

Abstract
The endogenous 24-h (circadian) rhythms exhibited by the cyanobacterium Synechococcus elongatus PCC 7942 and other organisms are entrained by a variety of environmental factors. In cyanobacteria, the mechanism that transduces environmental input signals to the central oscillator of the clock is not known. An earlier study identified ldpA as a gene involved in light-dependent modulation of the circadian period, and a candidate member of a clock-entraining input pathway. Here, we report that the LdpA protein is sensitive to the redox state of the cell and exhibits electron paramagnetic resonance spectra consistent with the presence of two Fe4S4 clusters. Moreover, LdpA copurifies with proteins previously shown to be integral parts of the circadian mechanism. We also demonstrate that LdpA affects both the absolute level and light-dependent variation in abundance of CikA, a key input pathway component. The data suggest a novel input pathway to the circadian oscillator in which LdpA is a component of the clock protein complex that senses the redox state of a cell.
Keywords: circadian clock, cyanobacteria, LdpA, protein interaction, redox
Introduction
Circadian rhythmicity is an endogenously controlled oscillation of physiological activities that recurs with a periodicity of roughly 24 h (Bünning, 1973). These rhythms can be distinguished from oscillations that are externally driven by their persistence when the organism is isolated from daily environmental cycles. This intrinsic rhythmicity foresees daily changes in the natural environment, and, thereby, allows anticipatory behavior that increases the fitness of an organism (Ouyang et al, 1998; Beaver et al, 2002). Circadian rhythms have been described for a wide variety of organisms, from bacteria to mammals (Dunlap et al, 2004). Even though the specific components that control circadian rhythms are not conserved across evolutionary domains and kingdoms, three major functional divisions are shared by all circadian systems: a central oscillator, a synchronizing input pathway, and an output pathway that conveys temporal information to downstream processes. The rhythmicity is generated by the central oscillator, which, in the cyanobacterium Synechococcus elongatus, consists of at least the KaiA, KaiB, and KaiC proteins (Ishiura et al, 1998). It has been proposed that the Kai proteins, along with other components of the clock, form a periodosome, a large heteromultimeric protein complex that is crucial for sustaining circadian rhythms (Kageyama et al, 2003; Golden, 2004). The components of the central oscillator receive a variety of environmental signals through input components of the clock, including CikA (Schmitz et al, 2000), LdpA (Katayama et al, 2003), and Pex (Kutsuna et al, 1998). The central oscillator passes temporal information to the genome through output components of the circadian system, which in cyanobacteria include the group 2 sigma factors (Nair et al, 2002), CpmA (Katayama et al, 1999), and a kinase that is closely associated with the central oscillator, SasA (Iwasaki et al, 2000).
The intrinsic periodicity of circadian oscillation (free-running period) is entrained to precisely 24 h by cues from the natural environment. In addition, the phasing of circadian rhythms can be reset by a variety of environmental factors (McClung, 2001; Wang and Sehgal, 2002), as is the case for human recovery from jet lag (Parry, 2002). The most well-studied resetting factors are light and temperature. Other resetting stimuli include addition of a variety of metabolically active substances, feeding and activity (for mammals), and hydration of dried seeds (for plants) (Wang and Sehgal, 2002).
In cyanobacteria, the phase of circadian rhythms can be reset by at least light pulses (Aoki et al, 1997), dark pulses (Schmitz et al, 2000), addition of a photosynthesis inhibitor (Katayama et al, 2003), or short-term overexpression of KaiC (Ishiura et al, 1998). Although some components of the input pathway have been identified, the means of delivering the input signal to the oscillator of the clock is still not clear. The bacteriophytochrome-related kinase CikA was identified as a key player in the input pathway, because inactivation of cikA abolishes the ability of the clock to be reset by a dark pulse (Schmitz et al, 2000). However, it is still not known what kind of environmental signal CikA receives. It is possible that, despite the absence of conserved amino-acid residues in the putative chromophore-binding domain, CikA binds a chromophore and is able to sense light directly. However, assays for in vivo chromophore binding have been negative (Mutsuda et al, 2003). Alternatively, CikA may sense changes in the environment through interaction with an unknown partner, which, in one way or another, senses changes in the surroundings.
We recently identified the ldpA (light-dependent period A) gene of S. elongatus as encoding a new component of the input pathway of the cyanobacterial circadian clock (Katayama et al, 2003). Unlike strains that lack CikA, mutants that lack LdpA still reset the clock in response to a dark pulse. Rather, ldpA mutants are insensitive to a light gradient that in wild-type cells modulates the circadian period by lengthening it at lower light intensities (Aschoff, 1981; Katayama et al, 2003). The ldpA gene encodes a protein predicted to contain iron-sulfur centers, which implies involvement of the protein in redox reactions. It was concluded that ldpA is involved in regulation of the circadian period by sensing specific changes in electron transport that are dependent on light intensity. Here, we report that LdpA carries redox-active centers consistent with two [Fe4S4]2+/1+ clusters, and that it copurifies with proteins that have been shown previously to be crucial for circadian control. LdpA is required for light-dependent modulation of CikA abundance, and contributes to CikA sensitivity to the redox state of the cell. The data suggest a novel mechanism of transduction of an environmental signal to the clock, in which LdpA is a component of the clock complex that is able to sense the redox state of the cell.
Results
LdpA contains redox-active iron-sulfur clusters
The ldpA sequence predicts a protein that carries two iron-sulfur clusters, one of which was suggested to be an Fe4S4 cluster and the other an Fe3S4 cluster (Katayama et al, 2003). To confirm the presence of the expected cofactors in the protein, we analyzed anaerobically purified recombinant LdpA protein using UV–visible absorption and electron paramagnetic resonance (EPR). The purified protein fraction had a brown color that is attributed to iron-sulfur-containing proteins, which supports the prediction that LdpA has the expected cluster(s). The UV–visible spectrum indicated the presence of a broad absorption peak around 420 nm (Figure 1A), typical for iron-sulfur clusters. Addition of 2 mM sodium dithionite, a reducing agent, quenched absorbance in this region, demonstrating that the iron-sulfur clusters are redox sensitive.
Figure 1.
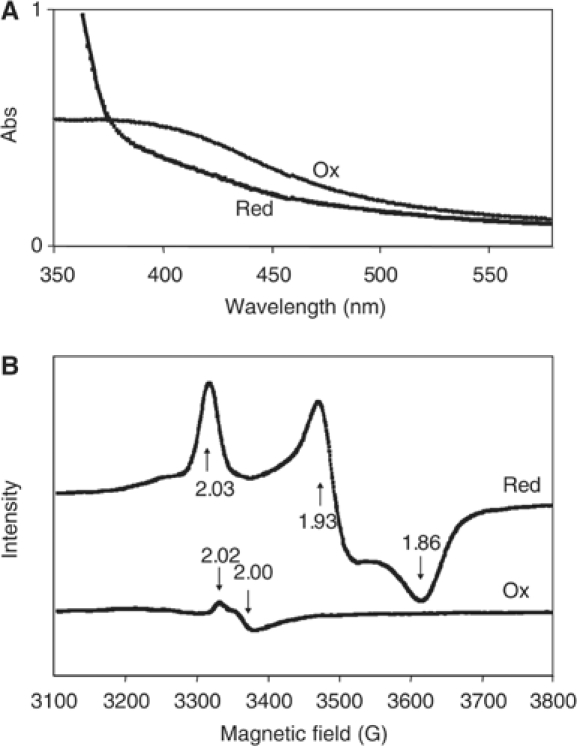
LdpA contains redox-active iron-sulfur clusters. (A) UV–visible absorption spectra of the purified recombinant protein. Ox, as-isolated reductant-free LdpA sample; Red, sample reduced with 2 mM dithionite. (B) 10 K X-band EPR spectra of the purified recombinant protein. Red, sample reduced with 2 mM dithionite; Ox, sample oxidized with thionin. Parameters are as described in Materials and methods.
