

Abstract
Cell-to-cell spread is a fundamental step in the infection cycle of Listeria monocytogenes that strictly depends on the formation of bacteria-induced protrusions. Since Listeria actin tails in the protrusions are tightly associated with the plasma membrane, we hypothesised that membrane–cytoskeleton linkers would be required for initiating and sustaining their formation and the subsequent cell-to-cell spread. We have found that ezrin, a member of the ezrin, radixin and moesin (ERM) family that functions as a key membrane–cytoskeleton linker, accumulates at Listeria protrusions. The ability of Listeria to induce protrusions and effectively spread between adjacent cells depends on the interaction of ERM proteins with both a membrane component such as CD44 and actin filaments. Interfering with either of these interactions or with ERM proteins phosphorylation not only reduces the number of protrusions but also alters their morphology, resulting in the formation of short and collapsed protrusions. As a consequence, Listeria cell-to-cell spread is severely impaired. Thus, ERM proteins are exploited by Listeria to escape the host immune response and to succeed in the development of the infection.
Keywords: actin cytoskeleton, ERM proteins, infection, Listeria monocytogenes, protrusion
Introduction
Listeria monocytogenes is a Gram-positive, facultative intracellular bacterium that causes food-borne infections, which can lead to abortion and diseases as severe as meningitis, septicaemias and gastroenteritis. At the cellular level, the infection cycle of Listeria is characterised by four major steps: adhesion to, and invasion of, host cells, escape from phagocytic vacuoles, actin-based intracellular movement and cell-to-cell spread. Except for the escape from phagocytic vacuoles, the other steps are characterised by an interplay between bacteria and the host cell actin cytoskeleton (Tilney and Portnoy, 1989; see also Vazquez-Boland et al, 2001; Portnoy et al, 2002).
Several studies have demonstrated that Listeria induces its own uptake into nonphagocytic cells and accomplishes to move within them by subverting the function of key cytoskeletal components such as Ena/VASP proteins and the Arp2/3 complex (see Frischknecht and Way, 2001; Vazquez-Boland et al, 2001; Cossart et al, 2003). These studies not only culminated in the reconstitution of Listeria motility in cell-free systems (Loisel et al, 1999), but have also been instrumental for understanding actin cytoskeleton dynamics in general. In contrast, the mechanisms underlying Listeria cell-to-cell spread have not yet been intensively studied. This process begins when motile bacteria approach the inner face of the plasma membrane, thus triggering the formation of finger-like structures called protrusions, which harbour the bacteria at their tips. Subsequently, these protrusions, due to the force generated by actin polymerisation, can penetrate into adjacent cells (Tilney and Portnoy, 1989; Robbins et al, 1999), leading to the direct cell-to-cell transfer of the bacteria that allows them to escape both the humoral and the cytotoxic T-cell host immune responses (see O'Riordan and Portnoy, 2002). Since Listeria actin tails are closely juxtaposed to the plasma membrane during protrusion formation and cell-to-cell spread, it is reasonable to conceive that actin cytoskeleton–membrane interactions contribute to the onset and progression of these processes.
Ezrin, radixin and moesin (ERM) proteins are a family of widely distributed membrane-associated proteins responsible for linking the plasma membrane to the underlying actin cytoskeleton (see Bretscher et al, 2002). The N-terminus of ERM proteins can interact with membrane components such as CD44, CD43, intercellular adhesion proteins (ICAMs), PtdIns (4,5) P2, and with the phosphoprotein EBP50 (Tsukita et al, 1994; Niggli et al 1995; Reczek et al, 1997; Serrador et al, 1997; Legg and Isacke, 1998; Yonemura et al, 1998). The C-terminal domain harbours a stretch of ∼30 amino acids that can bind to F-actin (Turunen et al, 1994; Pestonjamasp et al, 1995). ERM proteins exist in two functionally different states. In their inactive state, the C-terminal domain is thought to be associated with the N-terminal domain causing ERM proteins to acquire a ‘closed' conformation (Gary and Bretscher, 1995; Henry et al, 1995; Pearson et al, 2000). Upon activation by, for example, the phosphorylation of a critical threonine residue in the C-terminal domain of ERM proteins, the interactions between the N- and C-terminal domains are disrupted, thus exposing critical binding sites for the membrane–cytoskeleton interactions (see Bretscher et al, 2002). Merlin, a tumour suppressor protein closely related to ERM proteins, exhibits a similar N-terminus but lacks actin-binding sites in its C-terminus. Apart from these distinct structural properties, merlin requires, in contrast to ERM proteins, dephosphorylation to be active. Moreover, ERM proteins and merlin have antagonistic functional properties: ERM proteins promote cell growth, whereas merlin suppresses growth and inhibits signal transduction (Eldridge, 1981; Trofatter et al, 1993; Morrison et al, 2001 and our unpublished results; see also Bretscher et al, 2002).
ERM proteins have also been implicated in crucial steps of the life cycle of invasive bacteria that are characterised by the rearrangement of the actin cytoskeleton. Ezrin can be detected at the entry sites of the enteroinvasive bacterium Shigella flexneri in epithelial cells and the overexpression of its N-terminus impaired the ability of these bacteria to invade these cells (Skoudy et al, 1999). Furthermore, the hyaluronan (HA)-mediated interaction of Streptococcus pyogenes with host cells triggers ERM protein-dependent cytoskeletal changes and disruption of intercellular junctions, processes that precede their invasion of soft tissues (Cywes and Wessels, 2001). These changes are likely based on ERM protein dephosphorylation. Interestingly, ezrin also localises to Listeria protrusions but not to Listeria actin tails within the cell body (Sechi et al, 1997).
In light of the key role of ERM proteins as membrane–cytoskeleton linkers, we hypothesised that they may favour the formation of Listeria protrusions and the subsequent cell-to-cell spread by crosslinking the actin tails to the surrounding plasma membrane. Here, we demonstrate that the interaction of active (phosphorylated) ERM proteins with both membrane components and actin tails is essential for efficient protrusion formation and cell-to-cell spread.
Results
ERM proteins link actin tails to the plasma membrane in Listeria protrusions
Listeria protrusion formation and cell-to-cell spread can be ideally analysed in mature epithelial monolayers, which are expected to closely mimic the onset and development of these processes in vivo (see Temm-Grove et al, 1994, and references therein). However, protrusions inside mature epithelial monolayers are difficult to observe since the cells are tall and tightly packed against each other. We chose, therefore, to study protrusion formation in single or low-confluent cells, where they are easily detectable by light and electron microscopic techniques. Although the mechanisms underlying the formation of both types of protrusions may be different, their highly similar behaviour (see Robbins et al, 1999) makes the conclusions drawn here most likely appropriate for both types of protrusions.
The observation that ezrin accumulates at Listeria-induced protrusions but not at the actin tails within the cell body (Figure 1A and B; see also Sechi et al, 1997) suggests that active ERM proteins may associate with Listeria protrusions by simultaneously binding to the actin comet tails and to the membrane surrounding them. To test this possibility, we generated GFP-tagged amino- and carboxy-terminal domains of ezrin that can be defined as independent entities based on biochemical and structural data (see Pearson et al, 2000; Bretscher et al, 2002). To reduce the potential interference of GFP with the binding activity of both ezrin domains, the GFP moiety was cloned at the COOH and the NH2 ends of the amino- and carboxy-terminal domains of ezrin, respectively. In addition, to minimise a potential inhibitory effect of these constructs on protrusion formation, cells expressing low levels of both fusion proteins were analysed. According to previous observations (Algrain et al, 1993; Amieva et al, 1999), in uninfected HeLa cells, GFP-tagged amino- and carboxy-terminal domains of ezrin localised to the membrane and actin filaments, respectively (not shown), indicating that GFP did not alter their binding properties. In Listeria-infected HeLa cells, the GFP-tagged amino-terminal domain of ezrin localised to the membrane surrounding the protrusion but was not detectable along actin comet tails within the cytoplasm (Figure 1C), whereas the GFP-tagged carboxy-terminal domain of ezrin localised to the actin tails of motile Listeria (Figure 1D).
Figure 1.
