCTATCCGTATGATGTGCC GGATTATGCATGAA-3′ for the UNC5H2-HA Δ397–945 construct, and 5′-CACCATGAGGGCCCGGAGCGGG-3′ and 5′-GCTGAAGTCATTGCCTATCCGTATGATGTGCC GGATTATGCATGAA-3′ for the UNC5H2-HA Δ631–945 construct. The constructs encoding UNC5H1-HA and UNC5H3-HA were generated by a TOPO directional cloning strategy (Invitrogen) using pSEC-A-UNC5H1 or H3 as templates and the following primers: 5′-CACCATGGAGACAGACACA-3′ and 5′-TTCATGCATAATCCGGCACATCATACGGATAA CACTCGGCCTCCGA-3′ for the UNC5H1 construct, and 5′-CACCATGAGGAAAGGTCTG-3′ and 5′-TTCATGCATAATCCGGCACATCATACGGATAA TACTGTCCTTCTGC-3′ for the UNC5H3 construct. The constructs encoding UNC5H2-DD-Flag, DAPK-Flag and DAPK-DD-Flag were generated by cloning in pFlag3X the NotI–BamH1, NotI–EcoR1 and Not1–EcoR1, respectively, PCR fragment derived from pcDNA3.1-UNC5H2 or pcDNA3.1-DAPK as template and the following primers: 5′-GCGCGGCCGCGTCTGCCCCTGGCAAT-3′ and 5′-GCGGATCCTCAGCAATCGCCATCAGT-3′ for the UNC5H2-DD-Flag construct, 5′-TATGCGGCCGCAATGACCGTGTTC-3′ and 5′-ATAGAATTCTCACCGGGATAC-3′ for the DAPK-Flag construct, and 5′-GCGCGGCCGCGTCACTGACCAACACC-3′ and 5′-GCGGATCCTCACCGGGATACAACAGA-3′ for the DAPK-DD-Flag construct.
DAP-kinase activity assay
HEK 293T cells treated or not with purified netrin-1 (0.3 μg/ml), cultured or not in calcium-free DMEM (Invitrogen), were lysed for 1 h in 50 mM Tris pH 7.5, 1 mM EDTA, 1 mM EGTA, 0.5 mM Na3VO4, 0.1% 2-mercaptoethanol, 1% Triton X-100, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 10 mM sodium β-glycerol phosphate, 0.1 mM PMSF and 1 μg/ml each of aprotinin, pepstatin and leupeptin. Protein A-Sepharose and anti-Flag antibodies were used to immunoprecipitate DAP-kinase, and the immunocomplex was then washed with the lysis buffer. The Sepharose immunocomplex was then incubated with a reaction mixture consisting of 2 M MnCl2, 8 mM MgCl2, 50 mM HEPES pH 7.5, 20 μg DAPK substrate peptide KKRPQRRYSNVF (Tocris) and [γ-32P]ATP (15 μCi) in 50 μM cold ATP (Amersham) for 10 min at 30°C. 32P-labeled KKRPQRRYSNVF was then separated from free phosphate by loading the reaction sample into a P81 phosphocellulose paper (Euromedex) followed by three steps of 0.75% phosphoric acid and acetone washes. 32P incorporation was then read in a Beckmann scintillation counter. In the case of measurement of DAP-kinase activity in P5 rat, four hippocampi from P5 rats were dissected out, cut in small pieces, triturated in HBSS (Invitrogen) 0.65% glucose and further incubated with purified netrin for 1 h at 37°C. DAPK activity was then measured as before after DAP-kinase pull-down using the anti-DAP-kinase antibody described in Shani et al (2004).
Immunoprecipitation
Immunoprecipitation was carried out on HEK 293T cells transfected with various DAP-kinase- or UNC5H-expressing constructs as described previously (Forcet et al, 2001). Briefly, HEK 293T were lysed in 50 mM HEPES pH 7.6, 125 mM NaCl, 5 mM EDTA and 0.1% NP-40 in the presence of protease inhibitors, and further incubated with either anti-FlagM2, anti-HA, anti-phospho-serine antibody (Sigma) or anti-UNC5H2 (R&D system) and protein A-Sepharose (Sigma) to, pull down UNC5H2 or DAP-kinase. UNC5H2 interaction with DAP-kinase was detected by immunoblot. In the case of immunoprecipitation performed in mice brain embryos, the brains from E13 embryos were dissected out, cut in small species and triturated. They were further incubated in the lysate buffer described above and subjected to sonication. Immunoprecipitation was performed as above using anti-UNC5H2 antibodies or control antibodies and immunoprecipitates were immunoblotted using anti-DAP-kinase monoclonal antibody (Sigma).
Immunostaining
Transfected HEK 293T cells were fixed for 5 min with 2% formaldehyde and 2% sucrose and further permeabilized for 5 min with 10% sucrose and 0.5% NP-40. The primary antibody (anti-phospho-Ser308 DAPK; Sigma) was used diluted to 1/200. Coverslips were then mounted with vectashield+dapi (Vector Laboratories).
Cell death analysis
HEK 293T cell death was analyzed using Trypan blue staining procedure (Forcet et al, 2001). Apoptosis was also monitored by using the caspase-3 assay that utilizes the Ac-DEVD-AFC substrate (MBL, Japan). This activity was determined according to the manufacturer's instructions using a Ac-DEVD-CHO-pretreated lysate as a negative control. Caspase activation is presented as the ratio between the caspase activity of the sample and that measured in 293T cells transfected with pCMV; for all samples, the background remaining after inhibition by Ac-DEVD-CHO was subtracted. In the case of the measurement of cell death in MEF cells, transfected MEF cells were concentrated using the MACSSelectKk.I kit (Mylteny Biotech). Briefly, MEF cells were cotransfected with UNC5H2- or DCC-expressing construct and the pKk vector. Transfected cells were isolated with MACS MicroBeads conjugated to a monoclonal antibody directed against the surface marker encoded by pKk vector and were then further assessed for cell death determination.