To address the specific type(s) of cluster present in LdpA, EPR spectra of reduced and oxidized LdpA were recorded (Figure 1B). The spectrum of the reduced sample was dominated by a rhombic signal with g-values of 2.03, 1.93, and 1.86. Additional features of minor intensity also are apparent. The intensity of these spectral features collectively quantified to 2.4 spins/mol LdpA. Such gav=1.94 type signals are commonly associated with [Fe4S4]1+ clusters, and the spin intensity would suggest the presence of two such clusters in this protein rather than one Fe4S4 and one Fe3S4 cluster. Thionin-oxidized LdpA exhibited a signal with gav=2.01, typical of [Fe3S4]1+-containing proteins (Johnson et al, 1987, 1999); however, the intensity of this signal quantified to only 0.15 spins/mol LdpA, suggesting that this is a minor component, possibly arising from the decomposition of a small fraction of the Fe4S4 clusters.
Disruption or overexpression of ldpA affects period length in a kaiBC reporter strain
Previously, Katayama et al (2003) showed that disruption of ldpA shortens the circadian period of gene expression from two commonly used reporters, PpsbAI∷luxAB and PpurF∷luxAB. These promoter fusions represent two classes of gene regulation output from the circadian clock: psbAI is a prototypical class 1 gene, with a peak of expression at dusk, and purF represents a rare class whose peak expression is at dawn (Liu et al, 1996). Using either reporter, ldpA inactivation causes cells to become insensitive to a light gradient that would normally produce subtle changes in period length (Katayama et al, 2003). In the current work, the experiments were performed under a light intensity of 150 μE/m2 s, under which a slight shortening of period was observed in the previously described reporter strains (Katayama et al, 2003). To determine whether inactivation of ldpA also affects expression of the central clock genes, we inactivated ldpA in a strain that carries a fusion of bacterial luciferase reporter genes (luxAB) to the promoter of kaiBC, which naturally drives expression of the central oscillator components KaiB and KaiC. As shown in Figure 2, disruption of ldpA shortens period length in the kaiBC reporter strain by about 22 min: 24.47±0.09 h (n=6) for the mutant versus 24.85±0.22 h (n=6) for the wild-type strain.
Figure 2.
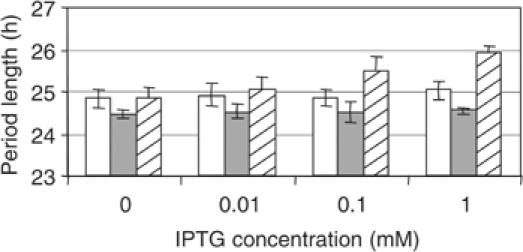
Disruption or overexpression of ldpA affects period length in a kaiBC reporter strain. Period length of the wild-type (AMC1004, open bars), ΩldpA (AMC1345, filled bars), and LdpA overexpression (AMC1347, hatched bars) reporter strains in the presence of the indicated concentrations of IPTG, as measured by bioluminescence assay (n=6, mean±SD).
We reasoned that if LdpA transmits a signal to the clock, then its overexpression, as well as its inactivation, should affect circadian rhythmicity. We constructed AMC1347, which carries an inactive native allele of ldpA (ΩldpA) and an ectopic allele, under control of an isopropyl β-D-thiogalactopyranoside (IPTG)-inducible promoter, that encodes His-tagged LdpA. In the absence of IPTG, the gene is expressed at levels sufficient to complement the short-period phenotype of the deletion mutant (Figure 2). Similar uninduced expression was previously shown to provide complementation of a cikA null strain by an ectopic cikA allele (Mutsuda et al, 2003). When increasing amounts of IPTG are added, period length is progressively increased, up to almost 26 h at 1 mM IPTG. Period lengths of the wild-type and the ΩldpA strains are not affected by IPTG (Figure 2). We conclude that the length of circadian period varies proportionately with the abundance of active LdpA. This range of periods corresponds to that exhibited by the wild-type strain under different light intensities (Katayama et al, 2003).
LdpA copurifies with circadian clock proteins
To identify a possible direct interaction of LdpA with the clock, we purified His-tagged LdpA from the cyanobacterium under low-stringency conditions, allowing copurification of proteins that interact with LdpA. Gross overexpression of clock proteins can cause arrhythmia in S. elongatus (Ishiura et al, 1998; Mutsuda et al, 2003). His-tagged LdpA was induced with 100 μM IPTG, the lowest concentration of inducer that yielded sufficient protein for purification; at this level of induction, circadian rhythmicity is robust and period is increased on average only 38 min (Figure 2). Normal clock function suggests that this level of expression does not indiscriminately affect clock proteins, thereby minimizing the probability of nonspecific binding of LdpA to other proteins. Because we did not know if LdpA interacts with any other protein at a particular time of the circadian cycle, we used unsynchronized cell cultures for affinity purification.
The total soluble protein fraction used for purification and the affinity-purified fraction that contains His-tagged LdpA and its putative interactants were subjected to immunoblot analysis using antisera raised against known circadian clock proteins. To identify proteins that may bind to the column nonspecifically, an extract from the wild-type strain, which does not carry the modified allele of ldpA, was subjected to the affinity purification procedure in parallel, and that eluate served as a negative control. LdpA copurified with KaiA (a central oscillator protein), CikA (a part of the input pathway of the clock), and SasA (a part of the output pathway that associates with the central oscillator) (Figure 3). These proteins were not detected in the wild-type fraction eluted at the same time under identical conditions. A group 2 sigma factor, RpoD4, not expected to be part of the periodosome, was not detected in the affinity-purified fractions that contain His-tagged LdpA. The current data do not address what proportions of the clock proteins are associated with LdpA.
Figure 3.
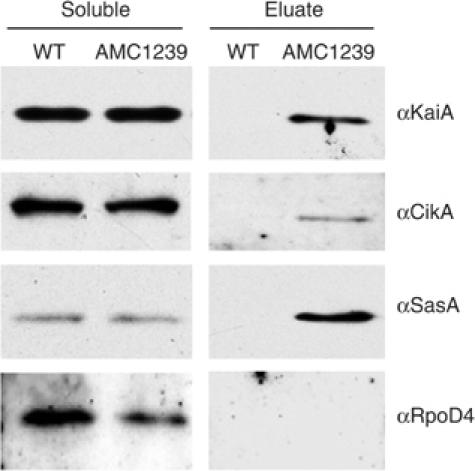
LdpA copurifies with circadian clock proteins. Immunoblot analysis of total soluble protein and affinity-purified fractions from wild-type (WT) and AMC1239 (His-tagged LdpA) strains. To induce LdpA production, 100 μM IPTG was added to unsynchronized cultures 24 h prior to collection. Cells were broken with glass beads, and the soluble fraction was purified. His-tagged LdpA was purified from the soluble fraction by affinity chromatography. Samples were subjected to SDS–PAGE and transferred to nitrocellulose membranes for immunoanalysis with the antisera indicated at the right of each panel. The immunoblots shown are from one of multiple independent experiments that gave essentially the same results. Because of differences in antisera sensitivities and film exposure times, the results are not quantitative among panels.