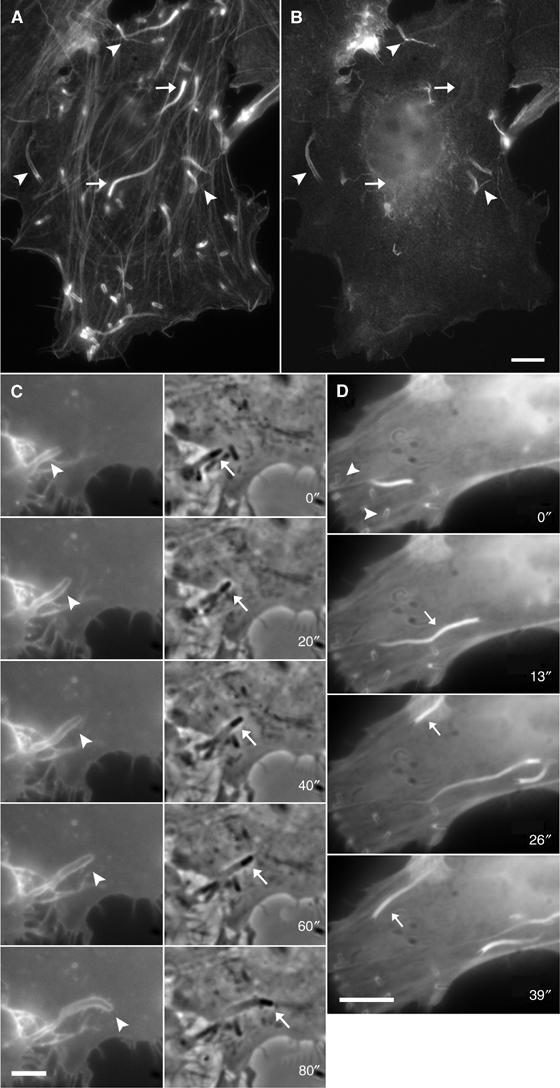
Localisation of ezrin and its GFP-tagged amino- and carboxy-terminal domains during Listeria-induced protrusion formation. HeLa cells were infected with Listeria, fixed and stained with fluorescent phalloidin (A) and a polyclonal antibody against ezrin (B). Listeria-induced protrusions are easily distinguished from intracellular actin tails since they appear slimmer and are connected to the cell periphery by a thin stalk (arrowheads in (A)). Ezrin can be detected at actin comet tails in protrusions (arrowheads in (B); corresponding actin labelling in (A)) but not along the actin tails in the cell body (arrows in (B); corresponding actin labelling in (A)). (C, D) Dynamics of the GFP-tagged amino-terminal (C) and the carboxy-terminal domain (D) of ezrin in HeLa cells infected with Listeria. Panels in (C) represent GFP fluorescence (left) and phase contrast images (right), respectively. The amino-terminal domain of ezrin localised at the membrane surrounding a Listeria-induced protrusion (arrowheads, left panels in (C)) harbouring the bacterium at its tip (arrows, right panels in (C)). The carboxy-terminal domain (D) of ezrin localises at the actin tails associated with motile Listeria (arrows) and can also be detected around nonmotile bacteria within the cell body (arrowheads). Scale bars: 5 μm (A–C); 10 μm (D).
Interaction of ERM proteins with the plasma membrane and actin filaments is essential for efficient protrusion formation
Since ERM protein domains expressed separately act as dominant-negative elements on ERM protein functions (Henry et al, 1995; De Joussineau et al, 2003), we reasoned that the expression of sufficiently high levels of ezrin domains would impair protrusion formation by interfering with endogenous ERM protein functions. Since the low magnification and resolution provided by the light microscope did not allow an unequivocal assessment of protrusion morphological features, we analysed these structures by scanning electron microscopy (SEM). Control Listeria protrusions had a slender shape and were connected to the cell surface by a thin stalk (facing arrowheads in Figure 2A). Conversely, the protrusions formed in HeLa cells expressing GFP-tagged NH2 or COOH ezrin had a less slender shape and were usually connected to the cell surface by a thick and distorted end (arrowhead in Figure 2B and C; see Table I).
Figure 2.
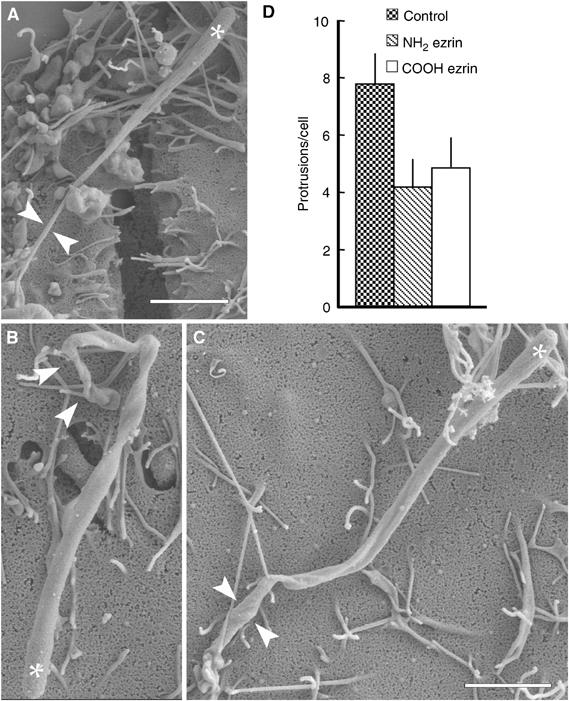
Overexpression of ERM protein domains impairs protrusion formation. (A–C) Listeria-infected HeLa cells expressing excess amounts of GFP-tagged amino- or carboxy-terminal domains of ezrin were fixed and processed for SEM. In nontransfected cells, these bacteria induced the formation of slender protrusions that are connected to the cell surface through a thin proximal portion (facing arrowheads in (A)), whereas in cells expressing the amino- (B) or carboxy-terminal (C) domain of ezrin, the protrusions had a distorted proximal portion that was thicker than that in control protrusions (arrowheads in (B, C)). White stars indicate the position of bacteria. Scale bars: 2 μm. (D) Quantification of the number of Listeria protrusions in control HeLa cells and in cells expressing GFP-tagged amino- or carboxy-terminal domain of ezrin. Following infection with Listeria, HeLa cells were fixed and then stained with fluorescent phalloidin to detect actin comet tails. The expression of both ezrin domains decreased the number of protrusions as compared to control cells. Error bars indicate one standard deviation from the mean.
Table 1.
Quantification of the number of normal and atypical protrusions related to the SEM analysisa
| Cells examined | Pseudopodia examined | Normal pseudopodia | Atypical pseudopodia | |
|---|---|---|---|---|
| HeLa cells | 10 | 17 | 15 (88%) | 2 (12%) |
| HeLa cells/NH2 ezrinb | 16 | 24 | 2 (8%) | 22 (92%) |
| HeLa cells/COOH ezrinb | 15 | 23 | 3 (13%) | 20 (87%) |
| RT4 cells | 35 | 52 | 48 (92%) | 4 (8%) |
| RT4 cells/CD44 tail wtc | 25 | 33 | 4 (12%) | 29 (88%) |
| RT4 cells/CD44 tail mutc | 19 | 38 | 32 (85%) | 6 (15%) |
| RT4 cells | 15 | 20 | 17 (85%) | 3 (15%) |
| RT4 cells/+1.1asmld | 15 | 26 | 4 (15%) | 22 (85%) |
| RT4 cells/ezrin T567Ae | 20 | 47 | 21 (45%) | 26 (55%) |
| RT4 cells/ezrin T567A/+1.1asmld | 26 | 56 | 28 (50%) | 28 (50%) |
| RT4 cells/ezrin T567De | 19 | 30 | 25 (83%) | 5 (17%) |
| RT4 cells/ezrin T567D/+1.1asmld | 23 | 43 | 35 (81%) | 8 (19%) |
| aTwo to three experiments were conducted to quantify the number of protrusions. | ||||
| bTransient expression of GFP-tagged ezrin domains. | ||||
| cStable expression of CD44 cytoplasmic tails. | ||||
| dOvernight treatment with 5 μg/ml 1.1asml anti-CD44 antibody. | ||||
| eStable expression of ezrin T567A or T567D point mutants. | ||||
We determined whether these morphological changes were accompanied by a reduction in the number of protrusions per cell. As the number of protrusions directly correlates with the number of motile bacteria, only cells that contained an equivalent number of motile bacteria (corresponding to the number of actin tails per cell) were analysed. Compared to control cells, the expression of either GFP-tagged NH2 or COOH ezrin caused a significant reduction in the number of protrusions per cell (Figure 2D; see Table II). Notably, the expression of ERM protein domains did not alter the morphology of Listeria actin tails within the cytoplasm (Figure 3A–C′) nor their length, which is proportional to the bacterial speed (see Theriot et al, 1992) (Figure 3D), indicating that intracellular Listeria motility per se was not affected in these assays. The specificity of our approach was further supported by the observation that protrusion formation was greatly impaired by the siRNA-induced downregulation of all endogenous ERM proteins in HeLa cells (Figure 4).
Table 2.