Acknowledgments
We thank C Guenebeaud and C Maisse for their help and discussion. We also thank H Arakawa for human UNC5H2 antibody. This work was supported by the Ligue Contre le Cancer (PM), the Schlumberger Fondation (PM), the NIH (PM) and by the Israel Science Foundation administered by the Israel Academy of Sciences and Humanities (AK). AK is an incumbent of Helena Rubinstein Chair of Cancer Research.
References
- Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB, Knowles BB (1997) The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature 386: 838–842 [DOI] [PubMed] [Google Scholar]
- Campbell DS, Holt CE (2003) Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37: 939–952 [DOI] [PubMed] [Google Scholar]
- Cohen O, Feinstein E, Kimchi A (1997) DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J 16: 998–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H, Spivak-Kroizaman T, Feinstein E, Kimchi A (1999) DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain. J Cell Biol 146: 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A (1995) Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev 9: 15–30 [DOI] [PubMed] [Google Scholar]
- Fearon ER, Cho KR, Nigro JM, Kern SE, Simons JW, Ruppert JM, Hamilton SR, Preisinger AC, Thomas G, Kinzler KW, Vogelstein B (1990) Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 247: 49–56 [DOI] [PubMed] [Google Scholar]
- Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E (1995) The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci 20: 342–344 [DOI] [PubMed] [Google Scholar]
- Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA (2002) Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci USA 99: 11025–11030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcet C, Stein E, Pays L, Corset V, Llambi F, Tessier-Lavigne M, Mehlen P (2002) Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation. Nature 417: 443–447 [DOI] [PubMed] [Google Scholar]
- Forcet C, Ye X, Granger L, Corset V, Shin H, Bredesen DE, Mehlen P (2001) The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc Natl Acad Sci USA 98: 3416–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K, Hinck L, Nishiyama M, Poo MM, Tessier-Lavigne M, Stein E (1999) A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 97: 927–941 [DOI] [PubMed] [Google Scholar]
- Inbal B, Cohen O, Polak-Charcon S, Kopolovic J, Vadai E, Eisenbach L, Kimchi A (1997) DAP kinase links the control of apoptosis to metastasis. Nature 390: 180–184 [DOI] [PubMed] [Google Scholar]
- Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH (2002) TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol 4: 51–58 [DOI] [PubMed] [Google Scholar]
- Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, Tessier-Lavigne M (1996) Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell 87: 175–185 [DOI] [PubMed] [Google Scholar]
- Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL, Tessier-Lavigne M (1997) Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature 386: 833–838 [DOI] [PubMed] [Google Scholar]
- Llambi F, Causeret F, Bloch-Gallego E, Mehlen P (2001) Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J 20: 2715–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlen P, Bredesen DE (2004) The dependence receptor hypothesis. Apoptosis 9: 37–49 [DOI] [PubMed] [Google Scholar]
- Mehlen P, Rabizadeh S, Snipas SJ, Assa-Munt N, Salvesen GS, Bredesen DE (1998) The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature 395: 801–804 [DOI] [PubMed] [Google Scholar]
- Raveh T, Droguett G, Horwitz MS, DePinho RA, Kimchi A (2001) DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat Cell Biol 3: 1–7 [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM (1997) Caspases: intracellular signaling by proteolysis. Cell 91: 443–446 [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS (2002) IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 3: 401–410 [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M (1996) Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87: 1001–1014 [DOI] [PubMed] [Google Scholar]
- Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M (1994) The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78: 409–424 [DOI] [PubMed] [Google Scholar]
- Shani G, Marash L, Gozuacik D, Bialik S, Teitelbaum L, Shohat G, Kimchi A (2004) Death-associated protein kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy to activate its cell death functions. Mol Cell Biol 19: 8611–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohat G, Spivak-Kroizman T, Cohen O, Bialik S, Shani G, Berrisi H, Eisenstein M, Kimchi A (2001) The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J Biol Chem 276: 47460–47467 [DOI] [PubMed] [Google Scholar]
- Tanikawa C, Matsuda K, Fukuda S, Nakamura Y, Arakawa H (2003) p53RDL1 regulates p53-dependent apoptosis. Nat Cell Biol 5: 216–223 [DOI] [PubMed] [Google Scholar]
- Thiebault K, Mazelin L, Pays L, Llambi F, Joly MO, Saurin JC, Scoazec JY, Romeo G, Mehlen P (2003) The netrin-1 receptors UNC5H are putative tumor suppressors controlling cell death commitment. Proc Natl Acad Sci USA 100: 4173–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WJ, Kuo JC, Yao CC, Chen RH (2002) DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J Cell Biol 159: 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Strickland P, Watanabe K, Hinck L (2003) UNC5H1 induces apoptosis via its juxtamembrane domain through an interaction with NRAGE. J Biol Chem 278: 17483–17490 [DOI] [PubMed] [Google Scholar]
- Yang X, Chang HY, Baltimore D (1998) Autoproteolytic activation of pro-caspases by oligomerization. Mol Cell 1: 319–325 [DOI] [PubMed] [Google Scholar]