We were not able to show consistently the presence of KaiB or KaiC in affinity-purified LdpA eluates, although they were occasionally detectable. We expect that this inconsistency arises from the lower sensitivity of our KaiB and KaiC antisera relative to that of the CikA, KaiA, and SasA antisera. For example, overnight exposures are required to detect KaiC with its respective antiserum in whole-cell extract, whereas 30 s exposures are sufficient to detect CikA, KaiA, or SasA with their respective antisera (data not shown).
LdpA forms a complex with clock proteins in a circadian fashion
Previously, it was shown that a large heteromeric protein complex that contains at least four proteins involved in circadian control forms in a circadian fashion (Kageyama et al, 2003). To see if interaction between LdpA and clock components cycles in a similar manner, we analyzed soluble and affinity-purified fractions collected at different time points from the entrained strain containing His-tagged LdpA. The amount of LdpA in total soluble and affinity-purified fractions increased with time due to culture growth and presence of IPTG in the medium (Supplementary Figure S1A). The amount of CikA and KaiA in the total soluble fractions increased during the first 12–16 h and then decreased, consistent with circadian oscillation as previously shown for KaiA (Kageyama et al, 2003). However, CikA and KaiA associated with LdpA did not follow the circadian pattern (Supplementary Figure S1A and B). The portion of CikA associated with LdpA was at its highest level initially when the cells came out of the dark (LL0.1), but stayed constant afterwards and did not show a circadian peak. A more striking pattern of LdpA association was seen for KaiA. The peak of KaiA in the total soluble fraction occurred around LL16, whereas the subset of KaiA associated with LdpA peaked reproducibly at LL8 (n=3 experiments; Supplementary Figure S1B). Specifically at LL8, a fraction of the population of KaiA associated with LdpA runs on SDS–PAGE as a smear above the band normally identified as KaiA. In some samples purified at LL8, almost all of the KaiA signal ran as a single band slightly higher than KaiA purified with LdpA at other time points (Supplementary Figure S1C).
Interaction of LdpA with CikA and KaiA in the absence of other clock proteins
To identify possible interacting partners of LdpA, we performed affinity purification of LdpA from cells that lack major components of the clock such as KaiA, KaiB, KaiC, or CikA. The absence of any single central oscillator protein (KaiA, KaiB, or KaiC) disrupted the formation of the core clock protein complex (Kageyama et al, 2003), but did not affect copurification of CikA with LdpA (Supplementary Figure S2). Likewise, in the absence of CikA, KaiA still copurified with LdpA.
Stability of LdpA is affected by the redox state of the cell
The presence of redox-active iron-sulfur clusters in LdpA suggests a role in redox sensing in the cell; in plants and cyanobacteria the status of the plastoquinone (PQ) pool is an important redox sensor (Li and Sherman, 2000; Surpin et al, 2002). To identify a possible mechanism of LdpA redox sensitivity, we used two electron transport inhibitors that affect the redox state of the PQ pool (Trebst, 1980): 3-(3,4-dichlorophenyl)-1,1-dimethyl-urea (DCMU) inhibits electron transport from the photosystem II reaction center chlorophyll to PQ, and, thereby, causes oxidation of the PQ pool; 2,5-dibromo-3-methyl-6-isopropyl-p-benzoquinone (DBMIB) blocks electron transport from PQ to the cytochrome b6f complex, and, thereby, causes reduction (saturation with electrons) of the PQ pool. A brief treatment (15 min) of DCMU applied to cells that contain His-tagged LdpA, at a concentration that completely blocks photosynthetic electron flow (10 μM), did not affect levels of any of the proteins checked by immunoblot analysis (Figure 4A). However, a 15-min treatment with an inhibitory concentration of DBMIB (10 μM) caused disappearance of LdpA and CikA; the level of KaiA decreased slightly. The abundance of D1 (a key photosystem II protein) and PsaC (a photosystem I iron-sulfur-containing protein) did not decrease in the presence of DBMIB, indicating that the inhibitor effect is not indiscriminate, and, notably, does not apply to all iron-sulfur proteins or proteins involved in electron transport. A 15-min treatment with an inhibitor of translation, chloramphenicol, did not change the amount of LdpA (Figure 4B) and CikA (data not shown), indicating that disappearance of these proteins in the presence of DBMIB is due to decreased stability, and not to a decrease in the rate of synthesis.
Figure 4.
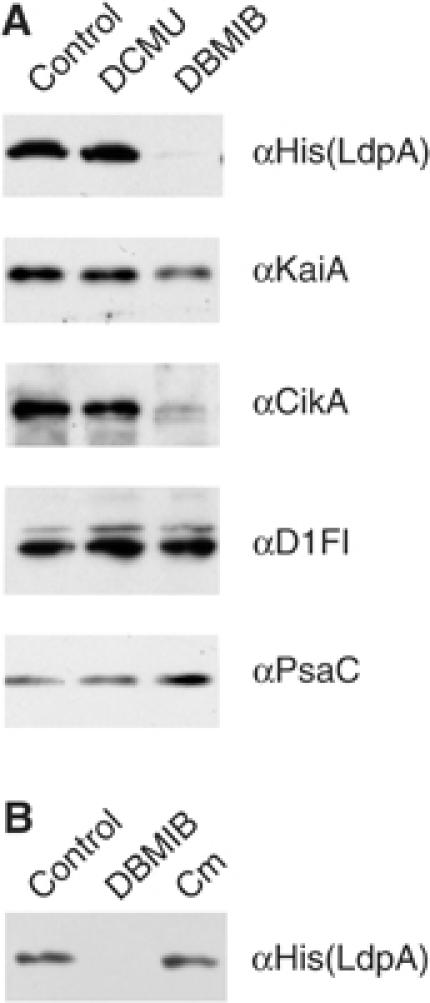
DBMIB affects stability of LdpA. Immunoblot analysis of whole-cell extract from AMC1239 (His-tagged LdpA). Antisera are indicated at the right of each panel. Whole-cell extract (10 μg) was loaded in each lane. (A) Cells were treated for 15 min with: ethanol (control, solvent used for inhibitors), DCMU (10 μM), or DBMIB (10 μM). (B) Cells were treated for 15 min with: ethanol (control, solvent used for inhibitors), DBMIB (10 μM), or chloramphenicol (Cm, 250 μg/ml).
LdpA affects CikA and KaiA abundance, and CikA sensitivity to cellular redox state
To see if LdpA is responsible for the decrease in CikA stability in the presence of DBMIB, we performed a dose-dependence experiment on the wild-type strain and a strain that lacks ldpA (Figures 5A and B). In the ldpA mutant, the steady-state level of CikA is reduced relative to that in wild type (Figure 5A); CikA stability was still responsive to DBMIB, but the sensitivity was reduced (as can be seen at 5 μM DBMIB). These data indicate that LdpA contributes to, but is not solely responsible for, sensitivity of CikA to DBMIB. The decrease in the amount of KaiA in the presence of DBMIB was not affected by the absence of LdpA (data not shown).
Figure 5.