Statistical analysis (Student's t-test ) of the data presented in this worka
| Data set #1 | Data set #2 | P-value | Significant difference | |
|---|---|---|---|---|
| Figure 2D | HeLa control (n=50) | HeLa NH2 ezrin (n=50) | <0.0001 | Yes |
| HeLa control (n=50) | HeLa COOH ezrin (n=50) | <0.0001 | Yes | |
| Figure 3D | HeLa control (n=50) | HeLa NH2 ezrin (n=50) | 0.0443 | Yes |
| HeLa control (n=50) | HeLa COOH ezrin (n=50) | 0.006 | Yes | |
| Figure 5A | RT4 control (n=60) | RT4 NF2 +HA (n=60) | <0.0001 | Yes |
| RT4 control (n=60) | RT4 NF2 −HA (n=102) | 0.7306 | No | |
| RT4 control (n=60) | RT4 L64P +HA (n=60) | 0.0013 | Yes | |
| Figure 5B | RT4 control (n=64) | RT4 NH2 NF2 (n=61) | <0.0001 | Yes |
| RT4 control (n=64) | RT4 COOH NF2 (n=61) | 0.5954 | No | |
| Figure 5D | RT4 control (n=120) | RT4 1.1asml (n=126) | <0.0001 | Yes |
| RT4 control (n=120) | RT4 5G8 (n=120) | 0.1915 | No | |
| Figure 6F | RT4 control (n=103) | RT4 CD44 tail wt (n=108) | <0.0001 | Yes |
| RT4 control (n=103) | RT4 CD44 tail mut (n=103) | 0.1422 | No | |
| Figure 7 | RPMC (n=90) | RPMC CD44wt (n=90) | <0.0001 | Yes |
| RPMC (n=90) | RPMC CD44mut (n=90) | 0.2753 | No | |
| Figure 8A | LLC-PK1 (n=100) | LLC-PK1 ezrin T/A (n=100) | <0.0001 | Yes |
| LLC-PK1 (n=100) | LLC-PK1 ezrin T/D (n=100) | 0.0017 | Yes | |
| Figure 8B | RT4 control −HA (n=100) | RT4 control +HA (n=100) | <0.0001 | Yes |
| RT4 ezrin T/A −HA (n=100) | RT4 ezrin T/A +HA (n=100) | 0.0242 | Yes | |
| RT4 ezrin T/D −HA (n=100) | RT4 ezrin T/D +HA (n=100) | 0.1694 | No | |
| RT4 control −HA (n=100) | RT4 ezrin T/A −HA (n=100) | <0.0001 | Yes | |
| RT4 control −HA (n=100) | RT4 ezrin T/D −HA (n=100) | 0.0054 | Yes | |
| RT4 control +HA (n=100) | RT4 ezrin T/A +HA (n=100) | 0.3343 | No | |
| RT4 control +HA (n=100) | RT4 ezrin T/D +HA (n=100) | <0.0001 | Yes | |
| Figure 8C | RT4 control −asml (n=22) | RT4 control +asml (n=28) | <0.0001 | Yes |
| RT4 ezrin T/A −asml (n=57) | RT4 ezrin T/A +asml (n=74) | 0.2491 | No | |
| RT4 ezrin T/D −asml (n=32) | RT4 ezrin T/D +asml (n=39) | 0.8670 | No | |
| RT4 control −asml (n=22) | RT4 ezrin T/A −asml (n=57) | <0.0001 | Yes | |
| RT4 control −asml (n=22) | RT4 ezrin T/D −asml (n=32) | 0.9216 | No | |
| RT4 control +asml (n=28) | RT4 ezrin T/A +asml (n=74) | 0.6632 | No | |
| RT4 control +asml (n=28) | RT4 ezrin T/D +asml (n=39) | <0.0001 | Yes | |
| aThe statistical analysis was carried out by comparing data set #1 with data set #2. | ||||
Figure 3.
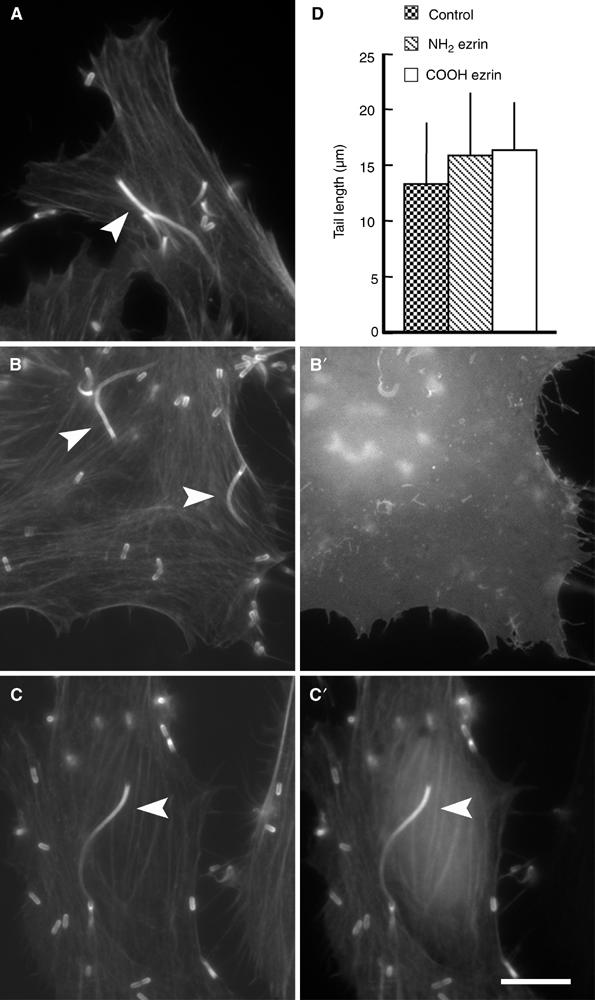
Overexpression of the amino- or carboxy-terminus of ezrin does not impair the motility of Listeria. Control HeLa cells and HeLa cells transfected with the GFP-tagged amino- or carboxy-terminal domain of ezrin were infected with Listeria, fixed and stained with fluorescent phalloidin. The actin tails induced by Listeria in HeLa cells expressing the amino- (B) or carboxy-terminus (C) of ezrin are morphologically indistinguishable from those induced by these bacteria in control cells (A). Panels A–C represent phalloidin staining, whereas panels B′–C′ represent GFP fluorescence. Scale bar: 5 μm. (D) Quantification of the length of Listeria actin tails (a parameter that is proportional to bacterial speed) in control HeLa cells and in HeLa cells expressing the amino- or carboxy-terminal domain of ezrin, showing that the expression of both ezrin domains does not impair Listeria motility. Error bars indicate one standard deviation from the mean.
Figure 4.
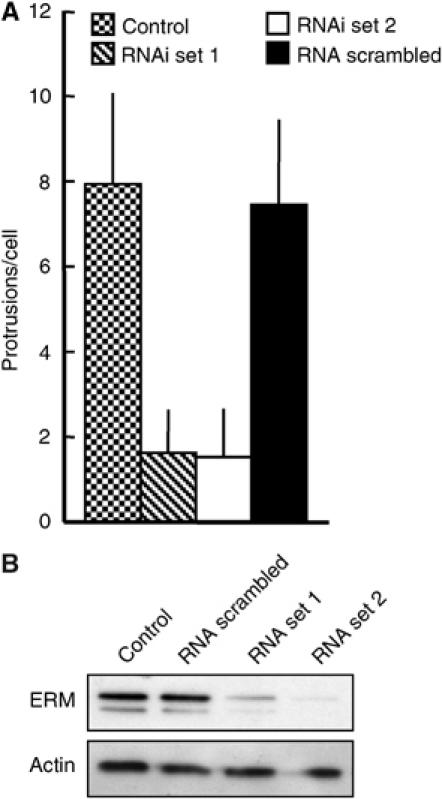
Downregulation of ERM proteins by siRNA impairs the formation of Listeria protrusions. (A) HeLa cells, treated for 30 h with two different siRNA sets specific for ezrin, moesin and radixin, a scrambled siRNA set or left untreated, were infected with Listeria for 6 h. Afterwards, cells were fixed and labelled with fluorescent phalloidin and an antibody for ERM proteins, and the number of protrusions per cells was determined. The treatment of the cells with both siRNA sets caused a marked impairment of protrusion formation compared to untreated cells and cells treated with a scrambled siRNA set. Error bars in A indicate one standard deviation from the mean. (B) Cell lysates were resolved by SDS–PAGE, blotted and probed with an ERM antibody. Both sets of siRNA duplexes caused a strong reduction in the levels of ERM proteins after 36 h, whereas the scrambled siRNA set had no effect. Actin served as the loading control.
Displacement and dephosphorylation of ERM proteins correlate with the impairment of protrusion formation
In another approach towards antagonising ERM protein localisation, we took advantage of the observation that the activation of merlin can displace ERM proteins from membrane components such as CD44 (Morrison et al, 2001). Since the COOH-terminus of merlin does not harbour F-actin-binding sequences, the merlin-induced displacement of ERM proteins would impair protrusion formation by interfering with ERM proteins–membrane, ERM proteins–actin interactions or both. In our system, the overexpression of merlin is under the tetracycline response promoter, and its activation can be induced by HA (Morrison et al, 2001). Active merlin decreased protrusion formation compared to control cells, cells in which merlin was inactive or cells that expressed an inactive merlin mutant (L64P) (Figure 5A and Supplementary data). Consistent with these data, the overexpression of the N-terminal domain of merlin (which, due to its structural similarity to ERM proteins, competes with them for binding to the membrane), but not the C-terminal domain of merlin (which is unable to bind to F-actin), reduced protrusion formation (see Supplementary data).
Figure 5.
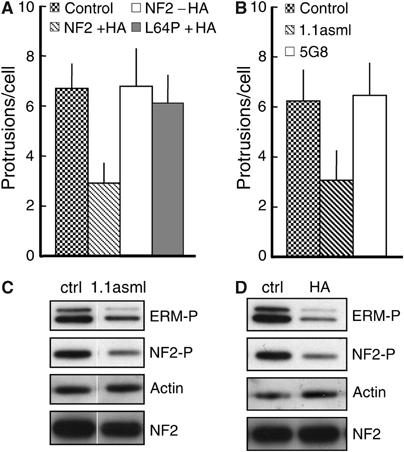
Displacement and dephosphorylation of ERM proteins correlate with the impairment of protrusion formation. (A) HA-induced activation of merlin impairs protrusion formation. Parental RT4 cells (control) and RT4 cells expressing wild-type merlin (NF2 WT 5/4) or a mutated variant of merlin (NF2 L64P) were infected with Listeria and processed for fluorescent microscopy. In RT4 cells treated with doxycycline and with HA to activate merlin (NF2 WT 5/4 +HA), protrusion formation was decreased as compared to untreated parental cells (control), RT4 cells expressing a mutated variant of merlin (NF2 L64P +HA) or RT4 cells not treated with HA (NF2 WT 5/4 −HA). (B) Specific engagement of CD44 reduces protrusion formation. After overnight treatment with anti-CD44 monoclonal antibodies 1.1asml or 5G8 and with doxycycline, RT4 cells were infected with Listeria and processed for fluorescence microscopy. As compared to control (untreated) cells, RT4 cells treated with the 1.1asml antibody supported less efficiently the formation of protrusions, whereas the treatment with the 5G8 antibody had no effect on this process. Error bars (in A, B and D) indicate one standard deviation from the mean. (C, D) Binding of HA or specific antibodies to CD44 induces NF2 and ERM protein dephosphorylation. Lysates from RT4 cells untreated or treated with HA or CD44 monoclonal antibody 1.1asml were resolved by SDS–PAGE and probed with antibodies against the phosphorylated form of ERM proteins (C) and NF2 (D). Both HA and the monoclonal 1.1asml induced a marked decrease in the level of phosphorylated ERM proteins and NF2 compared to control untreated samples. The third panel from top in (C, D) shows the actin loading control. Bottom panels in (C, D) indicate total NF2 showing that NF2 activation (dephosphorylation) does not cause NF2 degradation.
Since HA activates merlin (characterised by its dephosphorylation) predominantly by signalling through CD44 (Morrison et al, 2001), we determined whether the engagement of CD44 with specific antibodies could affect protrusion formation. The CD44 monoclonal antibody 1.1asml, which activates merlin as efficiently as HA (Morrison et al, 2001) did, indeed, reduce protrusion formation. By contrast, the CD44 monoclonal antibody 5G8, which does not activate merlin (Morrison et al, 2001), had no effect on protrusion formation (Figure 5B). The engagement of CD44 by HA or 1.1asml clearly reduced the phosphorylation of both merlin and ERM proteins without affecting their total abundance (Figure 5C and D), suggesting that the impairment of protrusion formation depends not only on the displacement of ERM proteins but also on their dephosphorylation (see below).
Overexpression of the ERM-binding site of CD44 impairs Listeria protrusion formation and morphology
Since ERM proteins need docking sites at the plasma membrane to function properly as membrane–cytoskeleton linkers, the displacement of ERM proteins from the membrane by overexpressing the ERM-binding site of the cytoplasmic tail of CD44 (Legg and Isacke, 1998; Morrison et al, 2001) would impair protrusion formation. In control RT4 cells, Listeria protrusions were morphologically similar to those induced in HeLa cells (Figure 6A; facing arrowheads in Figure 6C). In contrast, protrusions formed in RT4 cells expressing CD44 cytoplasmic tail were reduced in number (Figure 6F), much shorter (4.71±1.85 μm (n=27) versus 9.19±2.83 μm (n=47) in control cells) and linked to the cell surface by a thick and collapsed end (Figure 6B; arrowhead in Figure 6D). The expression of comparable levels of the CD44 cytoplasmic tail mutated in its ERM-binding site (see Legg and Isacke, 1998) did not affect both protrusion number and morphology (Figure 6E and F).
Figure 6.
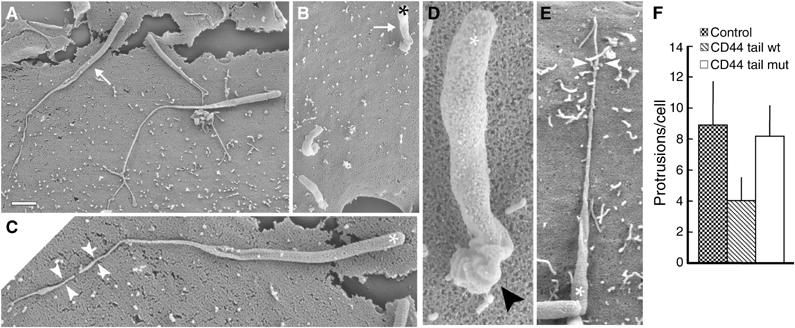
Impairment of Listeria protrusion formation by overexpression of the cytoplasmic tail of CD44. (A–E) SEM analysis of Listeria protrusions in parental RT4 cells (A, C), RT4 cells expressing wild-type CD44 cytoplasmic tail (B, D) and a mutated version of the CD44 cytoplasmic tail (E). In control cells and in cells expressing the mutated CD44 cytoplasmic tail, Listeria induced the formation of long protrusions (arrow in (A)), which are connected to the cell surface through a thin stalk (facing arrowheads in (C, E)). Conversely, in RT4 cells expressing wild-type CD44 cytoplasmic tail, the protrusions are shorter (arrow in (B)) and characterised by a thick and distorted proximal portion (arrowhead in (D)). Stars indicate the position of bacteria. Scale bar: 2 μm (A, B); 0.8 μm (C); 0.3 μm (D); 1 μm (E). (F) Quantification of the number of Listeria protrusions in control RT4 cells and in RT4 cells expressing wild-type or mutated forms of CD44 cytoplasmic tail. Expression of wild-type CD44 cytoplasmic tail impairs protrusion formation, whereas its mutated variant had no effect. Error bars indicate one standard deviation from the mean.
CD44 is sufficient to enhance protrusion formation but cannot rescue normal protrusion morphology in RPM-MC cells
Since CD44 localises to Listeria protrusions (see Supplementary data), and sequestration of ERM proteins away from the membrane proteins such as CD44 could impair protrusion formation, we sought to determine whether CD44 plays a role in Listeria protrusion formation. To this end, we used a CD44-negative melanoma cell line, RPM-MC (Thomas et al, 1992; Morrison et al, 2001). We reasoned that if CD44 functions by providing docking sites for ERM proteins at the plasma membrane, then its expression in RPM-MC cells would significantly enhance protrusion formation. In RPM-MC cells, Listeria induced a low number of distorted protrusions that was markedly enhanced by the expression of wild-type but not mutated CD44 (which does not bind ERM proteins) (Figure 7). However, in our RPM-MC system, CD44 could not restore normal protrusion morphology (see Supplementary data). Nevertheless, we can conclude that, at least in RPM-MC cells, CD44 contributes to protrusion formation by increasing the number of binding sites for ERM proteins.
Figure 7.