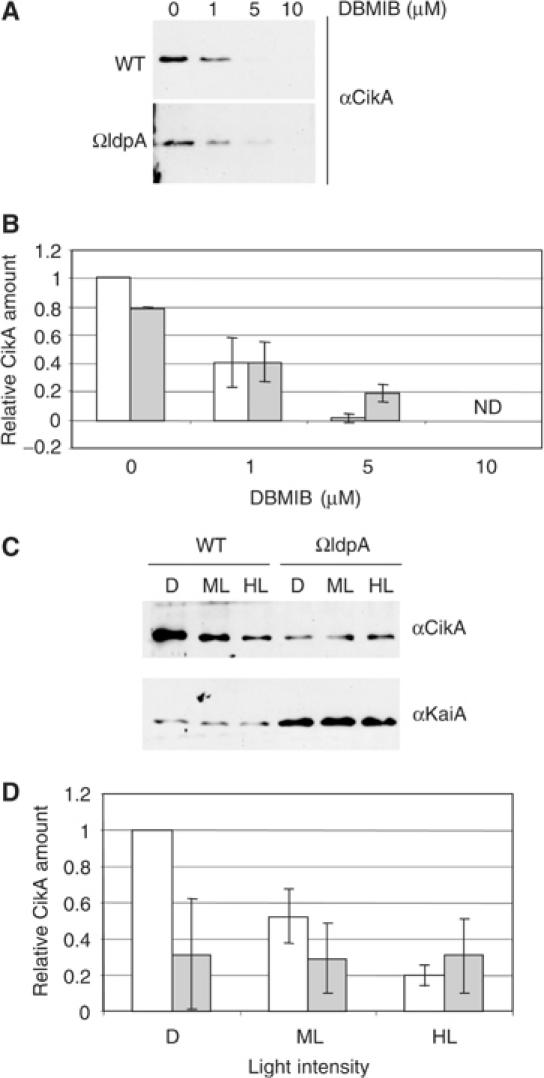
LdpA contributes to sensitivity of CikA to DBMIB, affects steady-state levels of both CikA and KaiA, and is necessary for light-dependent modulation of CikA abundance. (A, C) Immunoblot analysis of whole-cell extracts from the wild-type and ΩldpA (AMC1162) strains, prepared as described in Materials and methods. Antisera are indicated at the right of each panel. (A) Cultures were treated with DBMIB at the indicated concentrations for 15 min prior to harvesting. (C) Cultures were incubated for 6 h in darkness (D), or under medium (ML, 125 μE/m2 s) or high light intensity (HL, 250 μE/m2 s) prior to harvesting. (B, D) Quantification of data from experiments for which representative panels are shown in (A, C), respectively (n=3, mean±SD). Open bars, wild-type strain; filled bars, ΩldpA strain, ND, not detectable. The amount of CikA in the wild-type strain in the absence of DBMIB (B) or in the dark (D), to which other values are compared, is set at 1. The cultures for the upper panels (A, B) were grown under conditions different in aeration and light intensity from those for the lower panels (C, D); see Materials and methods for details.
DBMIB causes complete reduction of the PQ pool, an exaggeration of the condition cells face when placed in extremely high light. To see if exposure to high-light conditions affects the amount of native LdpA in cells, we attempted to raise antisera against LdpA. However, after multiple attempts, we were not able to generate antisera with a titer sufficient to detect LdpA in wild-type cyanobacterial protein fractions, even though the antisera can detect purified recombinant LdpA. To see if presence or absence of LdpA affects the amount of CikA and KaiA as a function of light intensity, we examined cultures exposed for 6 h to darkness, medium light intensity (125 μE/m2 s), and high light intensity (250 μE/m2 s). As shown in Figures 5C and D, the amount of CikA changes inversely with light intensity in the wild-type strain. However, when cells lack LdpA, CikA levels are relatively low and constant. CikA did not disappear under 250 μE/m2 s, a physiologically relevant light intensity that supports photosynthetic electron transport and would not entirely reduce the PQ pool as does 10 μM DBMIB. KaiA abundance is insensitive to light intensity in either strain, and is upregulated in the ldpA mutant.
Discussion
Light is considered to be the strongest stimulus for setting and entraining the circadian clock to keep it in synchrony with environmental cycles (McClung, 2001). In general, light is sensed by a photoreceptor, which passes the signal to the central oscillator of the clock by increasing or decreasing the amount of one of the components of the oscillator. In Neurospora crassa a flavin-binding LOV domain of WC-1 senses blue light (Cheng et al, 2003), which causes rapid activation of the protein, leading to an increase in the transcription rate of the frq gene, a key part of the oscillator (Crosthwaite et al, 1997). In Arabidopsis thaliana, the dark-dependent degradation of one of the central components of the clock, TOC1, requires its interaction with the ZTL protein, which, like WC-1, also contains a LOV domain (Mas et al, 2003). In Drosophila melanogaster, a cryptochrome homolog, dCry, senses light and affects degradation rates of Tim, a key component of the circadian oscillator (Ceriani et al, 1999).
Data presented here suggest that in S. elongatus, the clock receives at least some information about light not directly from a photoreceptor, but as a signal that reflects the redox state of the cell, which in photosynthetic organisms greatly depends on light intensity. We propose that by sensing the redox state of the PQ pool and changing the abundance of components of the clock, LdpA allows a fine-tuning of the length of the circadian period according to the metabolic state of the cell (Figure 6).
Figure 6.
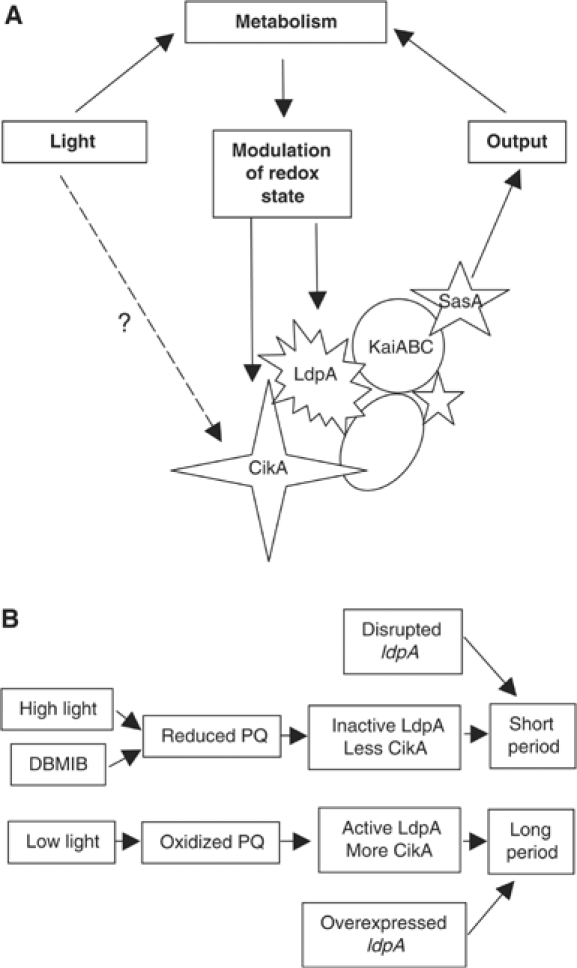
Proposed role of LdpA in the cyanobacterial clock. A detailed description is given in the text. (A) Possible role of LdpA in the periodosome, a protein complex that controls circadian rhythmicity. The specific placement of LdpA in the periodosome is preliminary and is based on data shown in Supplementary Figure S1. (B) Proposed mechanism of action of LdpA.