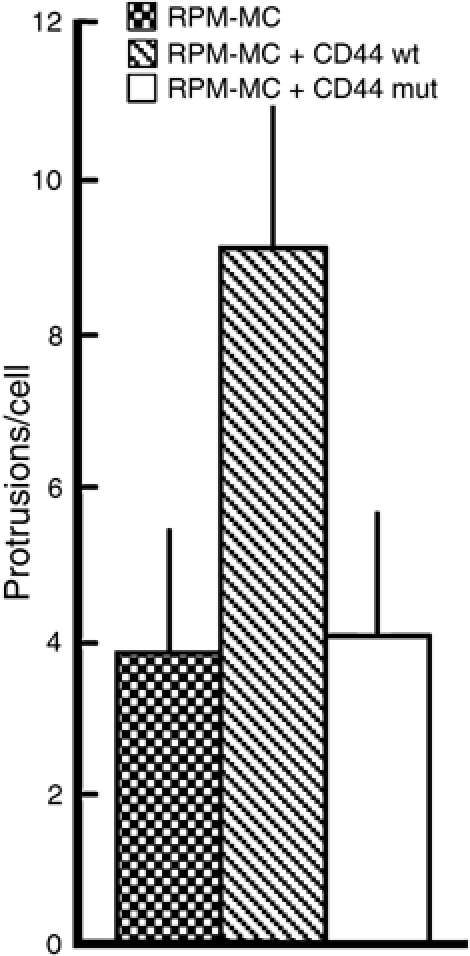
Expression of CD44 in RPM-MC cells is sufficient to enhance protrusion formation. Control RPM-MC cells and RPM-MC cells stably expressing CD44 or a CD44 variant mutated in its ERM-binding site were infected with Listeria, fixed and the number of protrusions per cell quantified. Expression of CD44 enhances protrusion formation compared to control cells and cells expressing the mutated variant of CD44. Error bars indicate one standard deviation from the mean.
Phosphorylation of threonine 567 within ezrin is crucial for efficient protrusion formation
To function as membrane–cytoskeleton linkers, ERM proteins must undergo an activation process, which primarily depends on the phosphorylation of a threonine residue (T567, T564 and T558 in ezrin, radixin and moesin, respectively) that is crucial for ERM protein activation and their interaction with actin filaments (Nakamura et al, 1999; see Bretscher et al, 2002).
Since we found that impaired protrusion formation corresponds to ERM protein dephosphorylation (Figure 5C and D), we sought to determine whether the phosphorylation of T567 in ezrin is crucial for protrusion formation. To this end, we used LLC-PK1 cells stably expressing ezrin mutants harbouring either a T567A or a T567D point mutation to mimic its constitutively inactive and active state, respectively (Gautreau et al, 2000). Since the ezrin T567A mutant localises at the cell membrane but binds less efficiently to actin filaments (Gautreau et al, 2000), the expression of this construct would impair protrusion formation by antagonising endogenous ERM protein actin-binding sites at the cell membrane. Moreover, because the phosphorylation of an equivalent threonine residue (T558) within moesin is required for the interaction of this protein with actin filaments (Nakamura et al, 1999), the ezrin T567D mutant would be expected to contribute to the interaction between membrane and actin tails during protrusion formation. The expression of ezrin T567D did not influence protrusion formation (Figure 8A), suggesting that enough active endogenous ERM proteins were already available at the plasma membrane to support efficiently this process. In contrast, the expression of the ezrin mutant T567A caused a significant reduction in the number of protrusions, indicating that the phosphorylation of T567 is required for protrusion formation (Figure 8A).
Figure 8.
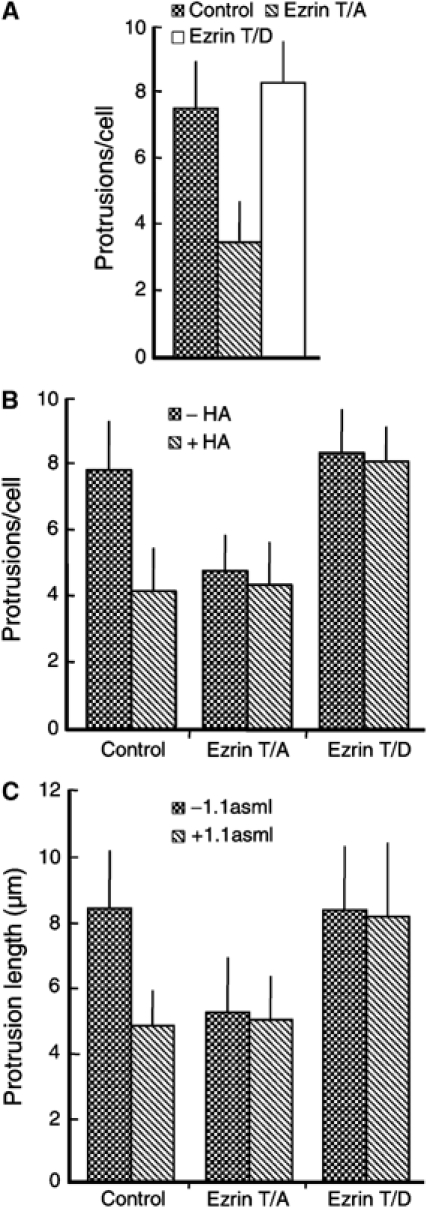
Phosphorylation-dependent activation of ERM proteins is critical for efficient protrusion formation. (A) Control LLC-PK1 cells and cells stably expressing the ezrin mutants T567A or T567D were infected with Listeria and processed for light microscopy. The expression of the ezrin mutant T567A, which mimics a constitutively inactive variant of this protein, reduces the number of protrusions compared to control LLC-PK1 cells and cells expressing the ezrin mutant T567D. (B) Control RT4 cells (WT) and cells stably expressing the ezrin mutants T567A (T/A) or T567D (T/D) were treated overnight with (+HA) or without (−HA) HA and then infected with Listeria. In control cells, the HA-induced activation of merlin reduces the number of protrusions per cell (see also Figure 5A). As expected, in RT4 cells expressing ezrin T567A, the number of protrusions induced by Listeria is reduced and this reduction is not enhanced by the merlin-driven displacement of ERM proteins from the plasma membrane. Conversely, the expression of ezrin T567D is able to counteract the reduction in the number of protrusions caused by merlin activation (compare WT with T567D). (C) Quantification of protrusion length for the experiment described in (B) showing that ezrin T567D counteracts the merlin-induced displacement of endogenous ERM proteins, thus resulting in the formation of protrusions of normal length, as determined by SEM. Error bars (A–C) indicate one standard deviation from the mean.
Constitutively active ezrin (T567D) restores efficient protrusion formation by antagonising merlin-induced displacement of ERM proteins from the plasma membrane
If active ERM proteins were required for supporting protrusion formation, then the expression of ezrin T567D (which constitutively localises at the cell membrane) would be sufficient to counteract the impairment of protrusion formation caused by the displacement of endogenous ERM proteins from the plasma membrane by active merlin. Accordingly, ezrin T567A would not be able to exert any counteracting function. In line with the above results (Figure 5C and D), the activation of merlin by either HA or the antibody 1.1asml caused a decrease in the number and length of protrusions induced by Listeria (Figure 8B and C). Ezrin T567D effectively counteracted the displacement of endogenous ERM proteins from the cell membrane, whereas ezrin T567A had no effect (Figure 8B).
Based on these observations, the expression of ezrin T567D should rescue normal morphological features of protrusions. In control RT4 cells, Listeria induced the formation of slender protrusions (Figure 9A; compare with Figure 6A), whereas the displacement of endogenous ERM from their membrane locations by active merlin (dox-treated RT4 tetNf2 cell line+HA) resulted in the formation of altered protrusions characterised by a thick and distorted distal portion (Figure 9B). According to the dominant-negative role of ezrin T567A (see Figure 8) and with respect to its decreased interaction with actin filaments (Gautreau et al, 2000), the protrusions formed in RT4 cells stably expressing this construct were similar to those observed in RT4 cells regardless of the state of merlin activation (Figure 9C and D; compare with Figure 6D). In contrast, the stable expression of ezrin T567D in RT4 tetNf2 cells clearly counteracted the displacement of endogenous ERM proteins by active merlin and fully restored the formation of normal protrusions (Figure 9E and F).
Figure 9.