A number of physiological responses in photosynthetic organisms are caused by changes in redox state, such as post-transcriptional processes in the chloroplast of plants and chloroplast redox control of nuclear gene expression (Barnes and Mayfield, 2003; Pfannschmidt et al, 2003). In the purple nonsulfur photosynthetic bacterium Rhodobacter sphaeroides 2.4.1 expression of a variety of genes is regulated by light intensity and oxygen (Roh et al, 2004). In the cyanobacterium Synechocystis sp strain PCC 6803, the redox state of the PQ pool controls expression of at least 140 genes (Hihara et al, 2003) including psbA genes, which encode a key component of the photosynthetic apparatus (Li and Sherman, 2000). Photoreceptors, such as phytochromes or cryptochromes, provide a direct way to sense light by absorbing photons with chromophores, whereas redox state serves as an indirect way to measure the metabolic status of the cell, which in cyanobacteria not only depends on light intensity, but also on a variety of other environmental factors, such as nutrients and temperature.
In this work, we show that LdpA is a redox-sensitive protein that appears to contain two Fe4S4 clusters (Figures 1 and 4). Bioinformatics comparisons had suggested that the predicted cofactor-binding motifs of LdpA were most similar to those of 7Fe8S ferredoxins, with one canonical 4Fe4S-binding motif and one atypical motif (Katayama et al, 2003). However, re-examination of alignments with ferredoxins that contain two 4Fe4S clusters shows the presence of sufficient and appropriately placed ligands for two 4Fe4S clusters, as biophysical data support here (Figure 1). The presence of iron-sulfur clusters in a protein implies its involvement in redox reactions, which include but are not limited to electron transfer, and sensing of small molecules such as O2, reactive oxygen species, and nitric oxide (Beinert and Kiley, 1999). Because LdpA is involved in adjusting circadian period length according to light intensity (which affects the redox state of the PQ pool) and its stability is greatly decreased in the presence of DBMIB (which reduces the PQ pool), we suggest that LdpA measures PQ redox status (Figure 6B). Specifically, we propose that when the PQ pool is reduced (under high light intensity or in the presence of DBMIB), LdpA is inactive due to its degradation, and the clock is locked into a default ‘short-period mode.' Mutants that lack ldpA are also locked in that mode. However, when the PQ pool is oxidized (under low-light conditions), LdpA is active and interacts with the central oscillator, which lengthens the period. Similarly, overexpression of LdpA (which presumably increases the amount of active LdpA) also lengthens the period.
Reduction of the PQ pool by DBMIB also decreased the stability of CikA, a protein that is crucial for circadian phase resetting in S. elongatus (Figure 4). Figures 5A and B show that LdpA contributes to the redox sensitivity of CikA, but is not solely responsible for CikA's ability to sense redox status. Because CikA does not have any recognizable motifs that suggest direct redox sensitivity, it is likely that CikA interacts with a protein (other than LdpA) that transduces a signal from the PQ pool to the clock through CikA. In wild-type cells, CikA abundance varies inversely with light intensity. In the absence of LdpA, the amount of CikA is not light sensitive, and remains locked at the high-light level. This is consistent with the shortened circadian period observed in cikA and ldpA mutants, and in the wild-type strain under high light. When LdpA is absent, the absolute decrease of CikA abundance and loss of its modulation by light are sufficient to explain the ldpA mutant phenotype of loss of ‘light-dependent period.' We propose that LdpA plays a dual role with CikA: LdpA destabilizes CikA when cells are grown under higher light intensities and stabilizes it under lower light intensities, causing an increase of CikA in the dark (Figure 5). Moreover, in the absence of LdpA, KaiA abundance is increased (Figure 5C), which indicates that LdpA affects not only a component of the input pathway (CikA), but also the central oscillator. The N-terminal domain of KaiA is a pseudo-receiver, which has been proposed to serve as a protein–protein interaction module that is an input device of the circadian clock (Williams et al, 2002). The data suggest that LdpA, KaiA, and CikA are functionally linked as an interactive network that supplies environmental information to the circadian oscillator. However, during the 6 h course of the light intensity exposures, both transcriptional and post-transcriptional mechanisms, including pathways not involving LdpA, may regulate circadian clock protein levels.
The idea that the metabolic state of the cell influences the circadian oscillator has been suggested before. For example, the redox state of ubiquitous cellular NAD cofactors regulates DNA-binding affinity of heterodimers Clock:BMAL1 and NPAS2:BMAL1, major components of the circadian clock in mammals (Rutter et al, 2001). The reduced form of the cofactor (NADH) promotes DNA binding of the heterodimers, and the oxidized form (NAD) decreases it. This observation is suggested to explain the fact that the mammalian clock can be reset by feeding (Stokkan et al, 2001).
One of the key findings of this study is that LdpA copurifies with SasA and KaiA (Figure 3), which have been suggested to form, along with KaiB and KaiC, a periodosome, a large heteromeric complex whose formation is important for timekeeping (Kageyama et al, 2003; Golden, 2004). Although it is possible that LdpA interacts individually with KaiA, SasA, and CikA, there is no indication that CikA, KaiA, and SasA contain any common protein interaction motifs that would be responsible for direct binding of each with LdpA. Also, yeast two-hybrid and in vitro protein interaction assays between LdpA protein and KaiA, KaiB, KaiC, and CikA proteins were negative (data not shown). The analysis of LdpA interactants from strains that lack major circadian clock proteins (Supplementary Figure S2) indicates that in the absence of central oscillator proteins (KaiA, KaiB, or KaiC), LdpA still interacts with CikA. Similarly, in the absence of CikA, LdpA copurifies with KaiA, so CikA is not necessary for interaction of LdpA with the central oscillator of the clock. Unlike mutants that lack CikA, mutants that lack LdpA are able to reset their circadian clock in response to a dark pulse, which indicates that CikA is able to deliver input information to the central oscillator in the absence of LdpA (Katayama et al, 2003). The most likely explanation that considers all of the data is that KaiA and CikA interact with each other through a yet unknown ‘linker' protein(s), which also interacts with LdpA (Figure 6A).
Copurification data indicate that only part of the population of CikA is associated with LdpA, and that the affinity of CikA to LdpA (or LdpA-containing protein complex) does not change significantly during the circadian cycle (Supplementary Figure S1). The difference in phasing among the peaks of KaiA in the affinity-purified fraction, KaiA in the soluble fraction, and LdpA indicates that the affinity between KaiA and LdpA (or LdpA-containing protein complex) is higher at LL8 (Supplementary Figure S1). Part of KaiA in the affinity-purified fraction at LL8 appears to be chemically modified or covalently bound to another protein. This higher molecular weight form of KaiA was not detectable in the total soluble fraction. We propose that purification of the LdpA–KaiA (modified) complex concentrates to detectable levels a small fraction of the KaiA population that is involved in complex formation and that contains the modified KaiA molecules.
In conclusion, this study has identified a novel input pathway to the central oscillator of the cyanobacterial circadian clock. We have demonstrated that LdpA is a redox-sensitive intrinsic component of the clock protein complex, and we suggest that LdpA senses a metabolic state of the cell and passes this information to the clock to adjust the period length accordingly. We have shown that LdpA affects the absolute, and light-dependent and redox-dependent, abundance of CikA, a key player of the environmental input pathway, as well as the absolute level of the clock protein KaiA, which is also implicated in the input pathway (Williams et al, 2002). The data provide insight into the molecular mechanism of the phenomenon known as Aschoff's rule, which describes an inverse relationship of light intensity and period length in diurnal organisms (Aschoff, 1981). Although first described more than 20 years ago, to our knowledge this is the first characterization of its mechanism of action at the molecular level. It remains to be seen if other organisms, particularly higher plants, have similar means for adjusting their period according to light intensity.
Materials and methods
Bacterial strains, culture conditions, and bioluminescence assay
Unless otherwise specified, wild-type and mutant strains of the cyanobacterium S. elongatus PCC 7942 were grown in BG-11M medium (Bustos and Golden, 1991) as 100-ml cultures shaken constantly at 250 r.p.m. at 30°C under 150 μE/m2 s of fluorescent light as described previously (Mutsuda et al, 2003). AMC703 and AMC704 (JL Ditty, SR Canales, SB Williams, and SS Golden, in preparation), which contain an in-frame deletion of kaiB or kaiC, respectively, as well as kaiBC∷luc (NS2), were grown in the presence of chloramphenicol (Cm, 10 mg/l). Media were supplemented with gentamycin (Gm, 2 mg/l) for cyanobacterial strains that carry interrupting cassettes in ldpA or cikA, with spectinomycin (Sp, 20 mg/l) for strains that carry an ldpA overexpression construct, and with kanamycin (Km, 5 mg/l) for AMC1353 to select for an interrupting cassette in kaiA. Wild-type and AMC1004 (kaiBC∷luxAB, NS2.1, Km; psbAI∷luxCDE, NS2.2, Cm) strains from our lab collection were used as hosts for transgenic analysis (Mutsuda et al, 2003).
Escherichia coli strains DH10B and RY3080 (BL21DE3 slyD∷Km) hosted plasmids and were cultivated in liquid or on agar-solidified LB medium in the presence of appropriate antibiotics as described previously (Sambrook et al, 1989).
The bioluminescence assay was performed as described previously (Nair et al, 2002).
Construction of mutant strains and DNA manipulations
Basic DNA manipulation was performed by standard procedures (Sambrook et al, 1989) using enzymes from New England Biolabs (Beverly, MA). PCR amplification was performed with PfuTurbo Hotstart DNA polymerase (Stratagene, La Jolla, CA). Oligonucleotide synthesis and sequencing (to verify recombinant modified alleles) were performed by the Gene Technologies Laboratory (Texas A&M University). Cyanobacterial transformations were performed as described earlier (Golden et al, 1986).
Strains that lack ldpA were constructed by insertion of an inactivation cassette (Golden, 1988). Plasmid pAM2188 (ldpA∷Gm) (Katayama et al, 2003) was introduced into cyanobacterial strains, resulting in AMC1162 (ΩldpA in wild-type background) and AMC1345 (AMC1004, ΩldpA).
To control expression of LdpA in E. coli and cyanobacterial cells, plasmid pAM3140 was constructed. The coding region of ldpA was amplified from genomic DNA using synthetic oligonucleotides (5′-GCGAATTCATGAGTAGTGCCTCATTCC-3′ and 5′-CGCGGATCCTCAATGGTGATGGTGATGGTGGG GGGTTCGTAGGAG-3′). The second oligonucleotide, corresponding to the C-terminal coding region of the gene, contains sequence that encodes a 6-histidine affinity tag (bold) followed by a stop codon (italics). The resulting PCR product was digested with EcoRI and BamHI (restriction sites underlined in respective oligonucleotides) and cloned into pAM2991, a vector for overexpression of genes under control of the IPTG-inducible Ptrc promoter in both E. coli and S. elongatus. The resulting plasmid (pAM3140, Ptrc∷ldpAHis) was used to transform RY3080 for overexpression in E. coli (strain AM3362). The same plasmid was used to transform a variety of S. elongatus strains, resulting in His-tagged LdpA-overexpressing AMC1239 (AMC1162, ΩldpA in wild-type background), AMC1347 (AMC1345, a kaiB reporter in ΩldpA background), AMC1352 (wild-type background), AMC1354 (AMC703, ΔkaiB), and AMC1355 (AMC704, ΔkaiC). AMC1352 (Ptrc∷ldpAHis in wild-type background) was transformed with pAM2969 (ΩkaiA) and pAM3341 (ΔcikA), yielding AMC1353 (ΩkaiA, Ptrc∷ldpAHis) and AMC1356 (ΔcikA, Ptrc∷ldpAHis), respectively.
Expression and purification of His-tagged LdpA in E. coli
His-tagged LdpA was purified from AM3362 using Ni-NTA affinity matrix (Qiagen, Valencia, CA) according to the manufacturer's recommendations. The host strain RY3080 was used as a negative control to ensure that nonspecific proteins that may bind to the column do not affect the UV–visible or EPR spectra. A culture (1500 ml) was grown aerobically at 37°C to an optical density of 0.6 at 600 nm (OD600=0.6). The culture was cooled to room temperature, expression of the transgene was induced for 2 h with 100 μM of IPTG, and cells were collected (about 7 g of cells for each strain). Cell breakage and Ni-NTA affinity purification were carried out inside Vacuum Atmospheres (HE453-2) and M-Braun (Labmaster 130) glove boxes that contain less than 1 p.p.m. oxygen as monitored continuously (Teledyne model 310 oxygen analyzer). The eluted fractions were analyzed by SDS–PAGE and stained with Coomassie brilliant blue (Sambrook et al, 1989). The fraction that contained the highest amount of LdpA (40 μM) and a corresponding fraction from the host strain, which did not carry His-tagged LdpA, were subjected to UV–visible spectra and EPR analysis (Guigliarelli and Bertrand, 1999).
UV–visible spectra and EPR analysis
EPR spectra were collected on a Bruker EMX spectrometer (Rheinstetten/Karlsruhe, Germany), under the following parameters: microwave power, 80 mW; microwave frequency, 9.42 GHz; modulation frequency, 100 kHz; modulation amplitude, 12 G; sweep time, 167 s; time constant, 327 ms; gain, 5.02 × 104. Anaerobically isolated LdpA was reduced with 2 mM dithionite or oxidized with excess of thionin, as judged by the presence of a persistent pale blue color. EPR signal intensity was quantitated as described using a 1.0 mM copper EDTA standard solution (Shin and Lindahl, 1992). The eluate fraction from RY3080 was used as a negative control and was also subjected to the UV–visible and EPR spectral analyses, but this sample did not show any significant peaks in either assay.
Expression of His-tagged LdpA and its purification from S. elongatus under low-stringency conditions
To purify proteins that interact with LdpA, Ni-NTA affinity chromatography purification (Qiagen, Valencia, CA) was performed under low-stringency conditions. LdpA was induced by adding 100 μM IPTG to cultures for 24 h. For initial experiments (Figure 3), 400 ml of unsynchronized cultures of the wild-type and AMC1239 strains was collected at an optical density of OD750=0.7. Cells were harvested at 1500 g for 10 min, and the pellet was washed by resuspending in 2 ml of IA (interactants) lysis buffer (50 mM NaH2PO4, 5 mM NaCl, pH 7.8) and repeating the spin. Pellets were resuspended in 200 μl of IA lysis buffer, frozen at −80°C, and quickly thawed at 37°C to allow partial cell breakage. For the remainder of the procedure, samples were kept on ice and in the presence of 1 mM phenylmethylsulfonyl fluoride, a protease inhibitor. Sufficient glass beads (212–300 μm) were added to leave 2–3 mm of cell suspension above the level of settled beads. Cells were broken by vigorous vortex mixing of each tube for 10 min in cycles of mixing for 30 s and cooling on ice for 30 s. A 100 μl portion of IA lysis buffer was added. Tubes were briefly mixed and spun at 1000 g, and the supernatant fraction was collected. To separate bulk protein from the beads, an additional 300 μl of IA lysis buffer was added; tubes were briefly mixed by vortexing, spun at 1000 g, and the supernatant fractions were collected again. The beads were washed in this manner three more times; the supernatant fractions of each sample were combined and spun for 10 s at 1000 g to pellet residual glass beads. The resulting green supernatant fractions were subjected to centrifugation at 100 000 g for 30 min at 4°C. Protein concentration of the resulting bright blue supernatant of each sample (total soluble protein fraction) was measured using the Lowry assay (Lowry et al, 1951). Soluble fractions were adjusted to the same concentration (12 mg/ml) with IA lysis buffer, and 1 ml was loaded on a column prepacked with 1 ml of Ni-NTA-agarose and equilibrated with IA lysis buffer. The column was washed seven times with 10 ml of IA wash buffer (50 mM NaH2PO4, 5 mM NaCl, 20 mM imidazole, pH 7.8). The proteins still bound to the column after washing were eluted with 2 ml of IA elution buffer (50 mM NaH2PO4, 5 mM NaCl, 250 mM imidazole, pH 7.8), and 0.5 ml fractions were collected. A 10 μl portion from the second 0.5 ml fraction, which contained the highest concentration of His-tagged LdpA, and a corresponding fraction from the wild-type strain were loaded on a 12.5% SDS–PAGE gel (Sambrook et al, 1989). Samples containing 2 μg protein from the total soluble protein fractions were also analyzed by SDS–PAGE.