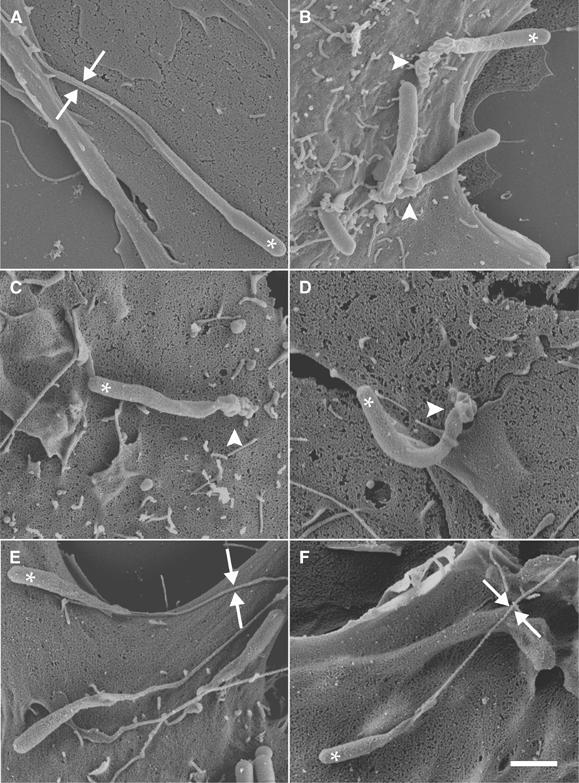
Expression of ezrin T567D in RT4 cells restores the normal morphological features of Listeria protrusion. Control RT4 cells (A) and cells treated overnight with the anti-CD44 monoclonal antibody 1.1asml (B) were infected with Listeria and processed for SEM. In control cells, Listeria induced the formation of normal protrusions (facing arrows in (A)), whereas in cells treated with the 1.1asml antibody, these bacteria formed short and distorted protrusions (arrowhead in (B); compare with Figure 6D). RT4 cells expressing the ezrin T567A (C, D) were treated as for (A, B). The expression of ezrin T567A in RT4 cells (C) caused the formation of distorted Listeria protrusions (arrowhead in (C)). The activation of merlin did not cause further changes in the morphological features of protrusions (arrowhead in (D); compare to (C)). In RT4 cells transfected with the constitutively active ezrin mutant T567D (E, F), Listeria induced the formation of normal protrusions (facing arrows in (E); compare with Figure 6A). The expression of ezrin T567D effectively counteracted the merlin-driven displacement of endogenous ERM proteins, thus resulting in the formation of protrusions having normal morphological features (facing arrows in (F)). White stars indicate the position of bacteria. Scale bar: 1 μm.
ERM proteins are essential for efficient cell-to-cell spread of Listeria
Since protrusion formation is essential for cell-to-cell spread of Listeria, it is conceivable that interfering with the recruitment of ERM proteins to protrusions would impair this process. Listeria cell-to-cell spread is best studied in cells that form polarised monolayers such as LLC-PK1, where it can invade discontinuous monolayers (e.g. discrete cellular islets) only through the baso-lateral membranes of their free bordering cells but is unable to directly infect cells residing in the centre of these islets (see Temm-Grove et al, 1994). As a consequence, the invasion of these centrally located cells can only take place via protrusion-dependent cell-to-cell spread.
To verify our hypothesis, LLC-PK1 cells were stably transfected with GFP-tagged CD44 tail (wild type or mutated) or with the ezrin mutant T567A. The expression of these constructs did not cause gross morphological changes of the cellular islets when compared to control cells by SEM (data not shown). Since the expression of the ezrin mutant T567D resulted in the formation of discontinuous monolayers often interrupted by holes (Gautreau et al, 2000) representing potential entry sites for Listeria, we did not test T567D in this assay. In control cells and in cells expressing the GFP-tagged CD44 tail mutant, Listeria invaded the more centrally located cells of an islet within 6 h after the beginning of the infection (Figure 10A and C). In contrast, the expression of either GFP-tagged CD44 tail wild-type or the ezrin mutant T567A slowed down the cell-to-cell spreading of Listeria, and the infection remained usually confined to the more peripheral cells of an islet (Figure 10B and D). The efficient cell-to-cell spread of Listeria correlated well with the formation of normal protrusions as determined by SEM (Figure 10a and c), whereas the impairment of cell-to-cell spread clearly correlated with deficient protrusion formation (Figure 10b and d).
Figure 10.
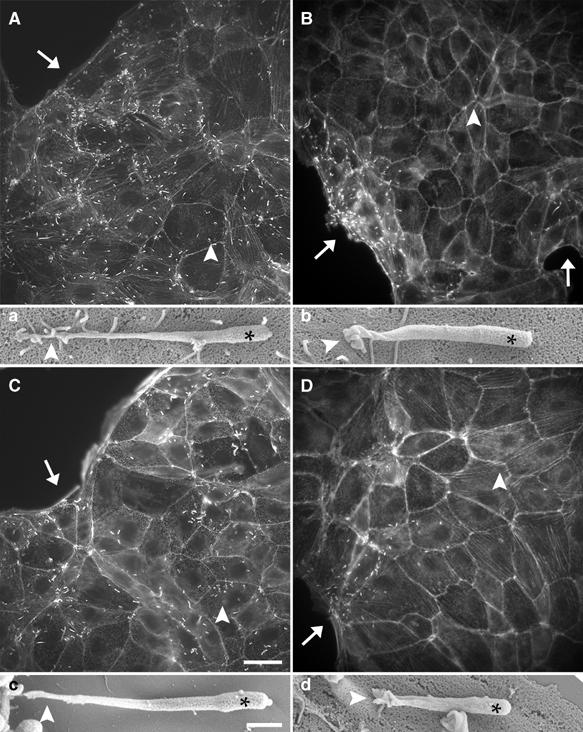
ERM proteins are essential for efficient cell-to-cell spread of Listeria. (A–D) Monolayer islet of untransfected LLC-PK1 cells or LLC-PK1 cells expressing GFP-tagged wild-type or mutated CD44 cytoplasmic tail or the ezrin mutant T567A were infected with Listeria. At 6 h after the beginning of the infection, cells were fixed and then stained with fluorescent phalloidin. In control cells (A) as well as in cells expressing the mutated CD44 tail (C), Listeria was able to invade the periphery (arrows in (A, C)) and the centre of a cellular islet (arrowheads in (A, C)). Conversely, in cells expressing wild-type CD44 tail or the ezrin mutant T567A, Listeria did not infect the cells in the centre of a cellular islet (arrowheads in (B, D)) but only the cells localised at the periphery of the islet (arrows in (B, D)). The ability of Listeria to spread from cell to cell correlated with the formation of normal protrusions (a, c), whereas the deficient cell-to-cell spread of these bacteria corresponded to the formation of defective protrusions (b, d). Scale bars: 20 μm (A–D); 1 μm (a–d).
Thus, the inhibition of ERM protein function severely impairs a fundamental step in the infection cycle of Listeria required by these bacteria for escaping the host immune response.
Discussion
Our findings highlight a novel strategy Listeria use to take advantage of actin–membrane interactions for successfully invading neighbouring cells. ERM proteins link Listeria actin tails to the plasma membrane and support the formation, and likely the stabilisation, of protrusions. In the absence of ERM function, both protrusion formation and the subsequent Listeria cell-to-cell spread are severely impaired, indicating that ERM proteins are essential for the effective progression of Listeria infection.
The impairment of protrusion formation is the direct consequence of interfering with ERM protein function and is not due to indirect morphological and cytoskeletal changes. This notion is supported by the observation that the ezrin mutant T567A impairs protrusion formation, but has no effect on cellular morphology (Gautreau et al, 2000). On the other hand, the ezrin mutant T567D, which is able to induce morphological changes in various cell lines (Gautreau et al, 2000; Pujuguet et al, 2003), can effectively rescue protrusion formation. Finally, the specific downregulation of ERM protein expression also results in the impairment of protrusion formation.
Listeria cell-to-cell spread requires that bacteria actively push the protrusions deep into the cytoplasm of adjacent cells in order to trigger their uptake (Robbins et al, 1999; our unpublished observations). Protrusion internalisation usually takes place after rather long periods of interaction with the recipient cell, and the half-life of actin filaments in protrusions is significantly longer than the average half-life of actin filaments in cytoplasmic actin tails (Robbins et al, 1999), suggesting that the structure of protrusions must be very stable in order to fulfil such process. Because the interaction of ERM proteins with the plasma membrane is essential for the stability of structures such as microvilli (Takeuchi et al, 1994; Crepaldi et al, 1997), we speculate that ERM proteins, by linking actin tails to the plasma membrane, confer stability onto forming protrusions resulting in a more efficient bacteria-induced propelling force against the resistance caused by the plasma membrane. Our hypothesis is supported by the observation that the displacement of ERM proteins from the membrane causes the formation of atypical protrusions that appear short, twisted and collapsed, indicating that the lack of ERM proteins not only reduces the number but also likely affects their stability. In addition, under the same experimental conditions, we found that Listeria cell-to-cell spread is severely impaired, suggesting that such atypical protrusions are not able to penetrate efficiently into recipient cells and to trigger their uptake.
The interaction of ERM proteins with CD44 is required for the formation and elongation of microvilli in various cell types (Yonemura and Tsukita, 1999) and the regulation of cell motility (Legg et al, 2002). Moreover, CD44 is also necessary for the internalisation of S. flexneri into cells (Lafont et al, 2002). Consistent with the role of CD44 in providing docking sites for ERM proteins at the membrane, we found that its expression in a CD44-negative melanoma cell line enhances protrusion formation. We note, however, that this effect does not correspond to the formation of morphologically normal protrusions, perhaps due to other intrinsic defects of these cells. Given the ability of Listeria to infect a wide range of cell types, it may be possible that CD44 and other ERM-binding membrane proteins such as CD43 and ICAM1–3 (Serrador et al, 1997, 1998; Yonemura et al, 1998) or other yet unknown membrane components contribute to protrusion formation in different cellular contexts.
The targeting of ERM proteins to the plasma membrane requires their activation, that is, ERM proteins acquire an open conformation that exposes previously masked binding sites for membrane components and actin filaments (see Bretscher et al, 2002). There is considerable evidence that phosphorylation of Thr residues in ERM proteins is critical for their activation (Nakamura et al, 1999; Fievet et al 2004). For example, the phosphorylation of Thr567 within ezrin plays a crucial role in the activation of ERM proteins, as indicated by the findings that an ezrin mutant in which Thr567 was exchanged by alanine to block its phosphorylation poorly associates with the actin cytoskeleton, impairs the development of tubular epithelial structures in LLC-PK1 cells (Gautreau et al, 2000) and inhibits the migration of cultured cells in wound closure assays (Ng et al, 2001). Accordingly, we found that the marked reduction of ERM protein phosphorylation in response to HA is accompanied by impaired protrusion formation. The unexpected result that the ezrin mutant T567A significantly impairs protrusion formation, although it has no detectable effect on the morphology and number of microvilli in LLC-PK1 cells (Gautreau et al, 2000), suggests that protrusion formation is more sensitive to expression levels of T567A than microvilli assembly. This possibility is consistent with the presence of several actin cytoskeleton-associated proteins such as villin and myosin I in the microvilli (Fath and Burgess, 1995) that can make these structures more stable and render them less sensitive to the expression of ezrin T567A.
Our findings provide novel clues about how Listeria exploit key ERM protein-dependent membrane–cytoskeleton interactions to propagate itself within, and avoid the immune response of, the host. They may also have implications in understanding the general mechanisms underlying the formation of membrane protrusions such as microvilli.
Materials and methods
Cloning of GFP-tagged proteins
Human ezrin cDNA was kindly provided by Professor Anthony Bretscher (Cornell University, Ithaca, NY). The NH2 (aa 1–296) and COOH (aa 475–585) fragments of ezrin were generated using the following primers (restriction sites are underlined):
NH2 ezrin: forward CCAAAGCTTGAAAATGCCGAAACCAATC, reverse GCTCCTCGATGGTCGACGGCTTCCTGC; COOH ezrin: forward GGAGCTGCAAAGCTTGATGACAGC, reverse CCTGGGTCGACTGTTGCAGGGCCTCG. NH2 ezrin and COOH ezrin were cloned into HindIII and SalI restriction sites of EGFP-N1 and EGFP-C2, respectively.
GFP-tagged CD44 cytoplasmic tail and its mutated variant were generated using the following primers: wild-type CD44 tail: forward CTAGTGGAAGCTTCGATGCGGCCATGAGTC, reverse CCAAAATAATGGGGTACCTGGTACACCCC; mutated CD44 tail: forward GGTTCCGCGAAGCTTCATGAGTCGAGC, reverse CCAAAATAATGGGGTACCTGGTACACCCC. PCR products were cloned into HindIII and KpnI restriction sites of EGFP-C2.
Bacterial culture, cell culture and infection
The wild-type weakly haemolytic L. monocytogenes strain EGD (serotype 1/2) was grown according to Sechi et al (1997). The growth conditions for PtK2 cells (ATCC, CCL 56), HeLa cells (ATCC, CCL 2), LLC-PK1 (kindly provided by Dr Monique Arpin, Curie Institute, Paris), RT4-D6-P2T and RPM-MC cells (kindly provided by Dr I Stamenkovic, MGH, Boston) have already been described by Sechi et al (1997), Gautreau et al (2000) and Morrison et al (2001). RPM-MC cell clones expressing high levels of full-length standard CD44 or its mutated form were isolated by limited dilution (Morrison et al, 2001).
Infection of cells was carried out according to Sechi et al (1997). The ability of L. monocytogenes to spread between adjacent cells was essentially analysed as described by Temm-Grove et al (1994). Prior to infection with Listeria, LLC-PK1 cells were grown for 48–72 h to allow the formation of discrete cellular islets. We analysed cellular islets, comparable in size, of control LLC-PK1 cells (untransfected and expressing CD44 tail mutated) and LLC-PK1 cells in which ERM proteins were inhibited (CD44 wild type and ezrin T567A). Moreover, we run the cell-to-cell spread assay for the same period of time (6 h). We analysed 40–50 cellular islets per each condition (three independent experiments). In control cells, Listeria invaded all the cells located in the centre of all islets we analysed. In cells where ERM protein function was inhibited, we never observed bacteria in the cells located in the centre of the islets. Occasionally (10–20% of total islets), we observed bacteria in the cells proximal (forming the second, and seldom the third, cellular row from the periphery of an islet) to the cells located at the periphery of the islets.
Cell transfection and selection
Cells were transiently transfected using FuGENE 6 (Boehringer Mannheim) as recommended by the supplier. PtK2 and HeLa cells expressing high levels of GFP-tagged wild-type or mutated CD44 cytoplasmic tail or NH2- and COOH-terminal ezrin domains, respectively, were sorted using the FACS Vantage cell sorter (Becton Dickinson). Because the tendency of LLC-PK1 cells to form aggregates precluded their sorting, LLC-PK1 cells expressing GFP-CD44 tail or its mutated variant were selected using 1.5 mg/ml G-418 as selection marker. Stable RT4 cell clones expressing doxycycline-inducible full-length merlin (RT4 tetNF2) and clones expressing NH2- or COOH-merlin were generated as described by Morrison et al (2001). RT4 cell clones stably expressing the ezrin mutant T567A or T567D were generated by cotransfection of these ezrin mutants with the pCEP4 hygromycin-resistant vector (Invitrogen; see Morrison et al, 2001).
Downregulation of ERM proteins by silencing RNA
The downregulation of ERM proteins in HeLa cells was carried out using short interfering RNA (siRNA) according to the procedure described by Elbashir et al (2001). The following siRNA duplexes were used (only the sense strand is reported). Set I: ezrin 5′-ccccaaagauuggcuuucc-3′ (position in the ORF 704–722), moesin 5′-aaaagccccggacuucguc-3′ (ORF 786–804), radixin 5′-gcaguuggaaagggcacaa-3′ (ORF 948–966); Set II: ezrin 5′-uccacuauguggauaauaa-3′ (ORF 140–158), moesin 5′-agaucgaggaacagacuaa-3′ (ORF 1058–1076); radixin 5′-cucgucugagaaucaauaa-3′ (ORF 752–775). As control, the following scrambled duplexes (derived from Set I duplexes) were used: ezrin 5′-ccgucacaucaauugccgu-3′, moesin 5′-acuagacgaaccgucgcuc-3′, radixin 5′-gaugcagcagcaugaagag-3′.
For the siRNA, HeLa cells (50% confluent in a 24-well dish) were treated with 60–100 pmol/well of each duplex. The scrambled duplexes were cotransfected with the EGFP-C1 plasmid (duplexes and plasmid ratio 10:1) to identify the cells that received the scrambled duplexes. The decrease in the level of ERM expression was determined by Western blot using the antibody C-19 (Santa Cruz, USA), which recognises all ERM proteins, after 36 h incubation with siRNA. HeLa cells were infected with Listeria after 30 h of treatment with siRNA (at this time point, most of ERM protein expression was downregulated, but the cells retained sufficient viability to be used for infection assays). Since cells that were negative to ERM labelling were never infected, Listeria protrusions were quantified in cells in which the expression of ERM proteins was as low as possible, as judged by antibody labelling.
Supplementary Material
Supplementary Fig 1
{kind=link}
Supplementary Fig 2
{kind=link}
Supplementary Fig 3
{kind=link}
Supplementary Movie 1
Supplementary Movie 2
Supplementary Data
Acknowledgments
We thank Professor Monique Arpin for critical reading of the manuscript and suggestions and for providing LLC-PK1 cell lines and cDNAs for the ezrin mutants T567A and T567D. We thank Professor Anthony Bretscher and Professor Claire Isacke for providing the cDNA for human ezrin and for CD44 tail domains, respectively. We also thank Brigitte Denker, Petra Hagendorff and Lothar Gröbe for technical assistance. JW was supported by the Deutsche Forschungsgemeinschaft and by the Fonds der Chemischen Industrie. HM and PH were supported by the Association for International Cancer Research (grant ref. 01-144).