The samples used for Supplementary Figure S1 were prepared similar to the samples for Figure 3, except that 100 ml of cells was collected for each time point. Also, the amount of cells was not adjusted to the same protein concentration between the samples; instead, the collected cells were disrupted under identical conditions, yielding a set of samples with protein concentration progressively increasing with each following time point, as the culture grew. Samples used for Supplementary Figure S2 were prepared similar to the samples for Figure 3, except that 100 ml of cells was collected for each sample, yielding 1 ml of the soluble fraction, which was adjusted to 3 mg protein/ml before loading on a column.
For immunoblot analysis, proteins were electrophoretically transferred to nitrocellulose membranes using a Trans-blot SD semi-dry transfer cell (Bio-Rad, Hercules, CA). Blots were blocked with TBS buffer supplemented with 0.1% Tween 20 and 1% milk, and then incubated for 2 h at room temperature with primary antisera raised against KaiA (1:7000 dilution), KaiB (1:1000 dilution), KaiC (1:1000 dilution), CikA (1:7000 dilution), SasA (1:28 000 dilution), RpoD4 (1:1000 dilution), D1 Form I (D1FI, 1:2000), or PsaC (1:5000). Antisera were diluted in TBS buffer supplemented with 0.1% Tween 20 and 1% milk. After washing, blots were incubated for 1 h at room temperature with a 1:7000 dilution of peroxidase-conjugated goat anti-rabbit IgG (Calbiochem, San Diego, CA) except for blots incubated with anti-KaiC antisera, which were incubated with a 1:7000 dilution of peroxidase-conjugated goat anti-chicken IgY (Aves Labs, Tigard, OR). Blotting with Penta-His antibody (Qiagen) was performed according to the manufacturer's recommendations. The signal was visualized using SuperSignal West Pico chemiluminescent substrate (Pierce, Rockford, IL). Blots were exposed to X-ray film and quantification was performed using Adobe Photoshop (Adobe Systems, San Jose, CA) and LabWorks (UVP, Upland, CA).
Treatment with inhibitors and light
Prior to treatment with electron transport inhibitors, expression of His-tagged LdpA (AMC1239 strain) was induced by adding 100 μM IPTG to cultures for 24 h. When indicated, DCMU or DBMIB was added to cyanobacterial cultures to a final concentration of 10 μM, or chloramphenicol to a final concentration of 250 μg/ml. Because the inhibitors are dissolved in ethanol, the same amount of ethanol was added to control samples. A 100 ml portion of cells at OD750=0.7 was treated for 15 min and pelleted for 10 min at 1500 g at 4°C. Cells were resuspended in 100 μl of ice-cold IA lysis buffer and kept on ice. Cells were broken by vigorous vortex mixing in the presence of glass beads for 5 min in cycles of mixing for 30 s and cooling on ice for 30 s. Ice-cold IA lysis buffer (100 μl) was added, the samples were briefly mixed by vortexing and spun at 1000 g to remove the beads, and the supernatant fractions were collected yielding whole-cell extract. A sample (6 μg protein) was loaded per lane of SDS–PAGE. Immunoanalysis was performed as described above.
For Figures 5A and B, the samples were treated in a similar manner, except that only 1 ml of cell at OD750=0.45 was treated with DBMIB. Cells were collected and resuspended in 70 μl of ice-cold IA lysis buffer, kept on ice, and broken in the presence of glass beads for 3 min in cycles of shaking for 1 min and cooling on ice for 1 min using MiniBeadbeater 8 (BioSpec Products, Bartlesville, OK). A 150 μl portion of ice-cold IA lysis buffer was added, the samples were briefly mixed by vortexing and spun at 1000 g to remove the beads, and the supernatant fractions were collected yielding whole-cell extract. A sample (6 μl) of this extract was loaded on each lane of SDS–PAGE.
For Figure 5C and D, 10 ml of cells at OD750=0.45 was evenly distributed in a Petri dish and exposed for 6 h to darkness, medium light intensity (125 μE/m2 s), or high light intensity (250 μE/m2 s). Cells were brought to the same optical density (OD750=0.45), and 1 ml was collected. The samples were treated identically to those used in Figures 5A and B (see above).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Acknowledgments
We thank Dr R Young for providing the RY3080 strain, Dr K-P Michel for anti-CikA antisera, Dr H Iwasaki for anti-SasA antisera, Dr DA Bryant for anti-PsaC antiserum, and J Xu for excellent technical assistance. We also thank the clocks journal club, and especially Dr D Bell-Pedersen, for insightful suggestions. The Gene Technologies Laboratory (Texas A&M University) provided sequencing and oligonucleotide services. This work was supported by a grant from the National Institutes of Health (GM62419) to SSG and the Robert A Welch Foundation (A-1170) to PAL.
References
- Aoki S, Kondo T, Wada H, Ishiura M (1997) Circadian rhythm of the cyanobacterium Synechocystis sp. strain PCC 6803 in the dark. J Bacteriol 179: 5751–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschoff J (1981) Freerunning and entrained circadian rhythms. In Handbook of Behavioral Neurobiology: Biological Rhythms, Aschoff J (ed) pp 81–93. NY: Plenum Press [Google Scholar]
- Barnes D, Mayfield SP (2003) Redox control of posttranscriptional processes in the chloroplast. Antioxid Redox Signal 5: 89–94 [DOI] [PubMed] [Google Scholar]
- Beaver LM, Gvakharia BO, Vollintine TS, Hege DM, Stanewsky R, Giebultowicz JM (2002) Loss of circadian clock function decreases reproductive fitness in males of Drosophila melanogaster. Proc Natl Acad Sci USA 99: 2134–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinert H, Kiley PJ (1999) Fe-S proteins in sensing and regulatory functions. Curr Opin Chem Biol 3: 152–157 [DOI] [PubMed] [Google Scholar]
- Bünning E (1973) The Physiological Clock. NY: Springer-Verlag [Google Scholar]
- Bustos SA, Golden SS (1991) Expression of the psbDII gene in Synechococcus sp. strain PCC 7942 requires sequences downstream of the transcription start site. J Bacteriol 173: 7525–7533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceriani MF, Darlington TK, Staknis D, Mas P, Petti AA, Weitz CJ, Kay SA (1999) Light-dependent sequestration of TIMELESS by CRYPTOCHROME. Science 285: 553–556 [DOI] [PubMed] [Google Scholar]
- Cheng P, He Q, Yang Y, Wang L, Liu Y (2003) Functional conservation of light, oxygen, or voltage domains in light sensing. Proc Natl Acad Sci USA 100: 5938–5943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosthwaite SK, Dunlap JC, Loros JJ (1997) Neurospora wc-1 and wc-2: transcription, photoresponses, and the origins of circadian rhythmicity. Science 276: 763–769 [DOI] [PubMed] [Google Scholar]
- Dunlap JC, Loros JJ, DeCoursey PJ (eds) (2004) Chronobiology: Biological Timekeeping. Sunderland, MA: Sinauer Associates Inc. [Google Scholar]
- Golden SS (1988) Mutagenesis of cyanobacteria by classical and gene-transfer-based methods. Methods Enzymol 167: 714–727 [DOI] [PubMed] [Google Scholar]
- Golden SS (2004) Meshing the gears of the cyanobacterial circadian clock. Proc Natl Acad Sci USA 101: 13697–13698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden SS, Brusslan J, Haselkorn R (1986) Expression of a family of psbA genes encoding a photosystem II polypeptide in the cyanobacterium Anacystis nidulans R2. EMBO J 5: 2789–2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigliarelli B, Bertrand P (1999) Application of EPR spectroscopy to the structural and functional study of iron-sulfur proteins. Adv Inorg Chem 47: 421–497 [Google Scholar]
- Hihara Y, Sonoike K, Kanehisa M, Ikeuchi M (2003) DNA microarray analysis of redox-responsive genes in the genome of the cyanobacterium Synechocystis sp. strain PCC 6803. J Bacteriol 185: 1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura M, Kutsuna S, Aoki S, Iwasaki H, Andersson CR, Tanabe A, Golden SS, Johnson CH, Kondo T (1998) Expression of a gene cluster kaiABC as a circadian feedback process in cyanobacteria. Science 281: 1519–1523 [DOI] [PubMed] [Google Scholar]
- Iwasaki H, Williams SB, Kitayama Y, Ishiura M, Golden SS, Kondo T (2000) A KaiC-interacting sensory histidine kinase, SasA, necessary to sustain robust circadian oscillation in cyanobacteria. Cell 101: 223–233 [DOI] [PubMed] [Google Scholar]
- Johnson MK, Bennett DE, Fee JA, Sweeney WV (1987) Spectroscopic studies of the seven-iron-containing ferredoxins from Azotobacter vinelandii and Thermus thermophilus. Biochim Biophys Acta 911: 81–94 [DOI] [PubMed] [Google Scholar]
- Johnson MK, Duderstadt RE, Duin EC (1999) Biological and synthetic [Fe3S4] clusters. Adv Inorg Chem 47: 1–82 [Google Scholar]
- Kageyama H, Kondo T, Iwasaki H (2003) Circadian formation of clock protein complexes by KaiA, KaiB, KaiC, and SasA in cyanobacteria. J Biol Chem 278: 2388–2395 [DOI] [PubMed] [Google Scholar]
- Katayama M, Kondo T, Xiong J, Golden SS (2003) ldpA encodes an iron-sulfur protein involved in light-dependent modulation of the circadian period in the cyanobacterium Synechococcus elongatus PCC 7942. J Bacteriol 185: 1415–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama M, Tsinoremas NF, Kondo T, Golden SS (1999) cpmA, a gene involved in an output pathway of the cyanobacterial circadian system. J Bacteriol 181: 3516–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuna S, Kondo T, Aoki S, Ishiura M (1998) A period-extender gene, pex, that extends the period of the circadian clock in the cyanobacterium Synechococcus sp. strain PCC 7942. J Bacteriol 180: 2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Sherman LA (2000) A redox-responsive regulator of photosynthesis gene expression in the cyanobacterium Synechocystis sp. strain PCC 6803. J Bacteriol 182: 4268–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tsinoremas NF, Golden SS, Kondo T, Johnson CH (1996) Circadian expression of genes involved in the purine biosynthetic pathway of the cyanobacterium Synechococcus sp. strain PCC 7942. Mol Microbiol 20: 1071–1081 [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275 [PubMed] [Google Scholar]
- Mas P, Kim WY, Somers DE, Kay SA (2003) Targeted degradation of TOC1 by ZTL modulates circadian function in Arabidopsis thaliana. Nature 426: 567–570 [DOI] [PubMed] [Google Scholar]
- McClung CR (2001) Circadian Rhythms in Plants. Annu Rev Plant Physiol Plant Mol Biol 52: 139–162 [DOI] [PubMed] [Google Scholar]
- Mutsuda M, Michel KP, Zhang X, Montgomery BL, Golden SS (2003) Biochemical properties of CikA, an unusual phytochrome-like histidine protein kinase that resets the circadian clock in Synechococcus elongatus PCC 7942. J Biol Chem 278: 19102–19110 [DOI] [PubMed] [Google Scholar]
- Nair U, Ditty JL, Min H, Golden SS (2002) Roles for sigma factors in global circadian regulation of the cyanobacterial genome. J Bacteriol 184: 3530–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Andersson CR, Kondo T, Golden SS, Johnson CH (1998) Resonating circadian clocks enhance fitness in cyanobacteria. Proc Natl Acad Sci USA 95: 8660–8664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry BL (2002) Jet lag: minimizing it's effects with critically timed bright light and melatonin administration. J Mol Microbiol Biotechnol 4: 463–466 [PubMed] [Google Scholar]
- Pfannschmidt T, Schutze K, Fey V, Sherameti I, Oelmuller R (2003) Chloroplast redox control of nuclear gene expression—a new class of plastid signals in interorganellar communication. Antioxid Redox Signal 5: 95–101 [DOI] [PubMed] [Google Scholar]
- Roh JH, Smith WE, Kaplan S (2004) Effects of oxygen and light intensity on transcriptome expression in Rhodobacter sphaeroides 2.4.1. Redox active gene expression profile. J Biol Chem 279: 9146–9155 [DOI] [PubMed] [Google Scholar]
- Rutter J, Reick M, Wu LC, McKnight SL (2001) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293: 510–514 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Schmitz O, Katayama M, Williams SB, Kondo T, Golden SS (2000) CikA, a bacteriophytochrome that resets the cyanobacterial circadian clock. Science 289: 765–768 [DOI] [PubMed] [Google Scholar]
- Shin W, Lindahl PA (1992) Function and CO binding properties of the NiFe complex in carbon monoxide dehydrogenase from Clostridium thermoaceticum. Biochemistry 31: 12870–12875 [DOI] [PubMed] [Google Scholar]
- Stokkan KA, Yamazaki S, Tei H, Sakaki Y, Menaker M (2001) Entrainment of the circadian clock in the liver by feeding. Science 291: 490–493 [DOI] [PubMed] [Google Scholar]
- Surpin M, Larkin RM, Chory J (2002) Signal transduction between the chloroplast and the nucleus. Plant Cell 14 (Suppl): S327–S338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebst A (1980) Inhibitors in electron flow: tools for the functional and structural localization of carriers and energy conservation sites. Methods Enzymol 69: 675–715 [Google Scholar]
- Wang GK, Sehgal A (2002) Signaling components that drive circadian rhythms. Curr Opin Neurobiol 12: 331–338 [DOI] [PubMed] [Google Scholar]
- Williams SB, Vakonakis I, Golden SS, LiWang AC (2002) Structure and function from the circadian clock protein KaiA of Synechococcus elongatus: a potential clock input mechanism. Proc Natl Acad Sci USA 99: 15357–15362 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2