References
- Algrain M, Turunen O, Vaheri A, Louvard D, Arpin M (1993) Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane-cytoskeletal linker. J Cell Biol 120: 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva MR, Litman P, Huang L, Ichimaru E, Furthmayr H (1999) Disruption of dynamic cell surface architecture of NIH3T3 fibroblasts by the N-terminal domains of moesin and ezrin: in vivo imaging with GFP fusion proteins. J Cell Sci 112: 111–125 [DOI] [PubMed] [Google Scholar]
- Bretscher A, Edwards K, Fehon RG (2002) ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 3: 586–599 [DOI] [PubMed] [Google Scholar]
- Cossart P, Pizarro-Cerda J, Lecuit M (2003) Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol 13: 23–31 [DOI] [PubMed] [Google Scholar]
- Crepaldi T, Gautreau A, Comoglio PM, Louvard D, Arpin M (1997) Ezrin is an effector of hepatocyte growth factor-mediated migration and morphogenesis in epithelial cells. J Cell Biol 138: 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cywes C, Wessels MR (2001) Group A Streptococcus tissue invasion by CD44-mediated cell signalling. Nature 414: 648–652 [DOI] [PubMed] [Google Scholar]
- De Joussineau C, Soule J, Martin M, Anguille C, Montcourrier P, Alexandre D (2003) Delta-promoted filopodia mediate long-range lateral inhibition in Drosophila. Nature 426: 555–559 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Eldridge R (1981) Central neurofibromatosis with bilateral acoustic neuroma. Adv Neurol 29: 57–65 [PubMed] [Google Scholar]
- Fath KR, Burgess DR (1995) Microvillus assembly. Not actin alone. Curr Biol 5: 591–593 [DOI] [PubMed] [Google Scholar]
- Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, Arpin M (2004) Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol 164: 653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht F, Way M (2001) Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol 11: 30–38 [DOI] [PubMed] [Google Scholar]
- Gary R, Bretscher A (1995) Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol Biol Cell 6: 1061–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau A, Louvard D, Arpin M (2000) Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J Cell Biol 150: 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry MD, Gonzalez Agosti C, Solomon F (1995) Molecular dissection of radixin: distinct and interdependent functions of the amino- and carboxy-terminal domains. J Cell Biol 129: 1007–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafont F, Tran Van Nhieu G, Hanada K, Sansonetti P, van der Goot FG (2002) Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44–IpaB interaction. EMBO J 21: 4449–4457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legg JW, Isacke CM (1998) Identification and functional analysis of the ezrin-binding site in the hyaluronan receptor, CD44. Curr Biol 8: 705–708 [DOI] [PubMed] [Google Scholar]
- Legg JW, Lewis CA, Parsons M, Ng T, Isacke CM (2002) A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat Cell Biol 4: 399–407 [DOI] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, Carlier MF (1999) Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature 401: 613–616 [DOI] [PubMed] [Google Scholar]
- Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, Gutmann DH, Ponta H, Herrlich P (2001) The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev 15: 968–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Huang L, Pestonjamasp K, Luna EJ, Furthmayr H (1999) Regulation of F-actin binding to platelet moesin in vitro by both phosphorylation of threonine 558 and polyphosphatidylinositides. Mol Biol Cell 10: 2669–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Parsons M, Hughes WE, Monypenny J, Zicha D, Gautreau A, Arpin M, Gschmeissner S, Verveer PJ, Bastiaens PI, Parker PJ (2001) Ezrin is a downstream effector of trafficking PKC–integrin complexes involved in the control of cell motility. EMBO J 20: 2723–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggli V, Andreoli C, Roy C, Mangeat P (1995) Identification of a phosphatidylinositol-4, 5-bisphosphate-binding domain in the N-terminal region of ezrin. FEBS Lett 376: 172–176 [DOI] [PubMed] [Google Scholar]
- O'Riordan M, Portnoy DA (2002) The host cytosol: front-line or home front? Trends Microbiol 10: 361–364 [DOI] [PubMed] [Google Scholar]
- Pearson MA, Reczek D, Bretscher A, Karplus PA (2000) Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell 101: 259–270 [DOI] [PubMed] [Google Scholar]
- Pestonjamasp K, Amieva MR, Strassel CP, Nauseef WM, Furthmayr H, Luna EJ (1995) Moesin, ezrin, and p205 are actin-binding proteins associated with neutrophil plasma membranes. Mol Biol Cell 6: 247–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Auerbuch V, Glomski IJ (2002) The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol 158: 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujuguet P, Del Maestro L, Gautreau A, Louvard D, Arpin M (2003) Ezrin regulates E-cadherin-dependent adherens junction assembly through Rac1 activation. Mol Biol Cell 14: 2181–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczek D, Berryman M, Bretscher A (1997) Identification of EBP50: A PDZ-containing phosphoprotein that associates with members of the ezrin–radixin–moesin family. J Cell Biol 139: 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JR, Barth AI, Marquis H, de Hostos EL, Nelson WJ, Theriot JA (1999) Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J Cell Biol 146: 1333–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi AS, Wehland J, Small JV (1997) The isolated comet tail pseudopodium of Listeria monocytogenes: a tail of two actin filament populations, long and axial and short and random. J Cell Biol 137: 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrador JM, Alonso-Lebrero JL, del Pozo MA, Furthmayr H, Schwartz-Albiez R, Calvo J, Lozano F, Sanchez-Madrid F (1997) Moesin interacts with the cytoplasmic region of intercellular adhesion molecule-3 and is redistributed to the uropod of T lymphocytes during cell polarization. J Cell Biol 138: 1409–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrador JM, Nieto M, Alonso-Lebrero JL, del Pozo MA, Calvo J, Furthmayr H, Schwartz-Albiez R, Lozano F, Gonzalez-Amaro R, Sanchez-Mateos P, Sanchez-Madrid F (1998) CD43 interacts with moesin and ezrin and regulates its redistribution to the uropods of T lymphocytes at the cell–cell contacts. Blood 91: 4632–4644 [PubMed] [Google Scholar]
- Skoudy A, Nhieu GT, Mantis N, Arpin M, Mounier J, Gounon P, Sansonetti P (1999) A functional role for ezrin during Shigella flexneri entry into epithelial cells. J Cell Sci 112: 2059–2068 [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Sato N, Kasahara H, Funayama N, Nagafuchi A, Yonemura S, Tsukita S (1994) Perturbation of cell adhesion and microvilli formation by antisense oligonucleotides to ERM family members. J Cell Biol 125: 1371–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temm-Grove CJ, Jockusch BM, Rohde M, Niebuhr K, Chakraborty T, Wehland J (1994) Exploitation of microfilament proteins by Listeria monocytogenes: microvillus-like composition of the comet tails and vectorial spreading in polarized epithelial sheets. J Cell Sci 107: 2951–2960 [DOI] [PubMed] [Google Scholar]
- Theriot JA, Mitchison TJ, Tilney LG, Portnoy DA (1992) The rate of actin-based motility of intracellular Listeria monocytogenes equals the rate of actin polymerization. Nature 357: 257–260 [DOI] [PubMed] [Google Scholar]
- Thomas L, Byers HR, Vink J, Stamenkovic I (1992) CD44H regulates tumor cell migration on hyaluronate-coated substrate. J Cell Biol 118: 971–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Portnoy DA (1989) Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109: 1597–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, Eldridge R, Kley N, Menon AG, Pulaski K, Haase VH, Ambrose CM, Munroe D, Bove C, Haines JL, Martuza RL, MacDonald ME, Seizinger BR, Short MP, Buckler AJ, Gusella JF (1993) A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 72: 791–800 [DOI] [PubMed] [Google Scholar]
- Tsukita S, Oishi K, Sato N, Sagara J, Kawai A (1994) ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J Cell Biol 126: 391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turunen O, Wahlstrom T, Vaheri A (1994) Ezrin has a COOH-terminal actin-binding site that is conserved in the ezrin protein family. J Cell Biol 126: 1445–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J (2001) Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14: 584–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Hirao M, Doi Y, Takahashi N, Kondo T, Tsukita S (1998) Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J Cell Biol 140: 885–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Tsukita S (1999) Direct involvement of ezrin/radixin/moesin (ERM)-binding membrane proteins in the organization of microvilli in collaboration with activated ERM proteins. J Cell Biol 145: 1497–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig 1
Supplementary Fig 2
Supplementary Fig 3
Supplementary Movie 1
Supplementary Movie 2
Supplementary Data